")
Back to Journals » Clinical Ophthalmology » Volume 16
Proposed Role for Internal Lens Pressure as an Initiator of Age-Related Lens Protein Aggregation Diseases
Authors Glazier AN
Received 7 April 2022
Accepted for publication 13 July 2022
Published 27 July 2022 Volume 2022:16 Pages 2329—2340
DOI https://doi.org/10.2147/OPTH.S369676
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Scott Fraser
Alan N Glazier
Optometry, KeplrVision, Rockville, MD, USA
Correspondence: Alan N Glazier, Email [email protected]
Abstract: The process that initiates lens stiffness evident in age-related lens protein aggregation diseases is thought to be mainly the result of oxidation. While oxidation is a major contributor, the exposure of lens proteins to physical stress over time increases susceptibility of lens proteins to oxidative damage, and this is believed to play a significant role in initiating these diseases. Accordingly, an overview of key physical stressors and molecular factors known to be implicated in the development of age-related lens protein aggregation diseases is presented, paying particular attention to the consequence of persistent increase in internal lens pressure.
Keywords: lens, protein aggregation, cataract, presbyopia, oxidation
Introduction
The physiological lens is a major component of the optical system of the mammalian eye. It functions optimally during youth when its flexibility and optical clarity are uncompromised. In order for the lens to maintain optimal function, it must remain transparent with a stable, high refractive index (averaging somewhere between 1.41 and 1.43) despite insults that threaten it, such as continued UV exposure and oxidative stress.1,2 The major proteins of the human lens responsible for maintenance of transparency and high refractive index are the Crystallin proteins.3,4 Crystallin proteins are synthesized only once and never replaced due to the absence of protein turnover. As the crystallin proteins begin an aging process even before birth and continue to accumulate various oxidative insults throughout life, the task of preserving high transparency and stable refractive index becomes problematical. The literature attributes oxidative insults caused by UV exposure as the major cause of post-translational modifications leading to protein aggregation.5,6 Protein aggregation of crystallins increases lens stiffness, a contributing factor to presbyopia and cataract, initiates light scattering7 and, ultimately, reduces lens transparency which defines the condition known as cataract, all of which have a major impact on public health5 100% of humans who survive into their 5th decade will be affected by lens stiffness manifesting as presbyopia. In the US alone, incidence of age-related nuclear cataract (ARN) is around 40% in those aged 75 years or older.8 Vision Loss due to cataract in the developing world remains ophthalmology’s major unsolved problem9 It is of interest that, while the most common type of cataract, ARN, occurs in an area of the lens which receives the highest lifetime exposure to UV, little scientific evidence exists showing UV plays a major role in inducing ARN.10 Clearly, there is more to the process than is understood.
The biochemical process underlying age-related lens protein aggregation diseases occurs over time11 yet the young eye has several defenses against oxidative damage that remains intact while the process ensues. The cornea has natural UV filters, a first line of defense, filtering out UV effectively to around age 40, yet damage to lens proteins begins years before these filters start to decline12–16 In cortical cataract (cataract that occurs in the cortex or periphery of the lens), light-induced oxidation is attributed as a cause, yet the lens cortex is protected by the iris, eyelids, pupil and aperture. Also, Glutathione, the major natural lens antioxidant, is gradually depleted from the nucleus while levels in the cortex remain normal,15,17–20 yet cortical cataract ensues even with Glutathione present. Because studies conclude it may only be after middle age that light promotes oxidation,21 based on the available research it is unlikely oxidation acts alone in initiating and contributing to the progression of age-related lens protein aggregation diseases; parallel processes are strongly associated with the cascade of damage that ensues from oxidative insults.22,23
While the cause of age-related lens protein aggregation diseases is believed to be multifactorial, there is debate as to which factors other than oxidation are significant comorbidities. Periodic elevation in temperature of the eye over time, chemical exposure, pressure and osmotic changes have been suggested to contribute to the process.24 As age-related lens protein aggregation diseases are a major cause of vision loss worldwide, and as the only treatment for cataract involves surgical removal of the opacified lens and its replacement with a prosthesis.25,26 There is a significant unmet medical need for therapeutic treatments for age-related lens protein aggregation diseases. Developing effective treatments requires an understanding of all factors that contribute to the initiation and continuation of the process. Because of this, lens protein aggregation is a subject of intense biophysical research. This Perspective presents the need to identify root causes in order to pave the way to develop drug targets to confront age-related lens protein aggregation diseases and reduce the global burden.25
The mammalian lens has a separate and distinct internal pressure than intraocular pressure (IOP). In young mouse lenses internal lens pressure is around 340 mmHg and in elderly mouse lens pressure almost doubles.27 This increase occurs linearly over time, yet very little research suggest it as having a significant impact on the structure and function of lens crystallins or and other lens proteins responsible for maintaining lens function and homeostasis such as gap junction proteins. As pressure in tissues elsewhere in the body has been shown to lead to precipitation and aggregation of proteins, cross-linking and other factors implicated in protein aggregation diseases20,28–34 it appears to have been largely overlooked as a factor in age-related lens protein aggregation diseases. I present an argument suggesting linearly increasing internal lens pressure is a contributor to stiffening, light scattering and opacification that characterize age-related lens protein aggregation diseases; that this pressure plays a significant yet understated, even overlooked factor in the multifactorial process behind initiation and progression of these diseases.
Lens Crystallin Proteins: Architecture, Stress and Damage
Crystallins are the predominant structural proteins of the lens.35 Their short-range ordered packing into a dense homogeneous phase is decisive for lens transparency.25 Lens nuclear proteins have the greatest longevity of any human proteins. They are synthesized only once and never replaced due to the absence of protein turnover.25 Lens crystallins must maintain an ordered and stable architecture despite the ongoing onslaught of insults they endure throughout a lifetime including continued UV exposure and other oxidative stress.1,2 In fact, they begin an aging process even before birth and continue to accumulate various insults throughout life.25 As lens fiber cells lack ribosomes and other organelles, the lens is not anatomically capable of repairing or replacing damaged crystallins.25,36,37 Unfortunately, the high crystallin concentrations needed to attain the glass-like refractive index required for the eyes optical system are conducive to an environment suitable for protein aggregation.25 Protein aggregation increases with age38 as once soluble lens proteins become insoluble (precipitate), leading to deterioration of lens architecture, lens stiffness (presbyopia), changes in refractive index, optical aberrations and, ultimately, opacity (cataract).39,40 Folding and unfolding of the crystallins is required to maintain lens transparency.41 There are three major types, α-Crystallin, β-Crystallin, and γ-crystallin.25,42,43 In their healthy form, crystallins are non-cross-linked and fold and unfold without being modified by oxidation. However, in response to stresses such as mutations, proteolysis, increase in disulfide bridges, glycosylation, phosphorylation, carbamylation, scission of tryptophan indole rings, deamidation of asparagine and glutamine, and racemization of aspartic acid residues,25,36,37 crystallins often lose their native folds and tend to denature and aggregate.
