")
Back to Archived Journals » Hypoxia » Volume 2
Prolyl hydroxylase domain enzymes: important regulators of cancer metabolism
Authors Yang M, Su H, Soga T, Kranc K, Pollard P
Received 6 May 2014
Accepted for publication 30 May 2014
Published 30 August 2014 Volume 2014:2 Pages 127—142
DOI https://doi.org/10.2147/HP.S47968
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Ming Yang,1 Huizhong Su,1 Tomoyoshi Soga,2 Kamil R Kranc,3 Patrick J Pollard1
1Cancer Biology and Metabolism Group, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, UK; 2Institute for Advanced Biosciences, Keio University, Mizukami, Tsuruoka, Yamagata, Japan; 3MRC Centre for Regenerative Medicine, University of Edinburgh, Edinburgh, UK
Abstract: The hypoxia-inducible factor (HIF) prolyl hydroxylase domain enzymes (PHDs) regulate the stability of HIF protein by post-translational hydroxylation of two conserved prolyl residues in its α subunit in an oxygen-dependent manner. Trans-4-prolyl hydroxylation of HIFα under normal oxygen (O2) availability enables its association with the von Hippel-Lindau (VHL) tumor suppressor pVHL E3 ligase complex, leading to the degradation of HIFα via the ubiquitin-proteasome pathway. Due to the obligatory requirement of molecular O2 as a co-substrate, the activity of PHDs is inhibited under hypoxic conditions, resulting in stabilized HIFα, which dimerizes with HIFβ and, together with transcriptional co-activators CBP/p300, activates the transcription of its target genes. As a key molecular regulator of adaptive response to hypoxia, HIF plays important roles in multiple cellular processes and its overexpression has been detected in various cancers. The HIF1α isoform in particular has a strong impact on cellular metabolism, most notably by promoting anaerobic, whilst inhibiting O2-dependent, metabolism of glucose. The PHD enzymes also seem to have HIF-independent functions and are subject to regulation by factors other than O2, such as by metabolic status, oxidative stress, and abnormal levels of endogenous metabolites (oncometabolites) that have been observed in some types of cancers. In this review, we aim to summarize current understandings of the function and regulation of PHDs in cancer with an emphasis on their roles in metabolism.
Keywords: prolyl hydroxylase domain (PHD), hypoxia-inducible factor (HIF), metabolism, mouse models, hydroxylation, 2-oxoglutarate-dependent dioxygenases
Introduction
Low oxygen (O2) and nutrient availability are often encountered in solid tumors due to abnormal vasculature and rapid proliferation of cancer cells.1 Adaptation to these microenvironment changes involves alterations in the expression of genes that regulate cellular energy production, biosynthesis, cell growth, and redox homeostasis. This response is, in part, mediated by the transcription factor hypoxia-inducible factor (HIF), a key regulator of the mammalian hypoxia response. The HIF signaling cascade encompasses diverse cellular pathways, some of which also intersect with tumorigenic processes.2,3
HIF is a heterodimeric transcription factor comprised of an O2-sensitive α subunit and a constitutively expressed β subunit, both of which are basic helix-loop-helix Per-ARNT-Sim domain proteins.4 The stability of HIF is primarily regulated by post-translational prolyl hydroxylation of HIFα, a reaction catalyzed by the HIF prolyl hydroxylase domain proteins (PHDs), a group of enzymes that act as O2 sensors in metazoans.5 PHD enzymes were first described roughly 10 years after the discovery of HIF.6–9 They are members of a large family of non-heme iron-dependent dioxygenases that utilize ferrous iron (Fe(II)) as a co-factor to catalyze the four-electron reduction of molecular O2. They incorporate each O2 atom, respectively, into the tricarboxylic acid (TCA) cycle metabolite 2-oxoglutarate (2OG) and the substrate polypeptide, forming succinate, carbon dioxide, and, in the case of PHDs, trans-4-hydroxylated prolyl products.4 The three main PHD isoforms, PHD1, PHD2, and PHD3 (also termed EGLN2, EGLN1, and EGLN3, respectively) have overlapping but unique tissue expression patterns. PHD2 is present in most tissues, whereas PHD1 is mainly in testes but also in brain, kidney, heart, and liver, and PHD3 showed highest expression in the heart.10 Both PHD2 and PHD3 are strongly induced by hypoxia.7 The three isoforms share high sequence homology in their C-terminal catalytic domains, but not in their N-termini, the function of which remains to be elucidated.8
There are three HIFα isoforms (HIF1α, HIF2α, and HIF3α). HIF1α is ubiquitously expressed; HIF2α expression is more cell-specific, such as in endothelial, parenchyma, and interstitial cells, whereas HIF3α is less well studied and has multiple splice variants, some of which lack the transactivation domain present in HIF1α and HIF2α.5,11–13 Under normal O2 availability, HIFα is constantly expressed and hydroxylated by PHDs on two conserved prolyl residues (Pro-405 and Pro-531 in HIF1α) present within an LXXLAP motif in its O2-dependent degradation domain. Trans-4-prolyl hydroxylation confers a >1,000 fold increase in binding affinity to the protein pVHL (von Hippel-Lindau tumor suppressor), which is part of an E3-ubiquitin ligase complex that mediates ubiquitination and degradation of HIFα by the 26S proteasome.14 Because O2 is an obligatory substrate for PHDs, HIFαs escape PHD-dependent hydroxylation under hypoxic conditions, translocate to the nucleus and dimerize with the β subunit (also termed ARNT), and through binding to hypoxia-response elements in promoter regions of target genes, activate the transcription of its target genes (Figure 1). Another layer of regulation is conferred through the hydroxylation of a specific asparaginyl residue (Asn-803 in HIF1α) in the C-terminal activation domain of HIFα, a post-translational modification catalyzed by another 2OG-dependent dioxygenase termed factor-inhibiting HIF (FIH). β-hydroxylation of Asn-803 blocks the interaction of HIFα with its transcriptional co-activators CBP (cAMP response element-binding protein)/p300, thereby inhibiting HIF activity. Unlike FIH, which has a relatively tight affinity for O2 (Km value ~90 μM), PHDs bind O2 loosely with Km values of each isoform in the range of 230–250 μM.4 This concentration is well above observed physiological O2 levels, making the PHDs sensitive to variations in local O2 in tissues and thus acting as O2 sensors. However, the multiple co-substrate and co-factor dependent nature of the PHDs raises the possibility that they also respond to metabolic alterations (Figure 1). Further, PHDs have been reported to have HIF-independent functions. In this review, we aim to summarize the current understandings of the roles of PHD enzymes in tumorigenesis with an emphasis on metabolism.
