")
Back to Journals » Journal of Experimental Pharmacology » Volume 13
Promising Treatment Options for Axial Spondyloarthritis: An Overview of Experimental Pharmacological Agents
Authors Tahir H , Byravan S , Fardanesh A, Moorthy A
Received 24 February 2021
Accepted for publication 22 May 2021
Published 2 July 2021 Volume 2021:13 Pages 627—635
DOI https://doi.org/10.2147/JEP.S262340
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Paola Rogliani
Hasan Tahir,1,2 Swetha Byravan,3 Armin Fardanesh,4 Arumugam Moorthy3,5
1Department of Rheumatology, Royal Free London NHS Foundation Trust, London, UK; 2Division of Medicine, University College London, London, UK; 3Department of Rheumatology, University Hospitals of Leicester NHS Trust, Leicester, UK; 4Department of General Medicine, Royal Free London NHS Foundation Trust, London, UK; 5College of Life Sciences, University of Leicester, Leicester, UK
Correspondence: Hasan Tahir
Department of Rheumatology, Royal Free London NHS Foundation Trust, London, UK, Division of Medicine, University College London, London, UK
Email [email protected]
Abstract: Axial spondyloarthritis (axSpA) is a chronic inflammatory condition that predominantly affects the axial skeleton. All patients receive conservative management measures which include physiotherapy, patient education and use of nonsteroidal anti-inflammatory drugs (NSAIDs). Those with significant active disease will require escalation of their treatment with the use of biologics. Currently, there are five approved TNF inhibitors and two approved IL-17 inhibitors for use in axSpA. However, despite this up to 40% of patients do not respond or are intolerant to current available treatment. This leaves a significant number of patients with uncontrolled disease and unmet need for additional therapies. Though many drug classes have been trialed for axSpA they show poor efficacy; however, over the last few years there are three which demonstrate much greater promise as novel therapies for axSpA, these include dual neutralization of IL-17A and IL-17F, Janus kinase (JAK) inhibitors, and granulocyte-macrophage colony-stimulating factor (GM-CSF) inhibitors. This article reviews the evidence for these novel emerging therapeutic options for axSpA.
Keywords: Axial spondyloarthritis, novel therapies, JAK inhibitors, IL-17 inhibitors, GM-CSF inhibitors
Plain Language Summary
- Axial spondyloarthritis (axSpA) is a type of inflammatory arthritis that involves inflammation of the joints in the spine.
- At present the management of this condition includes conservative measures of physiotherapy and use of nonsteroidal anti-inflammatory medications which helps control the symptoms but not the disease itself.
- The next step of management is use of a class of drugs called biological therapies but a significant proportion of patients with axSpA do not respond, or are intolerant to current available biological therapies, and those that do respond, not all achieve disease remission.
- There is, therefore, an unmet need for newer biological therapies with novel mechanisms of action for the treatment of axSpA.
- Of these newer therapies the drugs that have demonstrated the most promising results in study trials are ones that target specific proteins involved in the pathogenesis of axSpA. These include dual neutralization of IL-17A and IL-17F, Janus kinase (JAK) inhibitors, and granulocyte-macrophage colony-stimulating factor (GM-CSF) inhibitors.
Introduction
Axial spondyloarthritis (axSpA) is a chronic inflammatory disease that predominantly affects the the axial skeleton (sacroiliac joints and spine). AxSpA is a term that encompasses patients with having either ankylosing spondylitis (abbreviated as AS, also termed radiographic axSpA; rad axSpA) or nonradiographic axSpA (nr-axSpA). In clinical practice, distinction between these forms of axSpA in an individual patient has limited impact on management and may not be relevant, although the classification is of interest for epidemiologic and other investigative purposes. Studies have demonstrated that around 10–40% of patients will progress from nr-axSpA to rad axSpA over a period of 2–10 years.1
The ASAS-EULAR 2016 guidelines have 13 recommendations regarding the optimal management of axSpA. It advises that all patients with axSpA (axial or peripheral) should be educated about their condition and healthy lifestyle encouraged with regular exercise and stopping smoking. For pharmacological management it recommends starting with a nonsteroidal anti-inflammatory drug (NSAIDs) and moving onto biological therapy in those who are still symptomatic. Overall, the primary goal of management is to control symptoms and inflammation to maximize long-term related quality of life, prevent progressive structural damage and preserve function and social participation.2