Chaperone molecules protect lens proteins from damage during folding and unfolding.24,25 α-Crystallin, a member of the heat shock family of proteins (HSPs), is a chaperone molecule.44,45 During cataractogenesis, α-crystallin becomes a water-insoluble cross-linked aggregate, resulting in a decrease of lens chaperone activity.12 As longevity of crystallin proteins is correlated with long-term retention of folded structure and is attributed to efficient capture and refolding by chaperones,42 protection provided by α-Crystallin against oxidative insult declines. The exhaustion of α-crystallin chaperone, a nonrenewable protein resource, is likely to be the limiting factor in causing almost all age-related cataracts.25 Currently, all proposed models of lens opacification include structural modifications within the crystallins.46
Post Translational Modifications of Lens Crystallin Proteins
Precipitation of lens proteins occurs secondary to chemical modifications known as post-translational modifications (PTMs). Proteins of mature fiber cells (MF) do not turnover, thus the lens nucleus contains the oldest proteins. Given the extreme age of nuclear crystallin proteins, a variety of PTMs accumulate.47,48 PTMs lead to presbyopia and cataract.49 The major lens PTMs include disulfide bonding, methionine oxidation, deamidation, phosphorylation, and proteolysis.20,48,50–53 Deamidation is a chemical reaction in which an amide group in the side chain of amino acids asparagine or glutamine is removed or converted to another functional group. Deamidation is the most common PTM affecting the human lens.52,54 Phosphorylation, the attachment of a phosphate group to a molecule or an ion, is the most common PTM of the α-crystallins.54,55 Crystallin proteolysis, the enzymatic breakdown of proteins or peptides into amino acids, is significantly increased in cataract as compared with young lenses.56–58
Antioxidation Protection in the Lens
Extensive oxidation of lens proteins begins at the lens fiber cell membrane.16,47,59 Glutathione, the major natural lens antioxidant, is vital for maintenance of transparency.13 Glutathione, which exists in unusually high concentration in the lens, detoxifies oxidants formed by UVA.60 Since corneal UV filters decrease at approximately 12% per decade, increasing amounts of UVA reach nuclear proteins of older lenses, forming reactive species which bind to proteins. Glutathione intercepts reactive species, minimizing damage. A barrier or bridge forms in the middle of the lens around middle-age restricting Glutathione from reaching the center of the lens, leaving proteins in this region susceptible to oxidation and PTMs.50,60 The genesis of age-related lens protein aggregation diseases may be traced to the onset of this barrier.61 A decrease in levels of Glutathione is a typical finding associated with nearly all experimental cataract.28,50,62,63
Lens Stress: Physical Forces Which Misfold Proteins
The initiators of age-related lens protein aggregation diseases are what misfolds proteins, not what oxidizes them once misfolded. It is proposed that some “stressors” acting over time unfolds proteins, increasing susceptibility to damage by oxidation that is an initiator of age-related lens protein aggregation diseases (Figure 1).
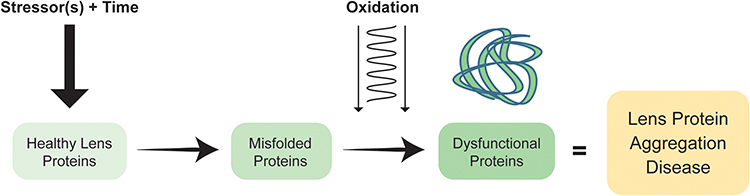 |
Figure 1 Schematic representation highlighting the role stressors play on the lens: over time, certain stressors initiate protein misfolding leading to dysfunctional proteins and the disease state. |
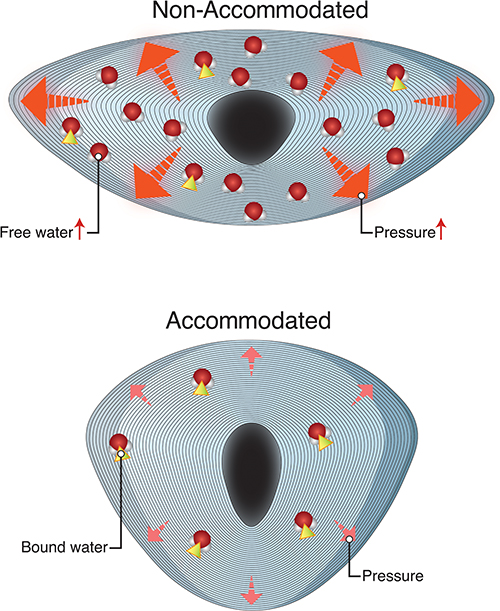 |
Figure 2 Representation of an accommodated and unaccommodated lens. Bottom figure – accommodated lens with most water molecules existing in the bound state. Top figure – unaccommodated lens fixed in its compressed configuration trapping free water within interstitium resulting in higher internal pressure. |
A review of literature suggests that increases in eye temperature lead to heat-based denaturing of lens proteins as a stressor contributing to the course of age-related protein aggregation diseases.
Heat
The absorption of infrared radiation by the lens results in development of cataracts.64 Goldman argues absorption of radiant energy by the iris conducting heat to the lens is responsible for cataracts.65–69 Studies suggest temperature of the anterior chamber varies by several degrees depending upon climate, with incidence of ARN greater in areas of higher temperature. However, structures anterior to the lens regulate temperature. Heat is absorbed by iris melanin and conducted away by iris blood circulation,70,71 conducted away by tear film evaporation, eyelid blood flow and minimized by reduction of the eye’s aperture through squinting and pupil regulation during high luminance situations. Also, natural convection of aqueous in the anterior chamber maintains temperature of the eye close to other body organs when subjected to thermal disturbance.69,72
Studies show exposure to even extreme heat causes minimal increases in eye temperature. Although few workers in an iron mill wore protection against heat, most were observed to “screw their eyelids up”, reducing corneal area exposed to radiant energy.69,73,74 While a radiation source of 1500 °C was responsible for increases in lens temperature of only 1–2 °C.52 A temperature increase of 1–2 °C is no more than what occurs in a normal eye when body-core temperature and ambient temperature are significantly above 37 °C.69
Based on these findings, researchers hypothesized if heat-induced denaturation of crystallin is not due to short-term exposure to high temperature, perhaps it is due to lower levels of elevated eye temperature over longer periods of time,65,75 yet the chaperone function of α-crystallin markedly improves with an increase in temperature,76 sufficiently compensating for the narrow range of temperatures to which the human eye is exposed.
Studies that suggest a relationship between age of onset of presbyopia and increase in ambient temperature66,77 failed to account for the concomitant increase in UV that occurs at latitudes approaching the equator.70,75 One study reported a positive relationship for cataract formation with increasing UV, latitude, and average hours of sunlight, however no relationship was found for increasing temperature.78
α-crystallin, a HSP, was identified in the lens subsequent to identification in cardiology where it was known to protect from heat shock.79 This may have led to the assumption that heat stress was a cause of age-related lens protein aggregation diseases. But HSP is a misnomer as HSPs protect crystallin protein misfolding from heat79 and other stresses including cold,80 UV,81 tissue remodeling,82 and pressure.83
Cumulative Exposure to UV
The literature reports cataracts appearing after exposure to UV in atomic bomb irradiation,84 whole-body irradiation,85,86 microwave,87 laser,88 x-ray89 and UV radiation.90,91 UV radiation, especially UVB, is an important risk for cortical cataracts,90,92–94 however, there is no evidence UVB is associated with ARN95–102 Based on available evidence, the following conclusions are drawn about exposure to UVB and cataract. (1) Exposure to UVB can cause lens opacities. (2) There is limited evidence exposure to solar UVB causes cortical opacities in humans. (3) Nuclear cataracts are not causally associated with exposure to solar UVB but there is an association between UVB and cortical opacities.100,103 The nucleus is directly along the visual axis and is exposed to UVB at a much higher amount than the cortex, yet UVB is implicated in cortical cataracts, not nuclear. If UVB cannot be implicated in cataract formation in the region of the lens receiving the most exposure to UVB and heat variations are not significantly at play, then some other stressors must contribute to initiation of age-related lens protein aggregation diseases.