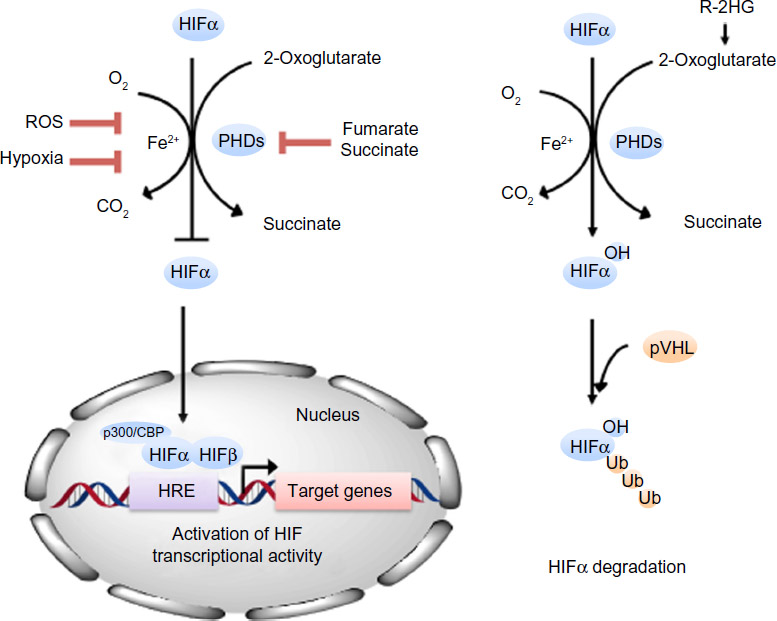 | Figure 1 PHDs are 2OG-dependent dioxygenases that regulate the stability of HIFα. In the presence of oxygen, PHDs post-translationally hydroxylate a prolyl residue in the NODDD and CODDD of HIFα subunits, which leads to its interaction with pVHL and ubiquitin-mediated degradation of HIFα by the 26S proteasome. Because PHDs have a low affinity for oxygen, they become inactive under hypoxic conditions, which allow HIFα to escape degradation, migrate to the nucleus, form a complex with the β subunit and transcriptional co-activators p300/CBP and regulate the transcription of its target genes by binding to hypoxia-responsive elements in their promoter regions. Fumarate and succinate structurally mimics 2OG (succinate is also a product of the reaction) and can competitively inhibit PHD activities when present at elevated concentrations, as observed in some cancer cells. R-2HG can enhance PHD activity and promote HIF degradation. In addition, ROS has also been suggested to modulate PHD activity. |
HIF-dependent regulation of metabolism
Elevated HIF1α and HIF2α levels have been observed in many human cancers and are often associated with poor prognosis.15 Particularly, a major proportion of clear cell renal cell carcinomas (ccRCCs) lose the function of VHL, which results in the inability to degrade HIFα in an O2-dependent manner.16 HIF1α and HIF2α have both overlapping and non-redundant functions.11 Overall, HIF1α is associated with metabolism and energy production (Figure 2), whereas HIF2α has a more pro-proliferative effect that is advantageous to tumor growth.17
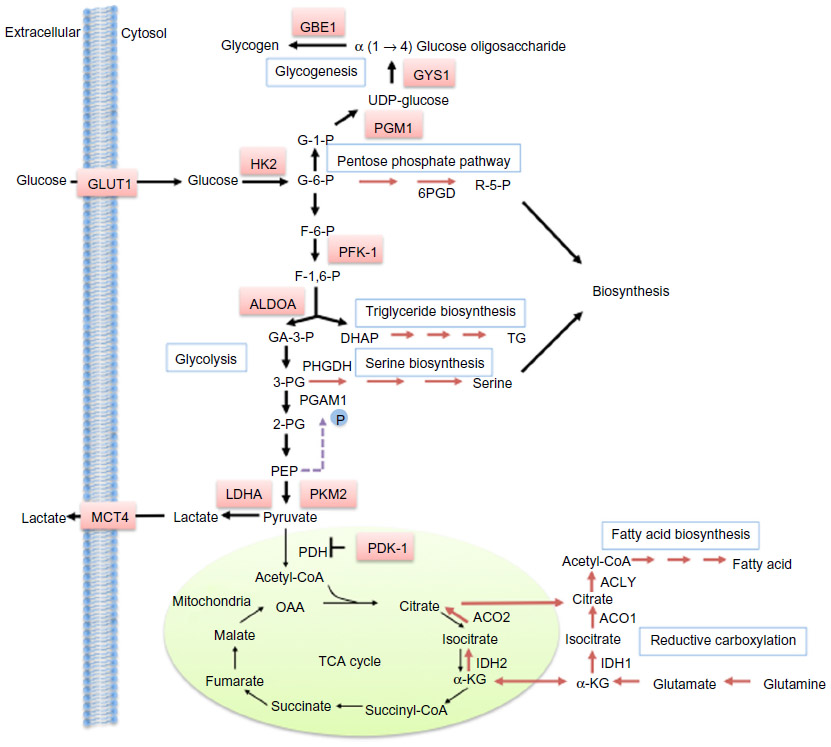 | Figure 2 HIF-dependent regulation of metabolism. |
Glycolysis
Many cancer cells exhibit enhanced capacity for anaerobic metabolism of glucose to produce lactate, even under sufficient O2 availability. This phenomenon was first described by Warburg nearly a century ago,18,19 and also forms the basis of diagnostic cancer imaging using the labeled glucose analogue fluorodeoxyglucose in position emission tomography.20 A high glycolytic flux allows for continued adenosine triphosphate (ATP) production independent of oxidative phosphorylation in the mitochondria, reduces the generation of reactive oxidative species (ROS), and provides glycolytic intermediates that could be utilized by biosynthetic pathways to support growth. HIF1α is known to upregulate glycolysis by activating the transcription of enzymes on the glycolysis pathway, such as 6-phosphofructo-1-kinase, hexokinase 2, and fructose-bisphosphate aldolase A. Additionally, HIF1α also regulates the auxiliary processes to glycolysis, such as increasing glucose uptake by activating the glucose transporter 1, and facilitating glucose carbon efflux via lactate by upregulating lactate dehydrogenase and monocarboxylate carrier MCT4.21–25
Pyruvate kinase (PK) catalyzes the final step in glycolysis, converting phosphoenolpyruvate (PEP) to pyruvate. The M2 isoform of pyruvate kinase (PKM2) is another glycolytic enzyme induced by HIF1α.26 PKM2 may be present as a highly active tetramer, or in a less active dimeric form, which is the case in many cancer cells. Due to the crucial position of PK on the glycolytic pathway, the presence of the inactive PKM2 dimer fine-tunes the glycolysis rate, causing accumulation of glycolytic intermediates, which can be channeled into biosynthetic pathways to produce nucleotides, phospholipids, and amino acids (reviewed by Vander Heiden et al).27 Thus, PKM2 has an anabolic effect by mediating glucose utilization in a manner that supports both bioenergetics and biosynthesis.
Recently, it has been shown that HIF1α also physically interacts with the PKM2 protein, resulting in increased transcriptional activity. Additionally, PHD3 catalyzes the hydroxylation of PKM2 on two prolyl residues at moderately hypoxic conditions, which strengthens the PKM2–HIF1α interaction and enhances HIF activity, forming a positive feedback loop for regulating glucose metabolism.26 A further layer of complexity is added by the fact that accumulation of the PKM2 substrate PEP promotes PEP-dependent histidine phosphorylation and thereby enhances the activity of phosphoglycerate mutase.28 Phosphoglycerate mutase is a glycolytic enzyme that catalyzes the conversion of 3-phosphoglycerate to 2-phosphoglycerate. 2-phosphoglycerate can stimulate the activity of phosphoglycerate dehydrogenase, the first and key regulatory enzyme in the serine biosynthesis pathway, whereas elevated 3-phosphoglycerate competitively inhibits the pentose phosphate pathway enzyme 6-phosphogluconate dehydrogenase, placing phosphoglycerate mutase at an important position in glucose metabolism.