Patients with symptoms due to active axSpA and an inadequate response to initial therapy, tumor necrosis factor inhibitors (TNFi) have demonstrated improved signs and symptoms and even inhibited radiological progression, though the evidence on this is conflicting.3,4 Five TNF inhibiting agents have been approved in Europe for patients with rad axSpA and four for nr-axSpA4 (see Table 1). Generally, patients who have active and early disease respond better to TNFi than those with advanced disease.5 However, despite the beneficial effects of TNF inhibition, up to 40% of patients do not respond (inadequate reduction of disease activity) or are intolerant and in those that do respond, not all achieve remission.6
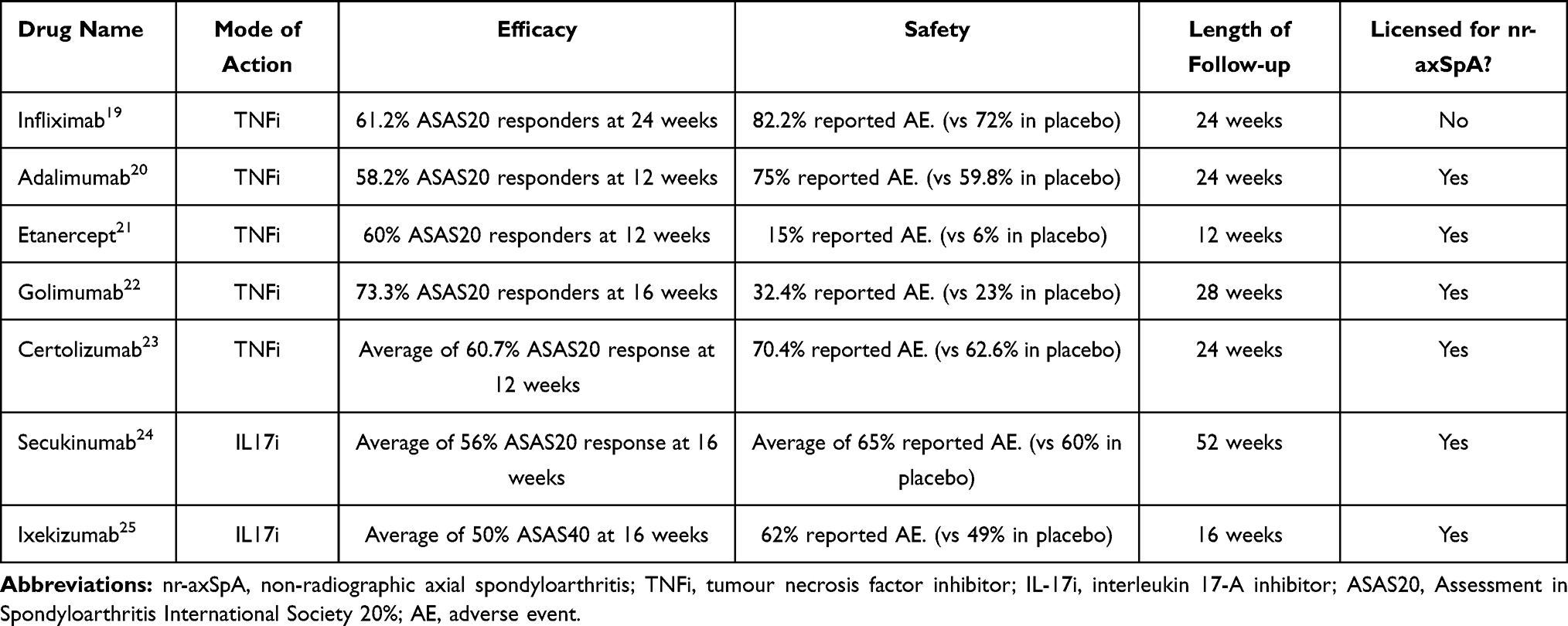 |
Table 1 Current Approved Biological Therapies for the Management of axSpA |
Over the last few years efficacy for several interleukin-17 inhibitors (IL-17i), in the management of axSpA has been proven.7,8 Currently there are two IL-17i available; secukinumab (SEC) and ixekizumab (IXE) which are both licensed for rad axSpA and nr-axSpA (see Table 1). TNFi and IL-17i have not been directly compared in patients with axSpA; however, both have similar levels of efficacy and similar safety profiles compared with placebo; and use of a biologic agent with a different mechanism of action provides an additional rationale for its use, especially in patients who have already not responded to either one or the other. That said unfortunately even with the availability of at least one IL-17i ∼20–30% of patients do not observe an improvement.9
Currently despite TNFi and IL-17i there is still a large proportion of axSpA patients whose disease is not controlled and, therefore, there remains a significant unmet need for additional approaches to therapy.
In recent years, numerous drug classes have been investigated for the treatment of axSpA, however, many of these drugs have failed to show any significant efficacy. These include the anti-IL12/23 inhibitors ustekinumab and risankizumab,10,11 the T cell co-stimulation inhibitor abatacept,12 IL-6 receptor inhibitors sarilumab and tocilizumab,13,14 the IL-1 receptor antagonist anakinra,15 the anti-CD20 antibody rituximab16 and the phosphodiesterase-4 inhibitor apremilast.17,18
This article reviews the evidence for the novel emerging therapeutic options for axSpA which have demonstrated promising results in trials and represents a prospective option for treatment. These include dual neutralization of IL-17A and IL-17F, Janus kinase (JAK) inhibitors, and granulocyte-macrophage colony-stimulating factor (GM-CSF) inhibitors.
Dual Neutralization of IL-17A and IL-17F
IL-17 is thought to be a key cytokine that plays a pivotal role in the pathogenesis of axSpA. Higher levels of serum IL-17 and circulating Th17 cells have been found in axSpA patients (especially in the facet joints of axSpA patients with greater IL-17-producing cells in comparison to those with osteoarthritis).26 Animal studies indicate that IL-17 blockade reduces receptor activator of nuclear factor κ B (RANK) ligand-dependent osteoclastogenesis upstream of TNFα.27
IL-17A shares greater than 50% structural homology with IL-17F and have similar pro-inflammatory function and signaling via the same receptor complex.28 IL-17A and IL-17F are both upregulated in a range of inflamed human tissues and cooperate with other pro-inflammatory cytokines, such as TNF, to amplify inflammatory responses.29 The impact of IL-17F together with IL-17A in pathological bone formation would indicate that neutralization of both these cytokines impedes this process to a larger degree than IL-17A alone.30 In addition, greater quantities of IL-17A and IL-17F have been found in the serum of patients with axSpA vs healthy controls, correlating with markers of systemic inflammation.31
Bimekizumab
Bimekizumab is a monoclonal antibody that selectively neutralize both IL-17A and IL-17F.32
The BE AGILE phase 11b study, was the first dose-ranging clinical study assessing the efficacy and safety of bimekizumab in patients with active axSpA.33 This was a 48-week double-blind controlled study involving 303 patients who were randomly assigned to receive bimekizumab at doses of 16 mg (n=61), 64 mg (n=61), 160 mg (n=60), or 320 mg (n=61), or placebo (n=60) every four weeks. At week 12 patients who received placebo, bimekizumab 16 mg or 64 mg were re-randomized to bimekizumab 160 or 320 mg every four weeks to week 48.