Internal Lens Pressure
Once proteins fold, various conditions pose a threat to their integrity. Temperature, pressure, osmotic changes and chemicals interfere with protein folding.24 Specifically, pressure has been shown to lead to precipitation and aggregation of proteins, cross-linking and other factors implicated in age-related lens protein aggregation diseases.19,28–34 Under high pressure, proteins are penetrated by water.82 Studies have shown hyperbaric oxygen accelerates aging in the nuclear region of the guinea pig lens, causing loss of cytoskeletal proteins, damage to plasma membranes and formation of protein disulfides.40,104,105 Such modifications are similar to those that occur in the nuclei of aging human lenses.106
Lipid membranes are extremely sensitive to pressure. Protein oxidation in the lens has been shown to initiate at the lipid membrane. Products of lipid oxidation in the lens increase with both age and development of cataract. Studies show that membrane derangement occurs in human cataractous lenses, indicating lipid oxidation and/or changes in the lipid membrane may be a cause of lens opacification (Figure 2).106
Sources Responsible for Elevating Internal Lens Pressure
The space between fiber cells is the interstitium, and fluid present within is interstitial fluid. Interstitial fluid circulates through the lens. An intracellular gradient of hydrostatic pressure drives fluid from center towards surface epithelial cells, pushing outwards against the lens capsule.107 Mathematically, hydrostatic pressure is the change in volume divided by the change in pressure; the more fluid that filters into the interstitium, the greater the volume of the interstitial space (Vi) and the hydrostatic pressure within that space (Pi). In young human lenses, the intracellular hydrostatic pressure gradient ranges from ∼340 mmHg in central fiber cells to 0 mmHg in surface cells, increasing with age.27
Water within the lens exists in two states; free water (water not bound to crystallin proteins) and bound water (water bound to crystallin proteins). During accommodation, liquid water is expulsed from its bound state with lens crystallin, becoming free water, decreasing osmotic pressure99,108 and moving from the lens to the surroundings, increasing lens osmolarity. As the ability to accommodate is lost, internal lens pressure increases causing the free-to-bound water ratio to decrease. The ability of the lens to respond reversibly to pressure decreases along with the decrease in accommodation, and, when accommodation is lost, an increase in free water ensues.108 Thus, with no remaining accommodation, the lens is fixed in its unaccommodated, compressed configuration with lower tendency for water movement out of the interstitium and a higher internal pressure. With aging, the ability of the lens to compensate for increased hydrostatic pressures decreases.109 When a response to pressure is irreversible, released water accumulates in lakes (Syneresis) witnessed in incipient and mature cataract. Total water content is much higher in cataractous lenses than normal lenses.109
Compression via Constraint of the Lens Capsule
The lens, confined by an elastic capsule, exists in a state in which small increases in fluid volume lead to large increases in pressure. Large increases in tissue interstitial pressure can lead to tissue damage and cellular death.109 The lens capsule is a strong basement membrane110 capable of shaping the lens and its curvature through accommodation.111 It completely encloses the lens.112–115 The elastic modulus of the capsule must be sufficiently higher than the lens substance to allow forces applied by the ciliary muscles to mold the lens shape. In fact, it is approximately two thousand times higher than the lens cortex and nucleus it surrounds.111,116,117
During accommodation, zonules apply stress with both parallel (stretching) and perpendicular (compressive) components. These stresses are transformed into a uniform stress approximately perpendicular to the lens surface. The transition from unaccommodated to accommodated state, includes a reduction of stresses perpendicular to the lens surface.118,119 Under uniaxial load, capsular elastic moduli at 10% strain increase with age until approximately age 35. Once the age of 35 has been exceeded capsule load is maximized.120 Continual production of lens fiber cells in the environment constrained by the capsule, fixed in a contained volume, contributes to continually increasing crowding and compaction. Fluidic changes combined with increasing pressure from the production of proteins in a confined space increase hydrostatic pressure, and consequently, the syneretic process increases resistance within the lens. Over time, lens stiffness121 and elastic modulus116 increase resulting from the continual accession of fiber cells.122 As the elastic modulus of the lens substance increases, more force must be transmitted through the capsule to mold its shape. This is thought to be the primary cause of Presbyopia.118,119
Pressure Triggers Oxidative Damage of Gap Junctions
Gap junctions are intercellular channels that mediate direct cell-to-cell transfer of ions and metabolites.122,123 The lens, lacking blood vessels, must use non-vascular mechanisms to move nutrients and waste products into and out of fiber cells in the lens center. This is achieved in part by an extensive network of gap junctions that join cells of the lens into an ionic and metabolic syncytium. In the lens, hundreds of gap junctions come together to form Gap Junction Channels (GJCs).124–127 GJCs are composed of the protein connexin and found in arrays called plaques. GJCs allow small molecules to pass directly between cytoplasm of two connected cells.128–130
Maintenance of metabolic activities of interior cells of the lens depends on this network of GJCs.131–134 An electrical gradient carried predominantly by Na+ flows into the lens and through extracellular spaces.134,135 The gradient drives Sodium (Na+) and Calcium (Ca+) into fiber cells, through them, and outward cell to cell via GJCs. Flow is concentrated in the equatorial region of the lens where fibers are differentiating and is termed coupling conductance. Central to age-related changes in the lens is a reduction in fiber cell gap junction coupling conductance,27 presumably due to oxidative damage to lens connexins.136,137 As fiber cell connexins are sensitive to oxidative damage, a downregulation of gap junction coupling with age causes depolarization of the intracellular voltage and increases in intracellular concentrations of Na+ and Ca+. Depolarization and increased intracellular Na+ concentration reduce the transmembrane driving force of fiber cell membrane Na+-dependent transporters. Collectively, these effects compromise systems that protect intracellular proteins and allow increased oxidative damage to crystallins. This down regulation impacts all factors involved with lens circulation.27 There is an approximate 50% decrease in junctional coupling observed during lens fiber maturation.138
As coupling conductance decreases, forces that drive lens circulation must increase to maintain circulating fluxes. This includes increases in intracellular hydrostatic pressure, diffusion for Na+, and voltage gradient. With age there is a reduction in the fiber cell transmembrane electrochemical potential for Na+ entry. Consequently, Na+ influx decreases and since it drives water flow out of the lens, water flow decreases, increasing gradients for intracellular Na+ concentration, voltage, and hydrostatic pressure.27,139 Since hydraulic conductivity depends on the number of open GJCs, reduced hydraulic conductivity and increased lens radius with age both cause intracellular hydrostatic pressure, pi, to increase.27
Intracellular hydrostatic pressure in the lens is expected to vary in proportion to the group of parameters:12
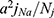 where j is the average density of fiber cell transmembrane influx of sodium (moles/cm), Nj is the number of open GJCs per area of fiber cell to cell contact (cm−2), and a is the lens radius (cm).140
where j is the average density of fiber cell transmembrane influx of sodium (moles/cm), Nj is the number of open GJCs per area of fiber cell to cell contact (cm−2), and a is the lens radius (cm).140
Based on this formula and knowing the number of open GJCs per area of fiber cell to cell contact decreases with age we can deduce that hydrostatic pressure in the lens increases with age.