In addition to promoting glycolysis, HIF also inhibits the oxidation of glucose carbon in the mitochondria by up-regulating pyruvate dehydrogenase kinase-1 and -3, which phosphorylates and thereby inactivates pyruvate dehydrogenase (PDH), the mitochondrial enzyme responsible for converting pyruvate to acetyl-CoA to prevent glycolysis-derived pyruvate from entering the tricarboxylic acid (TCA) cycle.29–31 Under hypoxia, mitochondrial respiration generates increased ROS, reasoning the diversion of pyruvate away from the TCA cycle, but perhaps another crucial advantage of metabolizing pyruvate through the lactate axis instead is the inherent need for the regeneration of co-factor nicotinamide adenine dinucleotide (NAD)+ in order to sustain a continued high glycolytic flux.
Mitochondrial activity
In addition to limiting pyruvate availability to the TCA cycle, HIF also regulates mitochondrial activity in a manner that decreases O2 consumption and promotes O2 conservation in order to maximize respiratory efficiency and limit ROS generation under low O2 tension. For example, HIF1α transcriptionally activates the mitochondrial protein NDUFA4L2 (NADH dehydrogenase [ubiquinone] 1 alpha subcomplex 4-like 2), which decreases O2 consumption by inhibiting electron transport chain Complex I activity.32 HIF also modulates cytochrome c oxidase (electron transport chain complex IV) by activating transcription of cytochrome c oxidase subunit IV-2 and ATP-dependent protease La, a mitochondrial protease that specifically degrades the cytochrome c oxidase subunit IV-1 isoform, leading to optimized respiration efficiency under hypoxia.33 Further, HIF1 regulates the expression of micro-RNA mir-210, causing downregulation of the Complex I subunit NDUFA4 (NADH dehydrogenase [ubiquinone] 1 alpha subcomplex), Complex II subunit SDHD (subunit D of succinate dehydrogenase [SDH] complex), the iron-sulfur cluster scaffold proteins 1/2 and the cytochrome c oxidase assembly protein COX10.34,35,36,37 Finally, the HIF target gene BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 impairs mitochondrial bioenergetics and promotes mitophagy, thereby conferring hypoxia tolerance.38,39
Glycogen and lipid metabolism
Significant lipid and glycogen accumulation occurs in some types of cancer, such as the VHL-defective ccRCC, giving its clear cell phenotype.40 HIF can enhance glycogen synthesis under hypoxia, leading to increased glycogen stores in both non-cancer and cancer cells. Both HIF1α and HIF2α induce the first enzyme in glycogenesis, phosphoglucomutase1,41 whereas HIF1α also upregulates other glycogenic enzymes, including glycogen synthase 1, glucose-1-phosphate uridylyltransferase and 1,4-α glucan branching enzyme.42
HIF can also influence lipid metabolism. For example, HIF1α specifically induces the phosphatidate phosphatase isoform lipin 1, which is an enzyme involved in triglyceride biosynthesis.43 Hypoxia induces the transcriptional factor sterol regulatory-element binding protein-1 through phosphorylation of protein kinase B and activation of HIF1α, leading to upregulation of fatty acid synthase.44 HIF1α upregulates peroxisome proliferator-activated receptor-γ to activate fatty acid uptake and glycerolipid biosynthesis,45 whereas constitutive activation of HIF2α resulted in severe hepatic steatosis associated with impaired β-oxidation, decreased lipogenic gene expression, and increased lipid storage capacity.46 Interestingly, in VHL-mutant RCC cells, HIF2α promoted glutamine catabolism to fuel lipid synthesis through the reductive carboxylation pathway, by which glutamine-derived 2OG is converted to citrate by the NADPH-dependent isocitrate dehydrogenase isoforms 1 or 2 (IDH1/2) and aconitase. Hif1α expression also induced reductive carboxylation in mouse neonatal epithelial kidney cells.47,48 Mechanistically, it was proposed that HIF expression induces low intracellular citrate levels, which enhances glutamine utilization through the reductive flux of the TCA cycle to support lipogenesis.
Antioxidative effect
Both HIF isoforms have been reported as upstream regulators of an antioxidative response in different cell types.49–51 HIF1α reduces mitochondria-dependent generation of ROS under hypoxia by promoting anaerobic metabolism of glucose. Increased flux through the pentose phosphate pathway, in part mediated by PKM2, produces NADPH, essential for countering oxidative stress. Hif2α-deficient mice exhibited multiple-organ dysfunctions and enhanced oxidative stress, and Hif2α was shown to regulate antioxidant enzymes, including superoxide dismutase 1, superoxide dismutase 2, glutathione peroxidase 1, and catalase.50 HIF2α knockdown in human lung adenocarcinoma cells also led to lower expression levels of antioxidant enzymes, including heme oxygenase 1, ceruloplasmin, crystallin, peroxiredoxin 3, and glutathione peroxidase 8.49
HIF-independent regulation by PHDs
Although HIF is the most well-known substrate, PHDs have been suggested to have other direct or indirect targets. Here we summarize a few findings in current literature that may be associated with tumorigenic processes.
Nuclear factor kappa-light-chain-enhancer of activated B cells pathway signaling
The transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which is involved in inflammatory and innate immune responses, is constitutively activated in some cancers.52 PHD1 and PHD3 can inhibit IκB kinase β (IKKβ), leading to decreased NF-κB signaling. It has been suggested that the regulation of IKKβ occurs via PHD-dependent hydroxylation of a putative LXXLAP motif present in the IKKβ proteins.53,54 However, PHD3 may also inactivate IKKβ by blocking its interaction with the heat shock protein 90 in a hydroxylation-independent manner.55 In line with this, PHD2 has been shown to downregulate angiogenesis and vasculogenesis by inhibiting angiogenin and interleukin 8 via the NF-κB pathway. Further, haplodeficient Phd2 mice exhibited activation of NF-κB signaling in tissue-resident M2-like macrophages, causing increased collateral arteriogenesis (Table 1).56
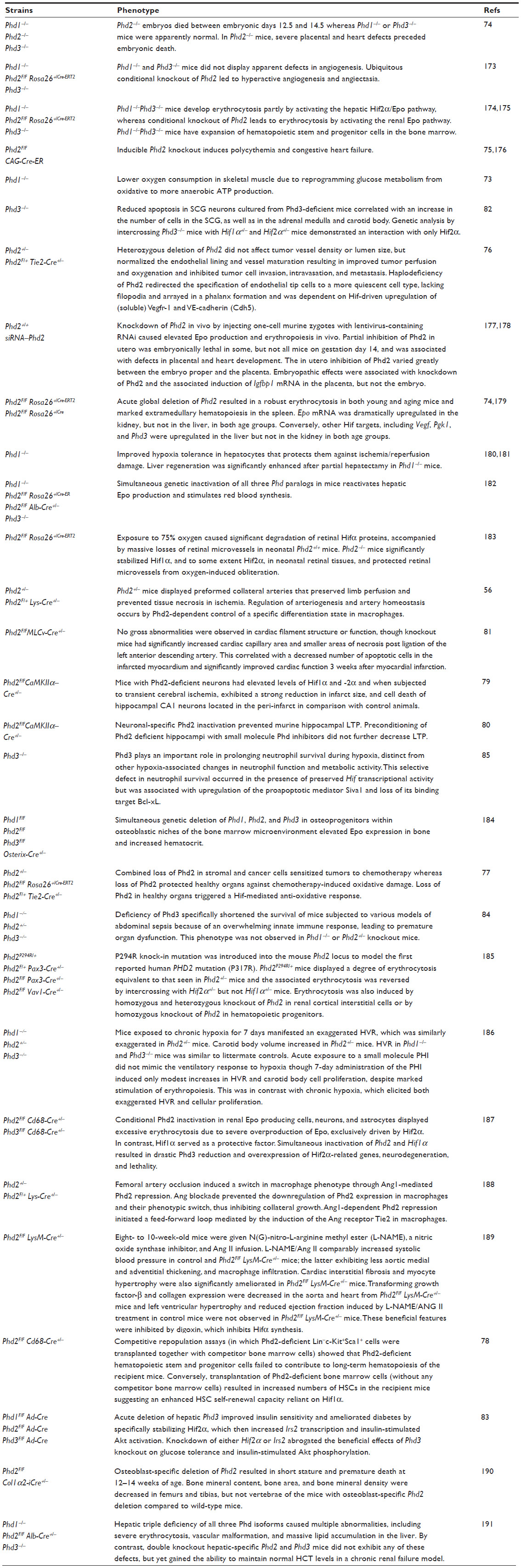 | Table 1 Phenotypes of Phd-deficient mice |
Activating transcription factor 4
Activating transcription factor 4 (ATF4) is a transcription factor that regulates genes involved in redox balance, amino acid metabolism, autophagy, and angiogenesis, promoting tumor survival under hypoxic and nutrient-deprived environments.57 It has been shown that ATF4 upregulation under hypoxia is dependent on PHD3 but independent of VHL-mediated ubiquitination.58 Another report demonstrated that both PHD1 and PHD3 interact with ATF4, and co-expression with either PHD isoform repressed ATF4 transcription activity. However, the authors also showed that ATF4 did not serve as a hydroxylation substrate for either PHD1 or PHD3 in vitro.59