The patients met modified New York criteria for AS, had an average Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score of about 6.5, an average total spinal pain score of about 7.0, and were permitted to have prior exposure to one TNFi.
At week 12, significantly more patients taking bimekizumab than placebo were Assessment in Spondyloarthritis International Society 40% (ASAS40) responders. The response rate improved with increasing dose, (6 mg: 29.5%, 64 mg: 42.6%, 160 mg: 46.7%, 320 mg: 45.9%) compared with placebo (13.3%). Rapid response to bimekizumab was observed in all groups but was highest in the 320 mg group with 50.8% achieving ASAS40 by week four. Similarly, significantly more bimekizumab treated patients achieved a 50% improvement in BASDAI score, with dose-response rates ranging from 23.0% in the 16 mg dose group to 47.5% in the 320 mg group, compared with 11.7% in the placebo group. The mean decreases in BASDAI score from baseline were a respective 1.7 and 2.9 points vs 1.0 points.
The primary endpoint was supported by all secondary end points at week 12. More patients in the bimekizumab achieved ASAS20 and ASAS5/6 which was highest in the 320 mg group (72.1% and 54.1% respectively).
In terms of patient reported outcomes (PROs) there was an improvement in spinal pain, morning stiffness and overall quality of life in all bimekizumab groups. A rapid reduction in C-Reactive Protein (CRP) was also observed and in 31 patients who had a follow-up MRI there was reduction in the Spondyloarthritis Research Consortium of Canada (SPARCC) SI joint scores and spine Berlin scores in the three highest dosed bimekizumab groups.
After week 12 all patients were re-randomized to receive either 160 or 320 mg of bimekizumab. There was a sustained rise in the number of patients achieving ASAS40 after week 12 and were maintained to week 48 in those remaining on the same dose of the drug (160 mg: 58.6%, 320 mg: 62.3%). Interestingly, in those who originally received placebo and then subsequently switched to bimekizumab, a similar proportion also achieved ASAS40 (160 mg: 54.2%, 320 mg: 50.0%).
Secondary endpoints also showed continued improvement including ASAS20 and ASAS5/6. BASDAI 50 demonstrated increasing rates up to week 48 with patients re-randomized to higher doses of bimekizumab achieving similar response to those were maintained on the same dose. Reductions in CRP was maintained in the 160 and 320 mg groups and those who were re-randomized also showed reduction and sustained improvement.
Improvements in PROs were demonstrated in both groups of patients, decrease in spinal pain, morning stiffness and Bath Ankylosing Spondylitis Functional Index (BASFI) were observed and there were similar reductions in fatigue across all dose groups. Patients also reported improvements in sleep which was sustained in the bimekizumab treated group and most importantly patients who were re-randomized from placebo to a bimekizumab group reported similar and significant improvement in ankylosing spondylitis quality of life (ASQoL).
There were no new safety findings and treatment-emergent adverse events (TEAE) were of mild-to-moderate intensity and comparable across the treatment groups, at 43.3% in the placebo group vs 29.3─47.5% in the bimekizumab treatment groups. Nasopharyngitis was the most frequent adverse event. Only six patients taking bimekizumab discontinued treatment due to adverse events. Oral candidiasis was reported by just 1.2% of patients given bimekizumab.
Overall, there was no unexpected safety finding compared with previous studies on bimekizumab.
This Phase IIb study demonstrated that bimekizumab provided rapid and sustained improvements in key efficacy measures in patients with rad axSpA with no unexpected safety findings.
This has prompted two Phase III trials; BE MOBILE 1 (NCT03928704), which is evaluating the efficacy of bimekizumab in patients with nr-axSpA and BE MOBILE 2 (NCT03928743) in patients with rad axSpA.34,35 Both are in the recruiting stages and estimated to complete by August 2022.
Targeting Janus Kinase Signaling
Cumulative data indicate that inhibition of JAK-mediated pathways may be a promising approach to treat inflammatory conditions such as chronic arthritis.36 JAK inhibitors are already licensed for the treatment of rheumatoid and psoriatic arthritis.
The result of activated JAK pathways is expression of various proteins including survival factors, cytokines, chemokines, and other molecules that enable leucocyte cellular trafficking and cell proliferation. Collectively they contribute to the pathophysiology of autoimmune driven inflammatory conditions.37 Thus, there has been a significant amount of interest in the JAK family, which has led to the development of JAK inhibitors that have various affinities for JAK targets (JAK1, JAK2, JAK3 and nonreceptor tyrosine-protein kinase; TYK2).