A study27 presents compelling data demonstrating that, in mouse lenses in which gap-junction coupling is increased, central pressure is lower, whereas if gap-junction coupling is reduced, central pressure is higher but surface pressure always remains zero. Thus, reduction in gap junction coupling is a direct cause of age-related increase in intracellular hydrostatic pressure in the nucleus of the lens.137 Further, their data indicate that the gap junction coupling in differentiating fibers (DF) and MF decreases with age, indicating the total number of open GJCs also decreases with age. Reduced hydraulic conductivity and increased radius with age both cause intracellular hydrostatic pressure to increase with age.27
Intralenticular Circulation, Conductance, and Calcium Homeostasis
Gap junction coupling of interior fiber cells to surface cells is an essential component of Ca2+ homeostasis. At the surface, Ca-ATPase and Na/Ca exchange transport out of the lens. This sets up circulation of Ca2+, where the path to surface depends on the presence of gap junctions.141 The MF deep within the lens do not have Ca-ATPase activity or Na/Ca exchange to transport Ca2+ out, yet are permeable to Ca2+, hence Ca2+ is continuously leaking into these cells.139 Ca2+ accumulates in MF until diffusion to the surface balances equilibrium. A loss of gap junction coupling cuts off the MF zone from this circulation. With time, ion gradients in uncoupled cells dissipate, intracellular Ca2+increases, proteolysis of cytoplasmic proteins (crystallin and connexins) occurs128 and denatured proteins aggregate and form light-scattering elements responsible for nuclear opacity.142,143 Several studies show a correlation between elevated Ca2+ and cataracts.144–147
It is hypothesized that this system creates a well-stirred intracellular environment in which active transport by peripheral cells maintains a homeostatic environment for cells in the MF zone. The appearance of cleaved connexins between the peripheral shell of DF and the inner core of MF has been correlated to a change in coupling conductance. Loss of coupling cuts off the MF zone from this circulation.143 I suggest the loss of coupling conductance cutting off the MF zone is the cause of the Glutathione Bridge, suggested as one of the major factors in cataractogenesis.
Pressure Effects on Cytoskeletal Proteins
The cytoskeleton of lens proteins may play an important role in Ca2+-induced transparency loss. At moderately increased Ca2+ levels, opacification occurs without major degradation of intracellular proteins and may be the result of Ca2+-stimulated interactions between the membrane-cytoskeletal network and crystallins and may even be reversible.142 Pressure has been shown to accelerate age-related loss of nuclear cytoskeletal proteins.148 Furthermore, pressure has been pinpointed as a cause of the disulfide cross-linking of MIP26 and cytoskeletal proteins spectrin and vimentin, implicated in cataracts.106
Conclusions
There is little conclusive literature indicating that UV plays a major role in inducing ARN,16 and its possibly after middle age that light even begins to promote lens oxidation.10,14 Cataract forms in areas of the lens blocked from exposure to short-wavelength light, eliminating it as a cause of cortical cataracts. The literature indicates oxidative processes taking place in the nucleus are not present in the cortex yet cataracts still form. As nuclear Glutathione decreases, Glutathione levels in the outer cortex remain normal yet cortical cataracts still form.17–20 Furthermore, UV damages only misfolded proteins, thus plays an intermediate role in the formation of age-related lens protein aggregation diseases; it is not an initiator of misfolding. The literature indicates that heat-stress is an unlikely initiator of age-related lens protein aggregation diseases. Research has established that increased pressure shifts proteins toward dissociation, and dissociated proteins are implicated as contributing to the formation of presbyopia and cataracts,90 and pressure converts proteins into partially folded segments,32,149–154 exposing them to oxidative damage. This leads to precipitation, aggregation, cross-linking and other factors implicated in the progression of age-related lens protein aggregation diseases.20,28,31,33,106
Evidence is presented proposing the barrier that forms in the middle of the lens in middle age impeding Glutathione from protecting the lens nucleus50,60 is suggested to be a direct result of linear increasing internal pressure of the lens with age.
Lens stiffening, opacification, water and Ca+ accumulation leading to distension of intracellular spaces, plasma membrane disruption, increased levels of disulfide bonding, decreased levels of soluble proteins, and loss of nuclear cytoskeletal proteins106,148 seen in cataract are likely influenced by linearly increasing internal lens pressure, not heat stress.
The number of open GJCs decreases with age causing hydrostatic pressure to increase, decreasing sodium influx leading to trouble moderating the increase in hydrostatic pressure.27,139 Also, at the point where the lens has lost all ability to accommodate, it is fixed in its compressed configuration with lower water movement out of the interstitial spaces creating higher internal pressure.109 With age, an increase in free water ensues108 and in the lens, small increases in fluid volume lead to large increases in pressure leading to tissue damage and cellular death.
In summary, the continual production of lens cell fibers in the environment constrained by the lens capsule contributes to continual crowding and compaction. These changes generate pressure and resistance leading to increased stiffness, elastic modulus, increased light scattering, and a less pliability decreasing the ability of the lens to accommodate116,118 an attribute of presbyopia. As pressure mounts, metabolic activity is compromised distorting lens architecture causing further light scatter, refractive changes, ultimately contributing to sclerosis and opacification of cataract.
It is proposed a root cause of age-related lens protein aggregation diseases is constant increasing internal lens pressure, which, over time, interrupts healthy folding and unfolding of lens proteins, disrupts lens fluid circulation and eliminates pathways for Ca2+ to leave the lens, increasing opportunities for oxidation to trigger PTMs that gradually contribute to age-related lens protein aggregation diseases.
Disclosure
Dr Alan Glazier reports patent 17/537,088 pending to Unassigned Licensee, patent 17/666,424 pending to Unassigned Licensee, patent 17/713,691 pending to Unassigned Licensee, and patent 17/718,673 pending to Unassigned Licensee. The author reports no other conflicts of interest in this work.
References
1. Khandekar J, Bari J, Shrikant S, Chary KVR. Structural studies on the individual domains of human gS-crystallin and its G57W mutant unfolds mechanistic insights into childhood cataracts. Biochem Biophys Res Commun. 2019;517:499–506. doi:10.1016/j.bbrc.2019.07.094
2. Davies MJ, Truscott RJ. Photo-oxidation of proteins and its role in cata- ractogenesis, J. Photochem Photobiol, B. 2001;63:114–125. doi:10.1016/S1011-1344(01)00208-1
3. Graw J. Genetics of crystallins: cataract and beyond. Exp Eye Res. 2009;88:173–189. doi:10.1016/j.exer.2008.10.011
4. Khandekar J, Bari J, Dheeraj D, Shrikant S, Chary KVR. A molecular dynamics perspective to identify precursors to aggregation in human γS-Crystallin unravels the mechanism of childhood cataracts. J Phys Chem B. 2019;123:1038.
5. Khandekar J, Bari J, Shrikant S, Chary KVR. Structure of G57W mutant of human γS-crystallin and its involvement in cataract formation. J Struct Biol. 2019;205:72–78. doi:10.1016/j.jsb.2019.02.003
6. Bari KJ, Sharma S, Chary KVR. Sequence specific 1H, 13C and 15N resonance assignments of a cataract-related variant G57W of human γS-crystallin. Biomol NMR Assign. 2018;12:51–55. doi:10.1007/s12104-017-9779-y
7. Hains PG, Truscott RJW. Post-translational modification in the nuclear region of young, aged and cataract human lenses. J Proteome Res. 2007;6:3935–3943.
8. Klein B, Klein R, Lee K. The Beaver Dam eye study. Arch Ophthalmol. 1998;115(2):299–325.
9. Gillies M, Brian G, La Nauze J. Modern surgery for global cataract blindness: preliminary considerations. Arch Ophthalmol. 1998;16(1):90–92.
10. Taylor HR. Epidemiology of age-related cataract. Eye. 1999;13(Pt 3b):445–448. doi:10.1038/eye.1999.119
11. Ray NJ. Biophysical chemistry of the ageing eye lens. Biophys Rev. 2015;7(4):353–368. doi:10.1007/s12551-015-0176-4
12. Stadtman ER. Protein oxidation and aging. Science. 1992;257(5074):1220–1224. doi:10.1126/science.1355616
13. Bova LM, Sweeney MH, Jamie JF, Truscott RJ. Major changes in human ocular UV protection with age. Invest Ophthalmol Vis Sci. 2001;42(1):200–205.