PHD3 and apoptosis
Kinesin-like protein KIF1β regulates neuronal apoptosis, and its loss-of-function mutations have been identified in neuroblastomas and pheochromocytomas.60 KIF1β induction is dependent on PHD3 hydroxylase activity, though it is unclear whether KIF1β is a direct substrate for hydroxylation.61 Recently it has been shown that HCLK2 (human homologue of the Caenorhabditis elegans biological clock protein Clk2) is hydroxylated by PHD3. HCLK2 hydroxylation activates the ataxia telangiectasia and Rad3-related checkpoint kinase 1 pathway and subsequent apoptosis in response to DNA damage.62 Another reported substrate for PHD3 is beta(2)-adrenergic receptor, a prototypic G-protein coupled receptor whose induction has been associated with apoptosis.63–65 Hydroxylation of beta(2)-adrenergic receptor leads to its interaction with the pVHL-E3 ligase complex and ubiquitin-mediated degradation.66
RNA polymerase II
PHD1 and PHD3 also hydroxylate the large subunit of RNA polymerase II (Rpb1) on Pro-1465 located within an LXXLAP motif, which is necessary for phosphorylation of Ser5 in the C-terminal domain of Rpb1.67 Hydroxylation leads to non-degradative ubiquitination and is necessary for oxidative stress-induced recruitment of Rpb1 to the chromatin-engaged fraction of VHL-positive RCC cells. Although PHD2 also interacted with Rpb1, it had an inhibitory effect on hydroxylation in this case. Hydroxylation of Pro-1465 was suggested to have an oncogenic effect as expression of wild-type Rpb1 stimulated kidney tumor growth, whereas expression of the P1465A mutant prevented it.68
Isoform specific effect of PHDs
Germline mutations in PHDs are not considered a frequent cause of hereditary pheochromocytoma or RCC,69 in which HIF upregulation is frequently demonstrated.70,71 Interestingly, PHD3 protein is undetectable in ccRCC primary tumors compared to pheochromocytoma and colorectal cancers,72 indicating context-dependent roles of PHD isoforms. Not surprisingly, Phd knockout mice also display some isoform-specific phenotypes (Table 1). Phd1 deficiency lowers O2 consumption and induces hypoxia tolerance in skeletal muscle by altering glucose metabolism from oxidative to anaerobic, thereby reducing ROS generation whilst maintaining a basal mitochondrial respiration rate. This protects Phd1-deficient myofibers against lethal ischemia.73 Mechanistically, loss of Phd1 activates the Pparα pathway, which upregulates pyruvate dehydrogenase kinase isozyme 4 (Pdk4) in a primarily Hif2α-dependent manner. Pdk4 restricts entry of glycolytic intermediates into the TCA cycle, reducing mitochondria respiration and resulting in hypoxia tolerance.73
Germline Phd2 knockout causes embryonic lethality due to placental and heart defects.74 Conditional deletion of Phd2 results in increased angiogenesis, polycythemia, and congestive heart failure in mice.75 Phd2 inactivation redirects endothelial tip cells to a more quiescent cell type to facilitate restoration of O2 supply, which is in part dependent on Hif2α-mediated upregulation of soluble vascular endothelial growth factor receptor-1 and vascular endothelial cadherin.76 Haplodeficiency of Phd2 normalized endothelial lining and vessel maturation in mouse stroma, and led to activation of the NF-κB pathway in macrophages, causing enhanced collateral arteriogenesis that prevented tissue necrosis in ischemia.56 Further, reduced Phd2 activity in hearts and kidneys amplifies the antioxidative response of both Hif isoforms and protects normal organ functions from side toxicity of chemotherapy-induced ROS.77 Conditional Phd2 knockout studies in the hematopoietic system revealed that Phd2 regulates hematopoietic stem cell maintenance under the steady state and stressful conditions.78 Neuronal-specific Phd2 knockout pre-conditions the forebrain against hypoxic/ischemic insults by inducing neuroprotective Hif target genes.79 In another study, neuronal Phd2 deletion led to impaired synaptic signaling in the hippocampus.80 Phd2 deletion in cardiomyocytes also protected mice from acute myocardial ischemic injury, potentially due to Hif1α-dependent tissue protection effects.81
Phd3 knockout in mice resulted in a hypofunctional sympathoadrenal system and reduced blood pressure.82 Acute deletion of Phd3 in the liver improved glucose tolerance and insulin sensitivity by specifically stabilizing Hif2α, which augments the expression of insulin receptor substrate-2 and leads to an increase in insulin-stimulated phosphorylation of protein kinase B and transcription factor Forkhead box protein O1.83 Loss of Phd3 activity also increased pro-inflammatory activity of macrophages through Hif1α- and NF-κB-dependent enhancement of the innate immune response, leading to premature organ dysfunction in septic Phd3-deficient mice.84 Further, Phd3-deficiency led to increased neutrophil apoptosis during hypoxia, an effect associated with the upregulation of the proapoptotic mediator Siva1 and loss of its binding target Bcl-xL.85
Regulation of PHD activity
PHDs depend on multiple co-substrates and co-factors for activity, and therefore are potential targets of regulation not only by O2, but also by other factors including 2OG availability, oxidative stress, and abnormal levels of endogenous metabolites that are structural mimics of 2OG. In addition to being a co-factor to dioxygenases, 2OG is also a key TCA cycle metabolite and is involved in intracellular nitrogen transport by acting as the nitrogen acceptor in transamination reactions. Gottlieb et al showed that amino acid starvation led to 2OG depletion and thereby inactivation of PHDs, the activity of which is required for amino acid-induced signaling of the mammalian target of rapamycin complex 1, raising the possibility that PHDs are also cellular nutrient sensors.86 On the other hand, it has been shown that purified PHD enzymes have a relatively tight affinity for 2OG. The Km values for 2OG are 1–2 μM for PHD1/2 and 12 μM for PHD3 in vitro,87 which are significantly lower than 2OG levels generally detected under normal physiology.88,89 The discrepancies in reported Km values of PHDs for 2OG may partly arise from the presence of endogenous 2OG that are pre-bound to the recombinant enzymes during purification. Therefore further evidence may be needed to support whether oscillations of intracellular 2OG concentration significantly alters PHD activities.