Several cytokines involved in the pathogenesis of axSpA are signaled via JAK pathways in combination with signal transducer and activator of transcription (STAT) intracellular transcription factors38 (Figure 1). JAK directly mediates cell signaling for a variety of extracellular cytokines including IFNγ, IL-7, IL-12, IL-15, IL-22, and IL-23. Also, the key pathogenic axis IL-23/IL-17 is under the influence of JAK signaling. The JAK2/TYK2 combination signals IL-23 and therefore JAK inhibition causes direct blockade of IL-23 as well as the other cytokines mentioned above and causes downstream indirect blockage of IL-17 and other important cytokines such as TNFα and IL-17.
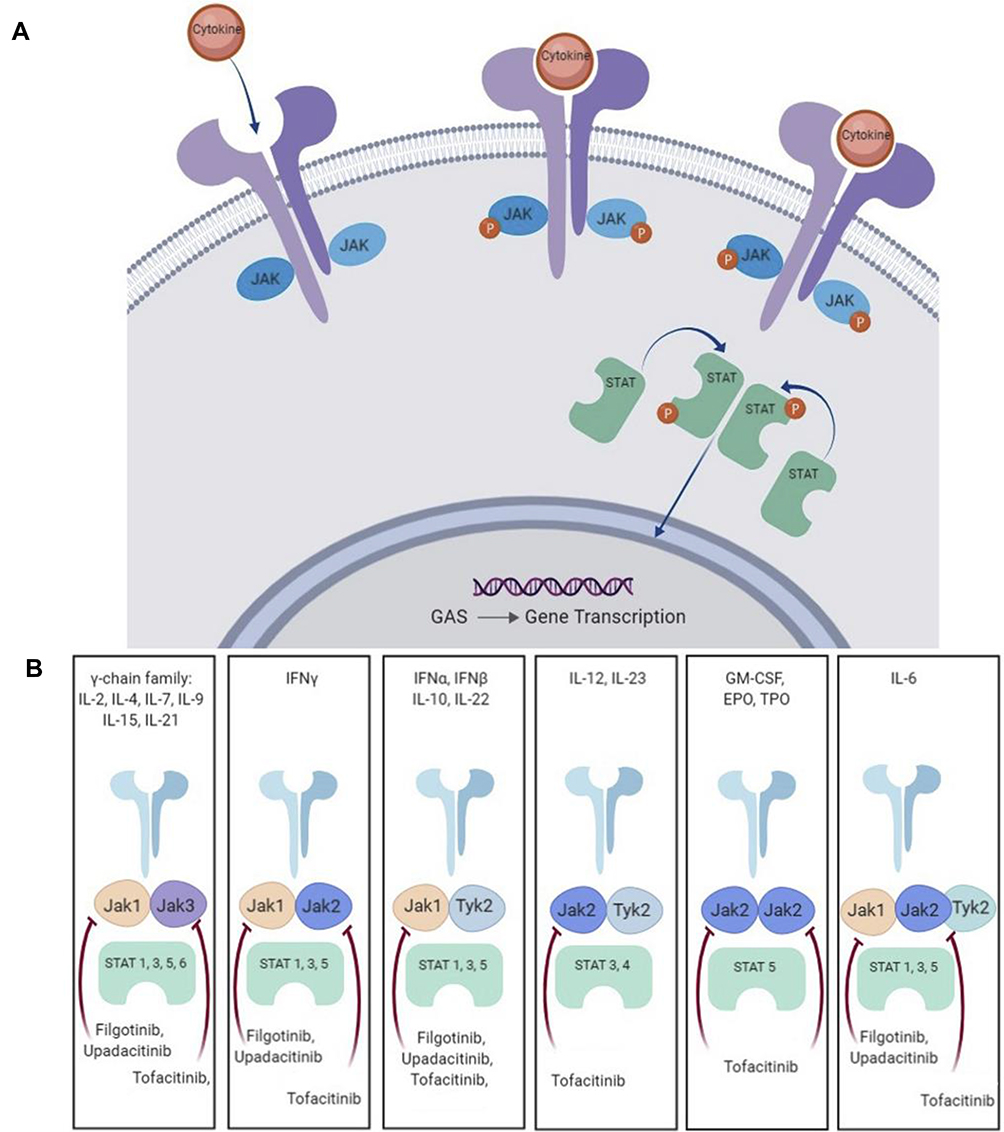 |
Figure 1 (A) The JAK-Stat signalling pathway. (B) Cytokine signally through JAK/STAT combination. Adapted from Bechman K, Yates M, Golloway J. The new entries in the therapeutic armamentarium: The small molecule JAK inhibitors. Pharmacol Res. 2020; 153: 104634.38 This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/). Adapted from Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285 (1–2):1–24. Feb 20. Copyright 2002, with permission from Elsevier.53 Adapted from Hodge AJ, Kawabata TT, Krishnaswami S, et al. The mechanism of action of tofacitinib – an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2016; 34 (2):318-328.54 Abbreviations: JAK, Janus kinase; TYK, tyrosine kinase; STAT, signal transducer and activator of transcription; IFN, interferon; IL, interleukin; EPO, erythropoietin; GM-CSF, granulocyte-macrophage colony-stimulating factor; TP0, thrombopoietin. |
Ultimately the inhibition of JAK pathways prevents expression of a variety of cytokines, chemokines and growth factors which cause bone loss, joint destruction, and proliferation of inflammatory cells. Therefore, this has resulted in great interest in the axSpA field as a potential target for therapy with subsequent development of biological therapies.