14. Linetsky M, Ortwerth BJ. Quantitation of the reactive oxygen species generated by the UVA irradiation of ascorbic acid-glycated lens proteins. Photochem Photobiol. 1996;63(5):649–655. doi:10.1111/j.1751-1097.1996.tb05669.x
15. McCarty CA, De Paola C, Livingston PM, Taylor HR. Reliability of a food frequency questionnaire to assess dietary antioxidant intake. Ophthalmic Epidemiol. 1997;4(1):33–39. doi:10.3109/09286589709058059
16. Truscott RJ. Age-related nuclear cataract-oxidation is the key. Exp Eye Res. 2005;80(5):709–725. doi:10.1016/j.exer.2004.12.007
17. Augusteyn RC. Protein modification in cataract: possible oxidative mechanism. In: Duncan G, editor. Mechanisms of Cataract Formation in the Human Lens. New York, NY: Academic Press; 1981:71–115.
18. Duncan G, Wormstone IM, Davies PD. The aging human lens: structure, growth, and physiological behaviour. Br J Ophthalmol. 1997;81(10):818–823. doi:10.1136/bjo.81.10.818
19. Harding JJ, Crabbe MJC. The lens: development, proteins, metabolism and cataract. In: Davson H, editor. The Eye. Orlando, FL: Academic Press; 1984:207–492.
20. Simpanya MF, Ansari RR, Suh KI, Leverenz VR, Giblin FJ. Aggregation of lens crystallins in an in vivo hyperbaric oxygen Guinea pig model of nuclear cataract: dynamic light-scattering and HPLC analysis. Invest Ophthalmol Vis Sci. 2005;46(12):4641–4651. doi:10.1167/iovs.05-0843
21. Linetsky M, Ortwerth BJ. The generation of hydrogen peroxide by the UVA irradiation of human lens proteins. Photochem Photobiol. 1995;62(1):87–93. doi:10.1111/j.1751-1097.1995.tb05243.x
22. Brennan LA, Kantorow M. Mitochondrial function and redox control in the aging eye: role of MsrA and other repair systems in cataract and macular degenerations. Exp Eye Res. 2009;88(2):195–203. doi:10.1016/j.exer.2008.05.018
23. Vinson JA. Oxidative stress in cataracts. Pathophysiology. 2006;13(3):151–162. doi:10.1016/j.pathophys.2006.05.006
24. Narberhaus F. Alpha-crystallin-type heat shock proteins: socializing minichaperones in the context of a multichaperone network. Microbiol Mol Biol Rev. 2002;66(1):64–93. doi:10.1128/MMBR.66.1.64-93.2002
25. Khandekar J, Bari J, Shrikant Sharma J. A perspective on biophysical studies of crystallin aggregation and implications for cataract formation. J Phys Chem B. 2020;124:11041–11054. doi:10.1021/acs.jpcb.0c07449
26. Rao GN, Khanna R, Payal A. The global burden of cataract. Curr Opin Ophthalmol. 2011;22:4–9. doi:10.1097/ICU.0b013e3283414fc8
27. Gao J, Wang H, Sun X, et al. The effects of age on lens transport. Invest Ophthalmol Vis Sci. 2013;54(12):7174–7187. doi:10.1167/iovs.13-12593
28. Padgaonkar V, Giblin FJ, Reddy VN. Disulfide cross-linking of urea-insoluble proteins in rabbit lenses treated with hyperbaric oxygen. Exp Eye Res. 1989;49(5):887–899. doi:10.1016/S0014-4835(89)80047-8
29. Cinar S, Cinar H, Chan HS, Winter R. Pressure-sensitive and osmolyte-modulated liquid-liquid phase separation of eye-lens gamma-crystallins. J Am Chem Soc. 2019;141(18):7347–7354. doi:10.1021/jacs.8b13636
30. Kauzmann W. Thermodynamics of unfolding. Nature. 1987;325(6107):763–764. doi:10.1038/325763a0
31. Padgaonkar VA, Leverenz VR, Fowler KE, Reddy VN, Giblin FJ. The effects of hyperbaric oxygen on the crystallins of cultured rabbit lenses: a possible catalytic role for copper. Exp Eye Res. 2000;71(4):371–383. doi:10.1006/exer.2000.0887
32. Silva JL, Oliveira AC, Vieira TC, de Oliveira GA, Suarez MC, Foguel D. High-pressure chemical biology and biotechnology. Chem Rev. 2014;114(14):7239–7267. doi:10.1021/cr400204z
33. Varma SD, Chand D, Sharma YR, Kuck JF
34. Bridgman PW. The coagulation of albumin by pressure. J Biol Chem. 1914;19:511–512. doi:10.1016/S0021-9258(18)88287-4
35. Benedek GB, Chylack LT
36. Brennan LA, McGreal-Estrada R, Logan CM, Cvekl A, Menko AS, Kantorow M. BNIP3/NIX is required for elimination of mitochondria, endoplasmic reticulum and golgi apparatus during eye lens organelle-free zone formation. Exp Eye Res. 2018;174:173–184. doi:10.1016/j.exer.2018.06.003
37. Dawes LJ, Shelley EJ, McAvoy JW, Lovicu FJ. A role for hippo/YAP-signaling in FGF induced lens epithelial cell proliferation and fibre differentiation. Exp Eye Res. 2018;169:122–133. doi:10.1016/j.exer.2018.01.014
38. Moreau KL, King JA. Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol Med. 2012;18(5):273–282. doi:10.1016/j.molmed.2012.03.005
39. Feng J, Smith DL, Smith JB. Human lens beta-crystallin solubility. J Biol Chem. 2000;275(16):11585–11590. doi:10.1074/jbc.275.16.11585
40. Swamy MS, Abraham EC. Lens protein composition, glycation and high molecular weight aggregation in aging rats. Invest Ophthalmol Vis Sci. 1987;28(10):1693–1701.
41. De Maio A. Heat shock proteins: facts, thoughts, and dreams. Shock. 1999;11(1):1–12. doi:10.1097/00024382-199901000-00001
42. Bloemendal H, de Jong W, Jaenicke R, Lubsen NH, Slingsby C, Tardieu A. Ageing and vision: structure, stability and function of lens crystallins. Prog Biophys Mol Biol. 2004;86(3):407–485. doi:10.1016/j.pbiomolbio.2003.11.012
43. Bode C, Tolgyesi FG, Smeller L, Heremans K, Avilov SV, Fidy J. Chaperone-like activity of alpha-crystallin is enhanced by high-pressure treatment. Biochem J. 2003;370(Pt 3):859–866. doi:10.1042/bj20021097
44. Andley UP, Song Z, Wawrousek EF, Bassnett S. The molecular chaperone alphaA-crystallin enhances lens epithelial cell growth and resistance to UVA stress. J Biol Chem. 1998;273(47):31252–31261. doi:10.1074/jbc.273.47.31252
45. Bagchi M, Katar M, Maisel H. Heat shock proteins of adult and embryonic human ocular lenses. J Cell Biochem. 2002;84(2):278–284. doi:10.1002/jcb.10023
46. Das P, King JA, Zhou R. Aggregation of γ-crystallins associated with human cataracts via domain swapping at the C- Terminal β-strands. Proc Natl Acad Sci U S A. 2011;108:10514–10519. doi:10.1073/pnas.1019152108
47. Siew EL, Opalecky D, Bettelheim FA. Light scattering of normal human lens. II. Age dependence of the light scattering parameters. Exp Eye Res. 1981;33(6):603–614. doi:10.1016/S0014-4835(81)80100-5
48. Hanson SR, Hasan A, Smith DL, Smith JB. The major in vivo modifications of the human water-insoluble lens crystallins are disulfide bonds, deamidation, methionine oxidation and backbone cleavage. Exp Eye Res. 2000;71(2):195–207. doi:10.1006/exer.2000.0868
49. Roosen-Runge F, Gulotta A, Bucciarelli S, et al. Crowding in the eye lens: modeling the multisubunit protein beta-crystallin with a colloidal approach. Biophys J. 2020;119(12):2483–2496. doi:10.1016/j.bpj.2020.10.035
50. Lampi KJ, Ma Z, Hanson SR, et al. Age-related changes in human lens crystallins identified by two-dimensional electrophoresis and mass spectrometry. Exp Eye Res. 1998;67(1):31–43. doi:10.1006/exer.1998.0481