The activity of PHDs can also be modulated by oncometabolites. SDH and fumarate hydratase (FH) catalyze sequential steps in the TCA cycle, and their inactivation results in accumulation of succinate and fumarate, respectively, both of which are structural analogues of 2OG (succinate is also a product of the 2OG-dependent dioxygenase-catalyzed reactions) and have been shown to inhibit the PHD enzymes at pathophysiological concentrations.90–94
Loss-of-function mutations in SDH are detected in paragangliomas and pheochromocytomas, as well as a number of other malignancies, whereas FH inactivation occurs primarily in hereditary leiomyomatosis and RCC.95 HIF stabilization is often observed in these tumors, potentially due to fumarate or succinate-mediated inhibition of PHDs.90,96–100 However, it has also been argued that, because PHDs have a tight binding affinity for 2OG, the activation of HIF in the SDH- or FH-deficient settings is likely to be due to inactivation of PHDs by oxidative stress, which is generated as a result of mitochondrial dysfunction.101,102
Gain of function mutations in IDH1 and IDH2 frequently occur in low-grade gliomas and acute myeloid leukemia, conferring a neomorphic enzyme activity that produces (R)-2-hydroxyglutarate (R-2HG), which is postulated to promote, rather than inhibit, the hydroxylase activities of PHD1 and PHD2, thereby diminishing HIF signaling (reviewed by Losman and Kaelin).103 Reduced HIF levels were associated with enhanced growth of IDH1 mutant human astrocytes.93 Consistent with this, PHD2 depletion in erythroleukemic cells transformed by the expression of IDH1 mutant abolished their growth factor independence and restored normal differentiation.104,105
ROS has also been assigned a role in regulating PHD activity. Potential mechanisms of ROS-induced PHD inactivation include oxidation of the ferrous iron (Fe[II]) to the ferric state (Fe[III]) or oxidation of active site residues.5 However, although peroxide treatment initially reduced PHD activity, prolonged exposure enhanced it.106 Bio-reductants such as ascorbate (vitamin C) or glutathione are necessary for maximal PHD activity in vitro, potentially through facilitating the reduction of ferric iron to its ferrous state.107,108 Treatment with ascorbate or the antioxidant N-acetylcysteine in vivo inhibited growth of Myc-mediated xenograft tumors by diminishing HIF1α levels in a PHD2 and VHL-dependent manner.109 The ROS scavenging agent Se-methylselenocysteine also displayed an antitumor effect in ccRCC xenograft tumors by promoting PHD2 activity and reducing HIF1α.72,110
There is controversy, however, as to whether PHDs are direct physiological targets of ROS. Genetic and pharmacological inhibition of electron transport chain has been shown to stabilize HIF, supporting a direct role of mitochondrial-generated ROS in HIF regulation.111–115 However, overexpression of thioredoxin reductase 1 blocked ROS generation under hypoxia but had no effect on HIF1α accumulation or transcriptional activity.116 In line with this, inhibition of mitochondria ATP synthase increased mitochondrial ROS production but did not lead to HIF1α stabilization.117,118 Further, it has been shown that the HIF asparaginyl hydroxylase FIH is markedly more sensitive to exogenous peroxides than PHDs,119 raising the possibility of differential regulation of HIF protein stability and transcriptional activity by hypoxia and oxidative stress. An alternative model of PHD regulation proposes that increased mitochondrial O2 consumption reduces cytosolic O2 availability to the PHDs, which can inhibit PHD activity and lead to HIF accumulation.120
Nitric oxide (NO) has also been proposed to regulate PHD activity. Biochemical and biophysical studies suggested that NO may compete with O2 for binding to the active site Fe(II), and can potentially interact with multiple cysteine residues in PHD2 in vitro.121 Although acute exposure to high doses of NO stabilized HIF1α, prolonged exposure or low doses reduced HIF1α accumulation in cells, potentially by modulating co-substrate and co-factor availability to PHDs and HIF1α-mediated feedback expression of PHD2.122
PHDs are also subject to regulation at the protein level. The seven in absentia homology 2 (Siah2) E3 ligase ubiquitin protein has been shown to preferentially target PHD3 for polyubiquitylation and proteasomal destruction.123 Siah2-dependent degradation of PHD3 increased under hypoxia conditions, whereas Siah2 null fibroblasts displayed reduced HIF1α expression levels.124 On the other hand, the FK506-binding protein 38 has been shown to negatively regulate PHD2 protein stability through interaction with the N-terminal MYND (myeloid, Nervy, and DEAF-1) finger domain of PHD2 in an ubiquitin-independent proteasomal pathway.125
Other 2-oxygenases and metabolism
The Fe(II) and 2OG-dependent dioxygenases are a large enzyme family comprised of over 60 members, many of which have yet unidentified functions. Here we summarize a few 2-oxygenases that may have implications in cancer.
Chromatin modifying 2-oxygenases
The ten-eleven translocation (TET) family of DNA modifying enzymes (comprising of TET1, TET2, and TET3) are 2OG-dependent dioxygenases that catalyse the hydroxylation of 5-methylcytosine to form 5-hydroxymethylcytosine,126 and the oxidation of 5-hydroxymethylcytosine to 5-formylcytosine and 5-carboxylcytosine.127,128 These reactions are considered intermediate steps in DNA demethylation, but interestingly, 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxylcytosine also exhibit replication dependent dilution and each has specific “reader” proteins,129–131 suggesting that these modifications may be functional during development. TET2 mutations frequently occur in hematological malignancies,132,133 and TET1 can act as a tumor suppressor in prostate and breast cancers and as an oncogene in myeloid/lymphoid leukemia-rearranged leukemia.134–136 Intriguingly, somatic mutations in TET2 and IDH occur in acute myeloid leukemia in a mutually exclusive manner.137 Further, 2HG-producing IDH mutant cancers frequently display a DNA hypermethylation phenotype,138–140 whereas R-2HG has been shown to inhibit TET2 activity in vitro.141 This evidence points to TET2 being a pathological target of the oncometabolite R-2HG. Similarly, a hypermethylation phenotype has been observed in SDH- and FH-mutant malignancies,142,143 indicating that the TET-mediated epigenome remodeling is potentially an important oncogenic mechanism in SDH and FH-deficient cancers.144
The JmjC domain-containing histone H3 lysine 27 demethylase UTX and histone H3 lysine 4 demethylase JARID1C are both 2OG-dependent dioxygenases. Inactivating somatic mutations in UTX have been identified in multiple tumor types, including myeloid leukemias, RCCs, breast and colorectal cancers, and glioblastoma.145 Inactivating mutations in JARID1C were identified in ccRCC, together with the histone H3 lysine 36 methyltransferase SET domain containing 2.146 The evidence highlights the importance of chromatin modifying enzymes in cancer regulation.