Tofacitinib
Tofacitinib is a first-in-class pan-JAK inhibitor with major inhibition of JAK3 and JAK1 and minor inhibition of JAK2. The drug inhibits the signal transduction of cytokines that promote the atypical autoimmune reaction in axSpA.39
In a 16-week Phase II proof of concept study, 208 patients with active rad axSpA were randomized to one of three tofacitinib groups (2, 5, or 10 mg) or placebo, which they received twice daily for 12 weeks and additional follow-up for four weeks.40 Patients were evaluated by MRI at baseline and after 12-weeks of treatment.
The primary endpoint was ASAS20 at week 12, which was 67.4%, 63.0% and 56.0% for 10 mg, 5 mg and 2 mg respectively and 40.1% for placebo group. Tofacitinib 10 mg had the highest response rate compared with the placebo than the other doses (27.3%).
In terms of secondary endpoints, all tofacitinib groups demonstrated improvement in ASAS40 and BASDAI50 of similar magnitude and were significant compared with placebo (∼42.3% and ∼43.6% respectively). Ankylosing Spondylitis Disease Activity Score (ASDAS) clinically important improvement (improvement ≥1.1 units) response rate was also significantly higher in all tofacitinib groups but not for ASDAS major response rates (improvement ≥2.0 units). A greater treatment effect was seen in those patients with a positive baseline MRI in all doses of tofacitinib and those with high combined CRP/MRI activity had greater treatment effect at 12 weeks compared with placebo, especially in the tofacitinib 5 mg group.
No new safety concerns were raised by patients treated with tofacitinib, and dose-dependent laboratory measures also appeared normal, resolving back to baseline values by week 16 of treatment. On the whole, tofacitinib 5 and 10 mg showed greater clinical and radiological improvement compared with placebo.
There is a completed Phase III trial on tofacitinib comparing 5 mg twice daily regimen with placebo, however, formal publication of results is still awaited (NCT03502616).41 Preliminary results reveal the primary and secondary endpoint of ASAS20 and ASAS40 respectively was met. 56.4% of patients in the tofacitinib group achieved ASAS20 compared with 29.4% of the placebo group and 40.6% in the tofacitinib group achieved ASAS40 compared with 12.5% in the placebo group.
The results for tofacitinib are encouraging but the data from the Phase III trial is required to further evaluate its efficacy.
Upadacitinib
Upadacitinib is a JAK inhibitor with preferential effects on JAK-1.
SELECT AXIS 1 was a Phase II/III double-blind, placebo-controlled two period study in which 187 patients with rad axSpA and an inadequate response to NSAIDs were randomly assigned 1:1 to oral upadacitinib 15 mg once daily or oral placebo for the 14-week period.42
The primary endpoint of ASAS40 was met with a clinical and statistical improvement with 52% in the upadacitinib group compared with 26% in the placebo group. There was also a significant improvement in BASDAI50 scores, SPARCC SI joint and spine scores especially (−6.93 vs −0.22). It also had a relatively rapid onset of response of approximately two weeks. Adverse events were reported in 62% of patients of the upadacitinib group compared with 55% in the placebo group with the most common being an increase in creatinine phosphokinase and the most common infection was nasopharyngitis. However, no new safety risks were detected, and the safety profile was consistent with previous studies.
Overall upadacitinib was found to be efficacious and well tolerated by patients. This has led to the development of a Phase III trial (SELECT AXIS 2) (NCT04169373), which is currently in its early stages and due to complete in 2023.43
Filgotinib
Filgotinib is another JAK inhibitor with preferential effects on JAK-1.
TORTUGA was a Phase II, randomized, double blind placebo-controlled study involving 116 patients with active axSpA with inadequate responses or intolerance to NSAIDs.44
Patients were randomized to receive either filgotinib 200 mg once daily or placebo for 12 weeks. The primary endpoint ASDAS was measured at week 12, which was significantly better in the filgotinib group compared with placebo (mean change from baseline −1.47 vs –0.57, least squares mean difference 0.85, 95%CI: −1.17 to −0.53).
Out of the three JAK inhibitors described the onset of response was quickest with filgotinib ∼1-week with significant improvement in ASDAS by the end of the first week.
Spinal mobility measured by Bath Ankylosing Spondylitis Metrology Index (BASMI) also demonstrated greater improvement with filgotinib at week 12 (mean change from baseline −0.75 vs −0.39). There was also a significant improvement in both SPARCC SI and spine scores. A post hoc analysis of the MRIs conducted at baseline and week 12 were re-evaluated which were observed to show a decrease in erosion scores and increase in backfill scores in the filgotinib group whereas in the placebo group the opposite was true supporting the structural effect filgotinib can have in axSpA.45
The quantity of adverse events was the same in both the treatment and placebo group (n=18). The most frequent was nasopharyngitis and there was one patient who discontinued the trial due to a case of pneumonia. Overall filgotinib was found to be safe and no new safety concerns were raised.
The phase II trial concluded that filgotinib was efficacious and that further investigation were warranted. Therefore, two Phase III trials were developed SEALION1-IR (NCT04483687) and SEALION2-NAÏVE [NCT04483700],46,47 but both have recently been suspended due to concerns from the US Food and Drug Administration (FDA) on the risk-benefit profile of the drug especially on sperm concentration.48
Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF)
GM-CSF is a pro-inflammatory cytokine that has a pleiotropic effect on myeloid cells including monocytes, macrophages, and dendritic cells and mediates their differentiation into a more inflammatory phenotype.49 In health GM-CSF plays an important role in injury or infection by mediating the interface between innate and adaptive immunity. GM-CSF is induced by other cytokines including IL-1β, IL-6 and TNFα, which are produced by a variety of cells.