51. Bloemendal H. The Lens Proteins. In. Molecular and Cellular Biology of the Eye Lens. New York: Wiley; 1981.
52. Hains PG, Truscott RJ. Post-translational modifications in the nuclear region of young, aged, and cataract human lenses. J Proteome Res. 2007;6(10):3935–3943. doi:10.1021/pr070138h
53. Wilmarth PA, Tanner S, Dasari S, et al. Age-related changes in human crystallins determined from comparative analysis of post-translational modifications in young and aged lens: does deamidation contribute to crystallin insolubility? J Proteome Res. 2006;5(10):2554–2566. doi:10.1021/pr050473a
54. Aquilina JA, Benesch JL, Ding LL, Yaron O, Horwitz J, Robinson CV. Phosphorylation of alphaB-crystallin alters chaperone function through loss of dimeric substructure. J Biol Chem. 2004;279(27):28675–28680. doi:10.1074/jbc.M403348200
55. Hains PG, Truscott RJW. Proteomic analysis of the oxidation of cysteine residues in human age-related nuclear cataract lenses. Biochim Biophys Acta. 2008;1784(12):1959–1964. doi:10.1016/j.bbapap.2008.07.016
56. Rao G, Santhoshkumar P, Sharma KK. Anti-chaperone betaA3/A1(102-117) peptide interacting sites in human alphaB-crystallin. Mol Vis. 2008;14:666–674.
57. Santhoshkumar P, Udupa P, Murugesan R, Sharma KK. Significance of interactions of low molecular weight crystallin fragments in lens aging and cataract formation. J Biol Chem. 2008;283(13):8477–8485. doi:10.1074/jbc.M705876200
58. David LL, Shearer TR. Role of proteolysis in lenses: a review. Lens Eye Toxic Res. 1989;6(4):725–747.
59. Garner MH, Spector A. Selective oxidation of cysteine and methionine in normal and senile cataractous lenses. Proc Natl Acad Sci U S A. 1980;77(3):1274–1277. doi:10.1073/pnas.77.3.1274
60. Giblin FJ. Glutathione: a vital lens antioxidant. J Ocul Pharmacol Ther. 2000;16(2):121–135. doi:10.1089/jop.2000.16.121
61. McGinty SJ, Truscott RJ. Presbyopia: the first stage of nuclear cataract? Ophthalmic Res. 2006;38(3):137–148. doi:10.1159/000090645
62. Calvin HI, Grosshans K, Blake EJ. Estimation and manipulation of glutathione levels in prepuberal mouse ovaries and ova: relevance to sperm nucleus transformation in the fertilized egg. Gamete Res. 1986;14(3):265–275. doi:10.1002/mrd.1120140310
63. David LL, Shearer TR. State of sulfhydryl in selenite cataract. Toxicol Appl Pharmacol. 1984;74(1):109–115. doi:10.1016/0041-008X(84)90276-X
64. Vogt A, Lüssi U. Weitere Untersuchungen über das Relief der menschlichen Linsenkernoberfläche. Graefes Arch Clin Exp Ophthalmol. 1919;100:157–167. doi:10.1007/BF01880469
65. Al-Ghadyan AA, Cotlier E. Rise in lens temperature on exposure to sunlight or high ambient temperature. Br J Ophthalmol. 1986;70(6):421–426. doi:10.1136/bjo.70.6.421
66. Heys KR, Friedrich MG, Truscott RJ. Presbyopia and heat: changes associated with aging of the human lens suggest a functional role for the small heat shock protein, alpha-crystallin, in maintaining lens flexibility. Aging Cell. 2007;6(6):807–815. doi:10.1111/j.1474-9726.2007.00342.x
67. Hockwin O, Kojima M, Muller-Breitenkamp U, Wegener A. Lens and cataract research of the 20th century: a review of results, errors and misunderstandings. Dev Ophthalmol. 2002;35:1–11.
68. Schwartz B. Environmental temperature and the ocular temperature gradient. Arch Ophthalmol. 1965;74:237–243. doi:10.1001/archopht.1965.00970040239022
69. Scott JA. The computation of temperature rises in the human eye induced by infrared radiation. Phys Med Biol. 1988;33(2):243–257. doi:10.1088/0031-9155/33/2/004
70. Mirnezami SA, Rajaei Jafarabadi M, Abrishami M. Temperature distribution simulation of the human eye exposed to laser radiation. J Lasers Med Sci. 2013;4(4):175–181.
71. Shafahi M, Vafai K. Human eye response to thermal disturbances. J Heat Transfer. 2011;133(12011):011009. doi:10.1115/1.4002360
72. Mishima S, Maurice DM. The oily layer of the tear film and evaporation from the corneal surface. Exp Eye Res. 1961;1:39–45. doi:10.1016/S0014-4835(61)80006-7
73. Coates JE, Keatinge GF. The incidence of lens changes in an iron rolling mill. Trans Ophthalmol Soc U K. 1955;75:653–665.
74. Wallace J, Sweetnam PM, Warner CG, Graham PA, Cochrane AL. An epidemiological study of lens opacities among steel workers. Br J Ind Med. 1971;28(3):265–271. doi:10.1136/oem.28.3.265
75. Sliney DH. Physical factors in cataractogenesis: ambient ultraviolet radiation and temperature. Invest Ophthalmol Vis Sci. 1986;27(5):781–790.
76. Reddy GB, Das KP, Petrash JM, Surewicz WK. Temperature-dependent chaperone activity and structural properties of human alphaA- and alphaB-crystallins. J Biol Chem. 2000;275(7):4565–4570. doi:10.1074/jbc.275.7.4565