FIH
In comparison to PHDs, whose catalytic activities are remarkably specific, FIH is a much more promiscuous enzyme, catalyzing the hydroxylation of not only HIF but also a large family of ankyrin repeat domain-containing proteins.147 In addition to being an asparaginyl hydroxylase, FIH can also catalyze the β-hydroxylation of aspartyl, histidinyl, serinyl, leucinyl, and isoleucinyl residues, although the biological significance of the non-asparaginyl substrates is unclear.148,149 Interestingly, the Fih null mice displayed a phenotype of dysregulated energy metabolism with no developmental defects or other classical features associated with canonical HIF functions.150 Fih overexpression in osteosarcoma cells has been shown to increase tumor growth in a xenograft mouse model, whereas its suppression did not influence tumor cell proliferation.151 In another study, FIH inhibition or knockdown in VHL-defective RCC cells increased HIF-dependent apoptosis and promoted survival of these cells.152 Taken together, the effects of FIH on metabolism is context-specific and may be both HIF-dependent and HIF-independent.
Fat mass and obesity-associated
The 2OG-dependent dioxygenase fat mass and obesity-associated protein (FTO) catalyzes RNA and single strand DNA demethylation in vitro.153–155 The expression of FTO is associated with increased risk for obesity and type 2 diabetes.154,156 Mouse models of Fto suggest its deficiency leads to postnatal growth retardation, increased energy expenditure, and a lean phenotype,157 whereas its overexpression results in increased food intake and obesity in mice.158 Amino acid deprivation in mouse and human cell lines downregulated FTO mRNA and protein levels, whereas FTO-deficient cells display reduced mammalian target of rapamycin complex 1 signaling potentially due to the inability to sense amino acids.159,160 Recently, it has also been suggested that FTO may be implicated in melanoma independent of body mass index.161
Ribosomal hydroxylases
Recently, a subset of 2-oxygenases has been shown to possess unprecedented enzymatic activities and catalyze post-translational hydroxylation of ribosomal proteins. The 2OG and Fe(II)-dependent oxygenase domain-containing protein 1 catalyzes the trans-3-prolyl hydroxylation of Pro-62 in the 40S ribosomal protein S23 and is postulated to have a role in the regulation of translation and growth.162–164 Jumonji domain-containing 4 is a 2-oxygenase that catalyzes the C4-lysyl hydroxylation of eukaryotic release factor 1 at Lys-63, a conserved residue located within its stop codon recognition domain, and eukaryotic release factor 1 hydroxylation has been suggested to promote translational termination efficiency.165 Myc-induced nuclear antigen with a molecular mass of 53 kDa (MINA53) and NO66 are known to interact with the Myc oncogenic pathway and are highly expressed in several cancers. Both MINA53 and NO66 have previously been assigned as histone lysine demethylases.166,167 However, direct evidence of their histone demethylase activity by mass spectrometry is lacking. Recently it was shown that MINA53 catalyzes the (2S, 3S)-3-histidyl hydroxylation of the 60S ribosomal protein Rpl27a at His-39, whereas NO66 hydroxylates the 60S ribosomal protein Rpl8 at His-216 with the same stereochemistry.168 Whether they play regulatory roles in translation or ribosomal biogenesis in vivo remains to be determined.
Conclusion
The identification and function of 2-oxygenases has rapidly expanded over the last decade, though the PHDs have remained a primary focus within this class of enzymes. Though mainly associated with the regulation of HIF, many studies have identified diverse and often contrasting roles in cellular metabolism for both PHD-mediated HIF-dependent and HIF-independent metabolic regulation. This can be explained, at least in part, by the range in specificity for HIF subunits and other substrates combined with tissue-specific expression and cellular localization of the different PHD isoforms. Both genetic and biochemical inhibition of the PHDs have been demonstrated to be of benefit in a plethora of clinical applications, including protective roles in ischemia/reperfusion injury, neural protection, support of macrophage function, enhanced neutrophil survival, protection against inflammatory bowel disease, and enhanced expression of epithelial barrier protection genes. Many of these functions are mediated by HIF1α, which plays a key role in the reprogramming of metabolism by activating the transcription of genes encoding glucose transporters and glycolytic enzymes. In contrast, PHD3-mediated regulation of HIF2α regulates hepatic lipid and glucose metabolism and provides a direct link between hypoxia and insulin signaling in diabetes.83 Further, HIF2α regulates erythropoietic responses to hypoxia in anemia inferring an important role in iron homeostasis.169–172
In summary, targeting of PHDs provides a feasible method of therapeutic intervention in cancer metabolism and has traction outside oncology. However, given the cell- and tissue-specific roles of the PHDs and their main substrate HIF, multiple factors should be considered in the design of either small compound inhibitors or genetic targeting, such as drug efficacy, toxicity, pharmacokinetics, and accurate delivery to the target tissue.
Acknowledgments
Cancer Research UK (A13349) has provided financial support to PJP, as has The European Research Council under the European Community’s Seventh Framework Programme (FP7/2007–2013)/ERC grant agreement no 310837. TS is financially supported by a Grant-in-Aid for scientific research on Innovative Areas, Japan (No 22134007), and the Yamagata Prefectural Government and City of Tsuruoka. KRK is a Cancer Research UK Senior Cancer Research Fellow. Work at KRK’s laboratory is funded by Cancer Research UK, Leukemia and Lymphoma Research, and the Kay Kendall Leukemia Fund.
Disclosure
The authors report no conflicts of interest in this work.
References
Harris AL. Hypoxia – a key regulatory factor in tumour growth. Nature Reviews Cancer. 2002;2(1):38–47. | |
Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. | |
Ratcliffe PJ. Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J Physiol. 2013;591 (Pt 8):2027–2042. | |
Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5(5):343–354. | |
Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402. | |
Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. | |
Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54. | |
Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–1340. | |
Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–468. | |
Lieb ME, Menzies K, Moschella MC, Ni R, Taubman MB. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem Cell Biol. 2002;80(4):421–426. | |
Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22. | |
Patel SA, Simon MC. Biology of hypoxia-inducible factor-2alpha in development and disease. Cell Death Differ. 2008;15(4):628–634. | |
Maynard MA, Qi H, Chung J, et al. Multiple splice variants of the human HIF-3 alpha locus are targets of the von Hippel-Lindau E3 ubiquitin ligase complex. J Biol Chem. 2003;278(13):11032–11040. | |
Min JH, Yang H, Ivan M, Gertler F, Kaelin WG Jr, Pavletich NP. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296(5574):1886–1889. | |
Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29(5):625–634. | |
Kaelin WG Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8(11):865–873. | |
Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014;2(1):3. | |
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. | |
Warburg O. The Metabolism of Tumours. London: Arnold Constable; 1930. | |
De Witte O, Lefranc F, Levivier M, Salmon I, Brotchi J, Goldman S. FDG-PET as a prognostic factor in high-grade astrocytoma. Journal of Neurooncology. 2000;49(2):157–163. | |
Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda). 2004;19:176–182. | |
Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12(2):149–162. | |
Ryan HE, Lo J, Johnson RS. HIF-1a is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17(11):3005–3015. | |
Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23(24):9361–9374. | |
Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. 2006;281(14):9030–9037. | |
Luo W, Hu H, Chang R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145(5):732–744. | |
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. | |
Vander Heiden MG, Locasale JW, Swanson KD, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329(5998):1492–1499. | |
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–185. | |
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3(3):187–197. | |
Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem. 2008;283(42):28106–28114. | |
Tello D, Balsa E, Acosta-Iborra B, et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011;14(6):768–779. | |
Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129(1):111–122. | |
Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10(4):273–284. | |
Favaro E, Ramachandran A, McCormick R, et al. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS One. 2010;5(4):e10345. | |
Chen Z, Li Y, Zhang H, Huang P, Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene. 2010;29(30):4362–4368. | |
Puisségur MP, Mazure NM, Bertero T, et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011;18(3):465–478. | |
Band M, Joel A, Hernandez A, Avivi A. Hypoxia-induced BNIP3 expression and mitophagy: in vivo comparison of the rat and the hypoxia-tolerant mole rat, Spalax ehrenbergi. FASEB J. 2009;23(7):2327–2335. | |
Rikka S, Quinsay MN, Thomas RL, et al. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011;18(4):721–731. | |
Fenner A. Kidney cancer: creating a molecular atlas of clear cell renal cell carcinoma genetics. Nat Rev Urol. 2013;10(9):489. | |
Pelletier J, Bellot G, Gounon P, Lacas-Gervais S, Pouysségur J, Mazure NM. Glycogen synthesis is induced in hypoxia by the hypoxia-inducible factor and promotes cancer cell survival. Front Oncol. 2012;2:18. | |
Pescador N, Villar D, Cifuentes D, et al. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PLoS One. 2010;5(3):e9644. | |
Mylonis I, Sembongi H, Befani C, Liakos P, Siniossoglou S, Simos G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J Cell Sci. 2012;125(Pt 14):3485–3493. | |
Furuta E, Pai SK, Zhan R, et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008;68(4):1003–1011. | |
Krishnan J, Suter M, Windak R, et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009; 9(6):512–524. | |
Rankin EB, Rha J, Selak MA, et al. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol Cell Biol. 2009;29(16):4527–4538. | |
Gameiro PA, Yang J, Metelo AM, et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013;17(3):372–385. | |
Metallo CM, Gameiro PA, Bell EL, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481(7381):380–384. | |
Bertout JA, Majmundar AJ, Gordan JD, et al. HIF2alpha inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc Natl Acad Sci U S A. 2009;106(34):14391–14396. | |
Scortegagna M, Ding K, Oktay Y, et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat Genet. 2003;35(4):331–340. | |
Mukhopadhyay CK, Mazumder B, Fox PL. Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J Biol Chem. 2000;275(28):21048–21054. | |
Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446(5):475–482. | |
Cummins EP, Berra E, Comerford KM, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103(48):18154–18159. | |
Fu J, Taubman MB. Prolyl hydroxylase EGLN3 regulates skeletal myoblast differentiation through an NF-kappaB-dependent pathway. J Biol Chem. 2010;285(12):8927–8935. | |
Xue J, Li X, Jiao S, Wei Y, Wu G, Fang J. Prolyl hydroxylase-3 is down-regulated in colorectal cancer cells and inhibits IKKbeta independent of hydroxylase activity. Gastroenterology. 2010;138(2):606–615. | |
Takeda Y, Costa S, Delamarre E, et al. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature. 2011;479(7371):122–126. | |
Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40(1):14–21. | |
Köditz J, Nesper J, Wottawa M, et al. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;110(10):3610–3617. | |
Hiwatashi Y, Kanno K, Takasaki C, et al. PHD1 interacts with ATF4 and negatively regulates its transcriptional activity without prolyl hydroxylation. Exp Cell Res. 2011;317(20):2789–2799. | |
Yeh IT, Lenci RE, Qin Y, et al. A germline mutation of the KIF1B beta gene on 1p36 in a family with neural and nonneural tumors. Hum Genet. 2008;124(3):279–285. | |
Schlisio S, Kenchappa RS, Vredeveld LC, et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008;22(7):884–893. | |
Xie L, Pi X, Mishra A, Fong G, Peng J, Patterson C. PHD3-dependent hydroxylation of HCLK2 promotes the DNA damage response. J Clin Invest. 2012;122(8):2827–2836. | |
Gu C, Ma YC, Benjamin J, Littman D, Chao MV, Huang XY. Apoptotic signaling through the beta-adrenergic receptor. A new Gs effector pathway. J Biol Chem. 2000;275(27):20726–20733. | |
Zaugg M, Xu W, Lucchinetti E, Shafiq SA, Jamali NZ, Siddiqui MA. Beta-adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation. 2000;102(3):344–350. | |
Shizukuda Y, Buttrick PM. Subtype specific roles of beta-adrenergic receptors in apoptosis of adult rat ventricular myocytes. J Mol Cell Cardiol. 2002;34(7):823–831. | |
Xie L, Xiao K, Whalen EJ, et al. Oxygen-regulated beta(2)-adrenergic receptor hydroxylation by EGLN3 and ubiquitylation by pVHL. Sci Signal. 2009;2(78):ra33. | |
Yi Y, Mikhaylova O, Mamedova A, et al. von Hippel-Lindau-dependent patterns of RNA polymerase II hydroxylation in human renal clear cell carcinomas. Clin Cancer Res. 2010;16(21):5142–5152. | |
Mikhaylova O, Ignacak ML, Barankiewicz TJ, et al. The von Hippel-Lindau tumor suppressor protein and Egl-9-Type proline hydroxylases regulate the large subunit of RNA polymerase II in response to oxidative stress. Mol Cell Biol. 2008;28(8):2701–2717. | |
Astuti D, Ricketts CJ, Chowdhury R, et al. Mutation analysis of HIF prolyl hydroxylases (PHD/EGLN) in individuals with features of phaeochromocytoma and renal cell carcinoma susceptibility. Endocr Relat Cancer. 2010;18(1):73–83. | |
Yang OC, Maxwell PH, Pollard PJ. Renal cell carcinoma: translational aspects of metabolism and therapeutic consequences. Kidney Int. 2013;84(4):667–681. | |
Pollard PJ, El-Bahrawy M, Poulsom R, et al. Expression of HIF-1alpha, HIF-2alpha (EPAS1), and their target genes in paraganglioma and pheochromocytoma with VHL and SDH mutations. J Clin Endocrinol Metab. 2006;91(11):4593–4598. | |
Chintala S, Najrana T, Toth K, et al. Prolyl hydroxylase 2 dependent and Von-Hippel-Lindau independent degradation of Hypoxia-inducible factor 1 and 2 alpha by selenium in clear cell renal cell carcinoma leads to tumor growth inhibition. BMC Cancer. 2012;12:293. | |
Aragonés J, Schneider M, Van Geyte K, et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008;40(2):170–180. | |
Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26(22):8336–8346. | |
Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG Jr. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008;111(6):3236–3244. | |
Mazzone M, Dettori D, Leite de Oliveira R, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136(5):839–851. | |
Leite de Oliveira R, Deschoemaeker S, Henze AT, et al. Gene-targeting of Phd2 improves tumor response to chemotherapy and prevents side-toxicity. Cancer Cell. 2012;22(2):263–277. | |
Singh RP, Franke K, Kalucka J, et al. HIF prolyl hydroxylase 2 (PHD2) is a critical regulator of hematopoietic stem cell maintenance during steady-state and stress. Blood. 2013;121(26):5158–5166. | |