Overexpression of GM-CSF and its receptor have been found in the synovial joints of those with axSpA and considered to stimulate joint damage by recruiting granulocyte and macrophage precursors from adjacent bone marrow and then inducing their differentiation into a more inflammatory phenotype. In addition, GM-CSF induces matrix metalloproteinases and osteoclasts via RANK ligand activation, which results in cartilage destruction and bone resorption.
GM-CSF receptors have also been found to be overexpressed on nerve endings in arthritic mice and, therefore, thought to contribute to arthritic pain. Neutralizing GM-CSF in arthritic mice with monoclonal antibodies resulted in significant alleviation of pain and improvement in joint inflammation.51 This has developed an interest in targeting GM-CSF in patients with axSpA to help reduce their disease burden.
Namilumab
Namilumab is a human monoclonal antibody targeting granulocyte macrophage-colony stimulating factor (GM-CSF).
A Phase IIa proof of concept trial (NAMASTE) is currently trailing the efficacy of namliumab in axSpA patients.51 Forty-two patients have been randomly assigned to receive the drug or placebo and the primary endpoint of ASAS20 is being assessed at week 12. Though this study has just been completed no results have been published but the results are eagerly awaited to assess the drug’s efficacy and to see if a phase III trial would be beneficial.
Namilumab has also been trialed for rheumatoid arthritis and has demonstrated statistically significant improvement in a phase 2 trial meetings its primary endpoint.52 The drug was thought to be safe overall and had an acceptable tolerability profile, with dyspnea, bronchitis and headaches being the most common TEAE.
Conclusion
There still remains an unmet need in the pharmacological management of axSpA with ~20–40% of patients not responding or intolerant to TNFi or IL-17i. Large numbers of trials examining potential targets have shown mixed results. Three new targets namely, dual neutralization of IL-17A and IL-17F, janus kinase (JAK) inhibitors, and granulocyte-macrophage colony-stimulating factor (GM-CSF) inhibitors have shown promise in phase II trials. Some of these are to progress to phase III trials whilst results of other completed phase III trials are eagerly awaited. It is hoped that these drugs will exhibit efficacy and safety in their phase III trials, and subsequently provide additional therapies for this condition and broaden the armamentarium of drugs for axSpA.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Protopopov M, Poddubnyy D. Radiographic progression in non-radiographic axial spondyloarthritis. Expert Rev Clin Immunol. 2018;14(6):525–533. doi:10.1080/1744666X.2018.1477591
2. Van der Heijde D, Ramiro S, Landewe R, et al. 2016 update of the ASAS-EULAR management recommendations for axial spondyloarthritis. Ann Rheum Dis. 2016;76:978–991. doi:10.1136/annrheumdis-2016-210770
3. Dubash S, McGonagle D, Marzo-Ortega H. New advances in the understanding and treatment of axial spondyloarthritis: from chance to choice. Ther Adv Chronic Dis. 2018;9(3):77–87. doi:10.1177/2040622317743486
4. Sieper J, Poddubnyy D. New evidence on the management of spondyloarthritis. Nat Rev Rheumatol. 2016;12(5):282–295. doi:10.1038/nrrheum.2016.42
5. Rudwaleit M, Listing J, Brandt J, Braun J, Sieper J. Prediction of a major clinical response (BASDAI 50) to tumour necrosis factor alpha blockers in ankylosing spondylitis. Ann Rheum Dis. 2004;63(6):665–670. doi:10.1136/ard.2003.016386
6. Tahir H. Therapies in ankylosing spondylitis—from clinical trials to clinical practice. Rheumatology. 2018;57(6):23–28. doi:10.1093/rheumatology/key152
7. Blair H. Secukinumab: a review in ankylosing spondylitis. Drugs. 2019;79(4):433–443. doi:10.1007/s40265-019-01075-3
8. Kiwalkar S, Beier S, Deodhar A. Ixekizumab for treating ankylosing spondylitis. Immunotherapy. 2019;11(15):1273–1282. doi:10.2217/imt-2019-0094
9. Baraliakos X, Braun J, Deodhar A, et al. Long-term efficacy and safety of secukinumab 150 mg in ankylosing spondylitis: 5-year results from the phase III MEASURE 1 extension study. RMD Open. 2019;5(2):e001005. doi:10.1136/rmdopen-2019-001005
10. Deodhar A, Gensler L, Siper J, et al. Three multicenter, randomized, double-blind, placebo-controlled studies evaluating the efficacy and safety of ustekinumab in axial spondyloarthritis. Arthritis Rheumatol. 2019;71(2):258–270. doi:10.1002/art.40728
11. Baeten D, Ostergaard M, Wei J, et al. Risankizumab, an IL-23 inhibitor, for ankylosing spondylitis: results of a randomised, double-blind, placebo-controlled, proof-of-concept, dose-finding phase 2 study. Ann Rheum Dis. 2018;77(9):1295–1302. doi:10.1136/annrheumdis-2018-213328
12. Song I-H, Heldmann F, Rudwaleit M, et al. Treatment of active ankylosing spondylitis with abatacept: an open-label, 24-week pilot study. Ann Rheum Dis. 2011;70(6):1108–1110. doi:10.1136/ard.2010.145946
13. Sieper J, Braun J, Kay J, et al. Sarilumab for the treatment of ankylosing spondylitis: results of a Phase II, randomised, double-blind, placebo-controlled study (ALIGN). Ann Rheum Dis. 2015;74(6):1051–1057. doi:10.1136/annrheumdis-2013-204963
14. Sieper J, Porter-Brown B, Thompson L, Harari O, Dougados M. Assessment of short-term symptomatic efficacy of tocilizumab in ankylosing spondylitis: results of randomised, placebo-controlled trials. Ann Rheum Dis. 2014;73(1):95–100. doi:10.1136/annrheumdis-2013-203559
15. Haibel H, Rudwaleit M, Listing J, Sieper J. Open label trial of anakinra in active ankylosing spondylitis over 24 weeks. Ann Rheum Dis. 2005;64(2):296–298. doi:10.1136/ard.2004.023176
16. Song I-H, Heldmann F, Rudwaleit M, Listing J, Appel H, Braun J. Different response to rituximab in tumor necrosis factor blocker-naive patients with active ankylosing spondylitis and in patients in whom tumor necrosis factor blockers have failed: a twenty-four-week clinical trial. Arthritis Rheum. 2010;62(5):1290–1297. doi:10.1002/art.27383
17. Pathan E, Abraham S, Van Rossen E, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in ankylosing spondylitis. Ann Rheum Dis. 2013;72(9):1475–1480. doi:10.1136/annrheumdis-2012-201915
18. Celgene. Celgene reports results from the Phase III posture study evaluating oral OTEZLA® in ankylosing spondylitis. [Internet]; 2014. Available from: https://ir.celgene.com/press-releases-archive/press-release-details/2014/Celgene-Reports-Results-from-the-Phase-III-POSTURE-Study-Evaluating-Oral-OTEZLA-in-Ankylosing-Spondylitis/default.aspx.