77. Miranda MN. Environmental temperature and senile cataract. Trans Am Ophthalmol Soc. 1980;78:255–264.
78. Taylor HR. The environment and the lens. Br J Ophthalmol. 1980;64(5):303–310.
79. Ritossa F. A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia. 1962;18(12):571–573. doi:10.1007/BF02172188
80. Matz JM, Blake MJ, Tatelman HM, Lavoi KP, Holbrook NJ. Characterization and regulation of cold-induced heat shock protein expression in mouse brown adipose tissue. Am J Physiol. 1995;269(1 Pt 2):R38–R47. doi:10.1152/ajpregu.1995.269.1.R38
81. Cao Y, Ohwatari N, Matsumoto T, Kosaka M, Ohtsuru A, Yamashita S. TGF-beta1 mediates 70-kDa heat shock protein induction due to ultraviolet irradiation in human skin fibroblasts. Pflugers Arch. 1999;438(3):239–244. doi:10.1007/s004240050905
82. Laplante AF, Moulin V, Auger FA, et al. Expression of heat shock proteins in mouse skin during wound healing. J Histochem Cytochem. 1998;46(11):1291–1301. doi:10.1177/002215549804601109
83. Salvador-Silva M, Ricard CS, Agapova OA, Yang P, Hernandez MR. Expression of small heat shock proteins and intermediate filaments in the human optic nerve head astrocytes exposed to elevated hydrostatic pressure in vitro. J Neurosci Res. 2001;66(1):59–73. doi:10.1002/jnr.1197
84. Nefzger MD, Miller RJ, Fujino T. Eye findings in atomic bomb survivors of Hiroshima and Nagasaki: 1963–1964. Am J Epidemiol. 1969;89(2):129–138. doi:10.1093/oxfordjournals.aje.a120922
85. Mole RH. Radiation effects in man: current views and prospects. Health Phys. 1971;20(5):485–490. doi:10.1097/00004032-197105000-00003
86. Zaret MM, Snyder WZ, Birenbaum L. Cataract after exposure to non-ionizing radiant energy. Br J Ophthalmol. 1976;60(9):632–637. doi:10.1136/bjo.60.9.632
87. Kurz GH, Einaugler RB. Cataract secondary to microwave radiation. Am J Ophthalmol. 1968;66(5):866–869. doi:10.1016/0002-9394(68)92801-8
88. Leibowitz HM, Luzzio AJ. Laser-induced cataract. Clinical observations. Arch Ophthalmol. 1970;83(5):608–612. doi:10.1001/archopht.1970.00990030608016
89. Worgul BV, Merriam GR
90. Brilliant LB, Grasset NC, Pokhrel RP, et al. Associations among cataract prevalence, sunlight hours, and altitude in the Himalayas. Am J Epidemiol. 1983;118(2):250–264. doi:10.1093/oxfordjournals.aje.a113632
91. Lerman S. Human ultraviolet radiation cataracts. Ophthalmic Res. 1980;12:303–314. doi:10.1159/000265094
92. Hollows F, Moran D. Cataract–the ultraviolet risk factor. Lancet. 1981;2(8258):1249–1250. doi:10.1016/S0140-6736(81)91490-2
93. Hodge WG, Whitcher JP, Satariano W. Risk factors for age-related cataracts. Epidemiol Rev. 1995;17(2):336–346. doi:10.1093/oxfordjournals.epirev.a036197
94. West SK, Duncan DD, Munoz B, et al. Sunlight exposure and risk of lens opacities in a population-based study: the Salisbury Eye Evaluation project. JAMA. 1998;280(8):714–718. doi:10.1001/jama.280.8.714
95. The Italian-American Cataract Study Group. Risk factors for age-related cortical, nuclear, and posterior subcapsular cataracts. Am J Epidemiol. 1991;133(6):541–553.
96. Charman WN. Ultraviolet radiation and cataract. Br J Ophthalmol. 1995;79(2):196. doi:10.1136/bjo.79.2.196
97. Collman GW, Shore DL, Shy CM, Checkoway H, Luria AS. Sunlight and other risk factors for cataracts: an epidemiologic study. Am J Public Health. 1988;78(11):1459–1462. doi:10.2105/AJPH.78.11.1459
98. Cruickshanks KJ, Klein BE, Klein R. Ultraviolet light exposure and lens opacities: the Beaver Dam Eye Study. Am J Public Health. 1992;82(12):1658–1662. doi:10.2105/AJPH.82.12.1658
99. Dolezal JM, Perkins ES, Wallace RB. Sunlight, skin sensitivity, and senile cataract. Am J Epidemiol. 1989;129(3):559–568. doi:10.1093/oxfordjournals.aje.a115168
100. Dolin PJ. Ultraviolet radiation and cataract: a review of the epidemiological evidence. Br J Ophthalmol. 1994;78(6):478–482. doi:10.1136/bjo.78.6.478
101. Leske MC, Chylack LT
102. Taylor HR, West SK, Rosenthal FS, et al. Effect of ultraviolet radiation on cataract formation. N Engl J Med. 1988;319(22):1429–1433. doi:10.1056/NEJM198812013192201
103. Seddon J, Fong D, West,SK, Valmadrid,CT. Epidemiology of risk factors for age-related cataract. Surv Ophthalmol. 1995;39(4):323–334. doi:10.1016/S0039-6257(05)80110-9
104. Ellis RJ, Minton AP. Protein aggregation in crowded environments. Biol Chem. 2006;387(5):485–497. doi:10.1515/BC.2006.064
105. Cavallotti C, Cerulli L. Age-Related Changes of the Human Eye. Totowa, NJ: Humana Press; 2008.
106. Borchman D, Giblin FJ, Leverenz VR, et al. Impact of aging and hyperbaric oxygen in vivo on Guinea pig lens lipids and nuclear light scatter. Invest Ophthalmol Vis Sci. 2000;41(10):3061–3073.
107. Chen Y, Gao J, Li L, et al. The ciliary muscle and zonules of zinn modulate lens intracellular hydrostatic pressure through transient receptor potential vanilloid channels. Invest Ophthalmol Vis Sci. 2019;60(13):4416–4424. doi:10.1167/iovs.19-27794
108. Bettelheim FA. Syneretic response to pressure in ocular lens. J Theor Biol. 1999;197(2):277–280. doi:10.1006/jtbi.1998.0855
109. Wang XW, Bettelheim FA. Distribution of total and non-freezable water contents of galactosemic rat lenses. Curr Eye Res. 1988;7(8):771–776. doi:10.3109/02713688809033208
110. Danysh BP, Czymmek KJ, Olurin PT, Sivak JG, Duncan MK. Contributions of mouse genetic background and age on anterior lens capsule thickness. Anat Rec. 2008;291(12):1619–1627. doi:10.1002/ar.20753
111. Fincham EF. The mechanism of accomodation. Br J Ophthalmol. 1937;43:e34.
112. Fisher RF. Changes in the permeability of the lens capsule in senile cataract. Trans Ophthalmol Soc U K. 1977;97(1):100–103.
113. Friedenwald JS. The permeability of the lens capsule to water, dextrose, and other sugars. Trans Am Ophthalmol Soc. 1930;28:195–211.
114. Friedenwald JS. The permeability of the lens capsule with special reference to the etiology of senile cataract. Arch Ophthalmol. 1930;3(2):182–193. doi:10.1001/archopht.1930.00810040060006
115. Lee CJ, Vroom JA, Fishman HA, Bent SF. Determination of human lens capsule permeability and its feasibility as a replacement for Bruch’s membrane. Biomaterials. 2006;27(8):1670–1678. doi:10.1016/j.biomaterials.2005.09.008
116. Fisher RF. The elastic constants of the human lens. J Physiol. 1971;212(1):147–180. doi:10.1113/jphysiol.1971.sp009315
117. Fisher RF. Elastic constants of the human lens capsule. J Physiol. 1969;201(1):1–19. doi:10.1113/jphysiol.1969.sp008739
118. Ziebarth NM, Borja D, Arrieta E, et al. Role of the lens capsule on the mechanical accommodative response in a lens stretcher. Invest Ophthalmol Vis Sci. 2008;49(10):4490–4496. doi:10.1167/iovs.07-1647
119. Danysh BP, Duncan MK. The lens capsule. Exp Eye Res. 2009;88(2):151–164. doi:10.1016/j.exer.2008.08.002
120. Krag S, Andreassen TT. Mechanical properties of the human lens capsule. Prog Retin Eye Res. 2003;22(6):749–767. doi:10.1016/S1350-9462(03)00063-6
121. Weeber HA, Eckert G, Soergel F, Meyer CH, Pechhold W, van der Heijde RG. Dynamic mechanical properties of human lenses. Exp Eye Res. 2005;80(3):425–434. doi:10.1016/j.exer.2004.10.010
122. Koretz JF, Cook CA, Kaufman PL. Accommodation and presbyopia in the human eye. Changes in the anterior segment and crystalline lens with focus. Invest Ophthalmol Vis Sci. 1997;38(3):569–578.