19. van der Heijde D, Dijkmans B, Geusens P, et al. Efficacy and safety of infliximab in patients with ankylosing spondylitis: results of a randomized, placebo‐controlled trial (ASSERT). Arthritis Rheum. 2005;52(2):582–591. doi:10.1002/art.20852
20. van der Heijde D, Kivitz A, Schiff MH, et al. Efficacy and safety of adalimumab in patients with ankylosing spondylitis: results of a multicenter, randomized, double‐blind, placebo‐controlled trial. Arthritis Rheum. 2006;54(7):2136–2146. doi:10.1002/art.21913
21. Calin A, Dijkmans BA, Emery P, et al. Outcomes of a multicentre randomised clinical trial of etanercept to treat ankylosing spondylitis. Ann Rheum Dis. 2004;63(12):1594–1600. doi:10.1136/ard.2004.020875
22. Deodhar A, Reveille J, Harrison D, et al. Safety and efficacy of golimumab administered intravenously in adults with ankylosing spondylitis: results through week 28 of the GO-ALIVE study. J Rheumatol. 2018;45(3):341–348. doi:10.3899/jrheum.170487
23. Landewé R, Braun J, Deodhar A, et al. Efficacy of certolizumab pegol on signs and symptoms of axial spondyloarthritis including ankylosing spondylitis: 24-week results of a double-blind randomised placebo-controlled Phase 3 study. Ann Rheum Dis. 2014;73(1):39–47. doi:10.1136/annrheumdis-2013-204231
24. Baeten D, Sieper J, Braun J, et al. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med. 2015;373(26):2534–2548. doi:10.1056/NEJMoa1505066
25. van der Heijde D, Wei J, Dougados M, et al. Ixekizumab, an interleukin-17A antagonist in the treatment of ankylosing spondylitis or radiographic axial spondyloarthritis in patients previously untreated with biological disease-modifying anti-rheumatic drugs (COAST-V): 16 week results of a phase 3 randomised, double-blind, active-controlled and placebo-controlled trial. Lancet. 2018;392(10163):2441–2451. doi:10.1016/S0140-6736(18)31946-9
26. Shen H, Goodall J, Gaston J. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum. 2009;60(6):1647–1656. doi:10.1002/art.24568
27. Koenders M, Lubberts E, Oppers-Walgreen B, et al. Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol. 2005;167(7):141–149. doi:10.1016/S0002-9440(10)62961-6
28. Yang X, Chang S, Park H, et al. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205(5):1063–1075. doi:10.1084/jem.20071978
29. Glatt S, Baeten D, Baker T, et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann Rheum Dis. 2018;77:523–532. doi:10.1136/annrheumdis-2017-212127
30. Shah M, Maroof A, Al-Hosni R, et al. Bimekizumab blocks T cell-mediated osteogenic differentiation of periosteal stem cells: coupling pathological bone formation to IL-17A and IL-17F signaling [abstract]. Arthritis Rheumatol. 2017;69(10):1936.
31. Gracey E, Yao Y, Green B, et al. Sexual dimorphism in the Th17 signature of ankylosing spondylitis. Arthritis Rheumatol. 2016;68(3):679–689. doi:10.1002/art.39464
32. Glatt S, Helmer E, Haier B, et al. First-in-human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL-17A and IL-17F, in mild psoriasis. Br J Clin Pharmacol. 2017;83(5):991–1001. doi:10.1111/bcp.13185
33. Heijde D, Gensler L, Deodhar A, et al. Dual neutralisation of interleukin-17A and interleukin-17F with bimekizumab in patients with active ankylosing spondylitis: results from a 48-week phase IIb, randomised, double-blind, placebo-controlled, dose-ranging study. Ann Rheum Dis. 2020;79(5):595–604. doi:10.1136/annrheumdis-2020-216980
34. UCB Biopharma SRL. A study to evaluate the efficacy and safety of bimekizumab in subjects with active nonradiographic axial spondyloarthritis (BE MOBILE 1); 2011. Available from: https://clinicaltrials.gov/ct2/show/NCT03928704.