123. Boswell BA, Le AC, Musil LS. Upregulation and maintenance of gap junctional communication in lens cells. Exp Eye Res. 2009;88(5):919–927. doi:10.1016/j.exer.2008.11.031
124. Duncan G. The site of the ion restricting membranes in the toad lens. Exp Eye Res. 1969;8(4):406–412. doi:10.1016/S0014-4835(69)80006-0
125. Eisenberg RS, Rae JL. Current-voltage relationships in the crystalline lens. J Physiol. 1976;262(2):285–300. doi:10.1113/jphysiol.1976.sp011596
126. Rae JL. The electrophysiology of the crystalline lens. Curr Top Eye Res. 1979;1:37–90.
127. Mathias RT, Rae JL. Cell to cell communication in the lens. In: Sperelakis N, Cole WC, editors. Cell Interactions and Gap Junctions. Boca Raton, Fla.: CRC Press; 1989.
128. Gong X, Li E, Klier G, et al. Disruption of alpha3 connexin gene leads to proteolysis and cataractogenesis in mice. Cell. 1997;91(6):833–843. doi:10.1016/S0092-8674(00)80471-7
129. Fleishman SJ, Unger VM, Yeager M, Ben-Tal N. A Calpha model for the transmembrane alpha helices of gap junction intercellular channels. Mol Cell. 2004;15(6):879–888. doi:10.1016/j.molcel.2004.08.016
130. Unger VM, Kumar NM, Gilula NB, Yeager M. Three-dimensional structure of a 129 gap junction membrane channel. Science. 1999;283(5405):1176–1180. doi:10.1126/science.283.5405.1176
131. Goodenough DA. The crystalline lens. A system networked by gap junctional intercellular communication. Semin Cell Biol. 1992;3(1):49–58. doi:10.1016/S1043-4682(10)80007-8
132. Rae JL, Bartling C, Rae J, Mathias RT. Dye transfer between cells of the lens. J Membr Biol. 1996;150(1):89–103. doi:10.1007/s002329900033
133. Robinson KR, Patterson JW. Localization of steady currents in the lens. Curr Eye Res. 1982;2(12):843–847. doi:10.3109/02713688209020020
134. Mathias RT, Rae JL, Baldo GJ. Physiological properties of the normal lens. Physiol Rev. 1997;77(1):21–50. doi:10.1152/physrev.1997.77.1.21
135. Donaldson P, Kistler J, Mathias RT. Molecular solutions to mammalian lens transparency. News Physiol Sci. 2001;16:118–123. doi:10.1152/physiologyonline.2001.16.3.118
136. Berthoud VM, Beyer EC. Oxidative stress, lens gap junctions, and cataracts. Antioxid Redox Signal. 2009;11(2):339–353. doi:10.1089/ars.2008.2119
137. Gao J, Sun X, White TW, Delamere NA, Mathias RT. Feedback regulation of intracellular hydrostatic pressure in surface cells of the lens. Biophys J. 2015;109(9):1830–1839. doi:10.1016/j.bpj.2015.09.018
138. DeRosa AM, Mui R, Srinivas M, White TW. Functional characterization of a naturally occurring Cx50 truncation. Invest Ophthalmol Vis Sci. 2006;47(10):4474–4481. doi:10.1167/iovs.05-1582
139. Parker NR, Jamie JF, Davies MJ, Truscott RJ. Protein-bound kynurenine is a photosensitizer of oxidative damage. Free Radic Biol Med. 2004;37(9):1479–1489. doi:10.1016/j.freeradbiomed.2004.07.015
140. Gao J, Sun X, Moore LC, Brink PR, White TW, Mathias RT. The effect of size and species on lens intracellular hydrostatic pressure. Invest Ophthalmol Vis Sci. 2013;54(1):183–192. doi:10.1167/iovs.12-10217
141. Gao J, Sun X, Martinez-Wittinghan FJ, Gong X, White TW, Mathias RT. Connections between connexins, calcium, and cataracts in the lens. J Gen Physiol. 2004;124(4):289–300. doi:10.1085/jgp.200409121
142. Erickson-Lamy K, Schroeder AM, Bassett-Chu S, Epstein DL. Absence of time-dependent facility increase (“washout”) in the perfused enucleated human eye. Invest Ophthalmol Vis Sci. 1990;31(11):2384–2388.
143. Gong X, Baldo GJ, Kumar NM, Gilula NB, Mathias RT. Gap junctional coupling in lenses lacking alpha3 connexin. Proc Natl Acad Sci USA. 1998;95(26):15303–15308. doi:10.1073/pnas.95.26.15303
144. Duncan G, van Heyningen R. Distribution of non-diffusible calcium and sodium in normal and cataractous human lenses. Exp Eye Res. 1977;25(2):183–193. doi:10.1016/0014-4835(77)90130-0
145. Hightower KR, Reddy VN. Calcium content and distribution in human cataract. Exp Eye Res. 1982;34(3):413–421. doi:10.1016/0014-4835(82)90087-2
146. Baruch A, Greenbaum D, Levy ET, et al. Defining a link between gap junction communication, proteolysis, and cataract formation. J Biol Chem. 2001;276(31):28999–29006. doi:10.1074/jbc.M103628200
147. Shestopalov VI, Bassnett S. Development of a macromolecular diffusion pathway in the lens. J Cell Sci. 2003;116(Pt 20):4191–4199.
148. Padgaonkar VA, Lin LR, Leverenz VR, Rinke A, Reddy VN, Giblin FJ. Hyperbaric oxygen in vivo accelerates the loss of cytoskeletal proteins and MIP26 in Guinea pig lens nucleus. Exp Eye Res. 1999;68(4):493–504. doi:10.1006/exer.1998.0630
149. Roche J, Caro JA, Norberto DR, et al. Cavities determine the pressure unfolding of proteins. Proc Natl Acad Sci USA. 2012;109(18):6945–6950. doi:10.1073/pnas.1200915109
150. Silva JL, Silveira CF, Correia Junior A, Pontes L. Dissociation of a native dimer to a molten globule monomer. Effects of pressure and dilution on the association equilibrium of arc repressor. J Mol Biol. 1992;223(2):545–555. doi:10.1016/0022-2836(92)90669-B
151. Vidugiris GJ, Royer CA. Determination of the volume changes for pressure-induced transitions of apomyoglobin between the native, molten globule, and unfolded states. Biophys J. 1998;75(1):463–470. doi:10.1016/S0006-3495(98)77534-4
152. Chapeaurouge A, Johansson JS, Ferreira ST. Folding of a de novo designed native-like four-helix bundle protein. J Biol Chem. 2002;277(19):16478–16483. doi:10.1074/jbc.M105232200
153. Kitahara R, Yamada H, Akasaka K, Wright PE. High pressure NMR reveals that apomyoglobin is an equilibrium mixture from the native to the unfolded. J Mol Biol. 2002;320(2):311–319. doi:10.1016/S0022-2836(02)00449-7
154. Trzesniak D, Lins RD, van Gunsteren WF. Protein under pressure: molecular dynamics simulation of the arc repressor. Proteins. 2006;65(1):136–144. doi:10.1002/prot.21034
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.