35. UCB Biopharma SRL A study to evaluate the efficacy and safety of bimekizumab in subjects with active ankylosing spondylitis (BE MOBILE 2); 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT03928743.
36. Vaddi K, Luchi M. JAK inhibition for the treatment of rheumatoid arthritis: a new era in oral DMARD therapy. Expert Opin Investig Drugs. 2012;21(7):961–973. doi:10.1517/13543784.2012.690029
37. Veale D, McGonagle D, McInnes I, et al. The rationale for Janus kinase inhibitors for the treatment of spondyloarthritis. Rheumatology. 2019;58(2):197–205. doi:10.1093/rheumatology/key070
38. Bechman K, Yates M, Golloway J. The new entries in the therapeutic armamentarium: the small molecule JAK inhibitors. Pharmacol Res. 2020;153:104634. doi:10.1016/j.phrs.2020.104634
39. Electronic medicines compendium (emc). Xeljanz; 2017. Available from: https://www.medicines.org.uk/emc/medicine/33167#gref.
40. Heijde D, Deodhar A, Wei J, et al. Tofacitinib in patients with ankylosing spondylitis: a phase II, 16-week, randomised, placebo-controlled, dose-ranging study. Ann Rheum Dis. 2017;76(8):1340–1347. doi:10.1136/annrheumdis-2016-210322
41. Pfizer. Pfizer announces positive phase 3 study results for XELJANZ® (tofacitinib) in ankylosing spondylitis (AS); 2020, Available from: https://www.businesswire.com/news/home/20201106005130/en/Pfizer-Announces-Positive-Phase-3-Study-Results-for-XELJANZ-%C2%AE-tofacitinib-in-Ankylosing-Spondylitis-AS.
42. Heijde D, Song I, Pangan A, et al. Efficacy and safety of upadacitinib in patients with active ankylosing spondylitis (SELECT-AXIS 1): a multicentre, randomised, double-blind, placebo-controlled, phase 2/3 trial. Lancet. 2019;394(10214):2108–2117. doi:10.1016/S0140-6736(19)32534-6
43. AbbVie. A Study to Evaluate Efficacy and Safety of Upadacitinib in Adult Participants With Axial Spondyloarthritis (SELECT AXIS 2); 2020. Available from: https://clinicaltrials.gov/ct2/show/record/NCT04169373?cond=Ankylosing+Spondylitis&intr=JAK+Inhibitors&draw=2&rank=2&view=record.
44. Heijde D, Baraliakos X, Gensler L, et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active ankylosing spondylitis (TORTUGA): results from a randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392(10162):2378–2387. doi:10.1016/S0140-6736(18)32463-2
45. Maksymowych WP, Ǿstergaard M, Landewé RBM, et al. THU0377: impact of Filgotinib on structural lesions in the sacroiliac joints at 12 weeks in patients with active axial spondyloarthritis: magnetic resonance imaging data from the double-blind, randomized TORTUGA trial. Ann Rheum Dis. 2020;79(1):417. doi:10.1136/annrheumdis-2020-eular.2553
46. Gilead Sciences. Study to evaluate the efficacy and safety of filgotinib in participants with active ankylosing spondylitis who have an inadequate response to biologic disease-modifying antirheumatic drug therapy (SEALION1-IR); 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT04483687?type=Intr&cond=Ankylosing+Spondylitis&draw=2&rank=48.
47. Gilead Sciences. Study to evaluate the efficacy and safety of filgotinib in participants with active ankylosing spondylitis who are naive to biologic disease-modifying antirheumatic drug therapy (SEALION2-NAÏVE); 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT04483700?type=Intr&cond=Ankylosing+Spondylitis&draw=2&rank=45.
48. Ankylosing Spondylitis News. Gilead suspends enrollment in phase 3 trials of filgotinib in AS; 2020. Available from: https://ankylosingspondylitisnews.com/2020/11/02/gilead-suspends-enrollment-phase-3-trials-filgotinib/?cn-reloaded=1.
49. Worth C, Bowness P, Al-Mossawi M. Novel therapeutic targets in axial spondyloarthritis. Curr Treatm Opt Rheumatol. 2018;4(2):174–182. doi:10.1007/s40674-018-0095-1
50. Greven D, Cohen E, Gerlag D, Campbell J, Woods J, Davis N. Preclinical characterisation of the GM-CSF receptor as a therapeutic target in rheumatoid arthritis. Ann Rheum Dis. 2015;74(10):1924–1930. doi:10.1136/annrheumdis-2014-205234
51. Izana Bioscience Ltd. Efficacy and safety of namilumab for moderate-to-severe axial spondyloarthritis (NAMASTE); 2021. Available from: https://www.clinicaltrials.gov/ct2/show/study/NCT03622658?cond=Namilumab&draw=2&rank=1.
52. Taylor P, Saurigny D, Vencovsky J, et al. Efficacy and safety of namilumab, a human monoclonal antibody against granulocyte-macrophage colony-stimulating factor (GM-CSF) ligand in patients with rheumatoid arthritis (RA) with either an inadequate response to background methotrexate therapy or an inadequate response or intolerance to an anti-TNF (tumour necrosis factor) biologic therapy: a randomized, controlled trial. Arthritis Res Ther. 2019;21(1):101.
53. Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285(1–2):1–24. Feb 20.
54. Hodge AJ, Kawabata TT, Krishnaswami S, et al. The mechanism of action of tofacitinib – an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2016;34(2):318–328.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.