Kunze R, Zhou W, Veltkamp R, Wielockx B, Breier G, Marti HH. Neuron-specific prolyl-4-hydroxylase domain 2 knockout reduces brain injury after transient cerebral ischemia. Stroke. 2012;43(10):2748–2756. | |
Corcoran A, Kunze R, Harney SC, Breier G, Marti HH, O’Connor JJ. A role for prolyl hydroxylase domain proteins in hippocampal synaptic plasticity. Hippocampus. 2013;23(10):861–872. | |
Hölscher M, Silter M, Krull S, et al. Cardiomyocyte-specific prolyl-4-hydroxylase domain 2 knock out protects from acute myocardial ischemic injury. J Biol Chem. 2011;286(13):11185–11194. | |
Bishop T, Gallagher D, Pascual A, et al. Abnormal sympathoadrenal development and systemic hypotension in PHD3-/- mice. Mol Cell Biol. 2008;28(10):3386–3400. | |
Taniguchi CM, Finger EC, Krieg AJ, et al. Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat Med. 2013;19(10):1325–1330. | |
Kiss J, Mollenhauer M, Walmsley SR, et al. Loss of the oxygen sensor PHD3 enhances the innate immune response to abdominal sepsis. J Immunol. 2012;189(4):1955–1965. | |
Walmsley SR, Chilvers ER, Thompson AA, et al. Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J Clin Invest. 2011;121(3):1053–1063. | |
Durán RV, MacKenzie ED, Boulahbel H, et al. HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene. 2013;32(38):4549–4556. | |
Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem. 2007;282(7):4524–4532. | |
Tennant DA, Frezza C, MacKenzie ED, et al. Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death. Oncogene. 2009;28(45):4009–4021. | |
Thirstrup K, Christensen S, Møller HA, et al. Endogenous 2-oxoglutarate levels impact potencies of competitive HIF prolyl hydroxylase inhibitors. Pharmacol Res. 2011;64(3):268–273. | |
Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8(2):143–153. | |
O’Flaherty L, Adam J, Heather LC, et al. Dysregulation of hypoxia pathways in fumarate hydratase-deficient cells is independent of defective mitochondrial metabolism. Hum Mol Genet. 2010;19(19):3844–3851. | |
Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. | |
Hewitson KS, Liénard BM, McDonough MA, et al. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J Biol Chem. 2007;282(5):3293–3301. | |
MacKenzie ED, Selak MA, Tennant DA, et al. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol. 2007;27(9):3282–3289. | |
Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest. 2013;123(9):3652–3658. | |
Pollard PJ, Brière JJ, Alam NA, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14(15):2231–2239. | |
Pollard P, Wortham N, Barclay E, et al. Evidence of increased microvessel density and activation of the hypoxia pathway in tumours from the hereditary leiomyomatosis and renal cell cancer syndrome. J Pathol. 2005;205(1):41–49. | |
Pollard PJ, Wortham NC, Tomlinson IP. The TCA cycle and tumorigenesis: the examples of fumarate hydratase and succinate dehydrogenase. Ann Med. 2003;35(8):632–639. | |
Baysal BE. On the association of succinate dehydrogenase mutations with hereditary paraganglioma. Trends Endocrinol Metab. 2003; 14(10):453–459. | |
Gimenez-Roqueplo AP, Favier J, Rustin P, et al. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet. 2001;69(6):1186–1197. | |
Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28(2):718–731. | |
Sullivan LB, Martinez-Garcia E, Nguyen H, et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51(2):236–248. | |
Losman JA, Kaelin WG Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27(8):836–852. | |
Koivunen P, Lee S, Duncan CG, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483(7390):484–488. | |
Losman JA, Looper RE, Koivunen P, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339(6127):1621–1625. | |
Niecknig H, Tug S, Reyes BD, Kirsch M, Fandrey J, Berchner-Pfannschmidt U. Role of reactive oxygen species in the regulation of HIF-1 by prolyl hydroxylase 2 under mild hypoxia. Free Radic Res. 2012;46(6):705–717. | |
Flashman E, Davies SL, Yeoh KK, Schofield CJ. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem J. 2010;427(1):135–142. | |
Nytko KJ, Maeda N, Schläfli P, Spielmann P, Wenger RH, Stiehl DP. Vitamin C is dispensable for oxygen sensing in vivo. Blood. 2011;117(20):5485–5493. | |
Gao P, Zhang H, Dinavahi R, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12(3):230–238. | |
Chintala S, Tóth K, Cao S, et al. Se-methylselenocysteine sensitizes hypoxic tumor cells to irinotecan by targeting hypoxia-inducible factor 1alpha. Cancer Chemother Pharmacol. 2010;66(5):899–911. | |
Bell EL, Chandel NS. Mitochondrial oxygen sensing: regulation of hypoxia-inducible factor by mitochondrial generated reactive oxygen species. Essays Biochem. 2007;43:17–27. | |
Bell EL, Chandel NS. Genetics of mitochondrial electron transport chain in regulating oxygen sensing. Methods Enzymol. 2007;435:447–461. | |
Mansfield KD, Guzy RD, Pan Y, et al. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005;1(6):393–399. | |
Guzy RD, Hoyos B, Robin E, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1(6):401–408. | |
Brunelle JK, Bell EL, Quesada NM, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1(6):409–414. | |
Naranjo-Suarez S, Carlson BA, Tsuji PA, Yoo MH, Gladyshev VN, Hatfield DL. HIF-independent regulation of thioredoxin reductase 1 contributes to the high levels of reactive oxygen species induced by hypoxia. PLoS One. 2012;7(2):e30470. | |
Gong Y, Agani FH. Oligomycin inhibits HIF-1alpha expression in hypoxic tumor cells. Am J Physiol Cell Physiol. 2005;288(5):C1023–C1029. | |
Riby JE, Firestone GL, Bjeldanes LF. 3,3′-diindolylmethane reduces levels of HIF-1alpha and HIF-1 activity in hypoxic cultured human cancer cells. Biochem Pharmacol. 2008;75(9):1858–1867. | |
Masson N, Singleton RS, Sekirnik R, et al. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep. 2012;13(3):251–257. | |
Yang J, Staples O, Thomas LW, et al. Human CHCHD4 mitochondrial proteins regulate cellular oxygen consumption rate and metabolism and provide a critical role in hypoxia signaling and tumor progression. J Clin Invest. 2012;122(2):600–611. | |
Chowdhury R, Flashman E, Mecinovic J, et al. Studies on the reaction of nitric oxide with the hypoxia-inducible factor prolyl hydroxylase domain 2 (EGLN1). J Mol Biol. 2011;410(2):268–279. | |
Berchner-Pfannschmidt U, Tug S, Kirsch M, Fandrey J. Oxygen-sensing under the influence of nitric oxide. Cell Signal. 2010;22(3):349–356. | |
Nakayama K, Gazdoiu S, Abraham R, Pan ZQ, Ronai Z. Hypoxia-induced assembly of prolyl hydroxylase PHD3 into complexes: implications for its activity and susceptibility for degradation by the E3 ligase Siah2. Biochem J. 2007;401(1):217–226. | |
Nakayama K, Frew IJ, Hagensen M, et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell. 2004;117(7):941–952. | |
Barth S, Edlich F, Berchner-Pfannschmidt U, et al. Hypoxia-inducible factor prolyl-4-hydroxylase PHD2 protein abundance depends on integral membrane anchoring of FKBP38. J Biol Chem. 2009;284(34):23046–23058. | |
Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466(7310):1129–1133. | |
Ito S, Shen L, Dai Q, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333(6047):1300–1303. | |
He YF, Li BZ, Li Z, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333(6047):1303–1307. | |
Inoue A, Shen L, Dai Q, He C, Zhang Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011;21(12):1670–1676. | |
Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151(7):1417–1430. | |
Spruijt CG, Gnerlich F, Smits AH, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152(5):1146–1159. | |
Abdel-Wahab O, Mullally A, Hedvat C, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114(1):144–147. | |
Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–2301. | |
Hsu CH, Peng KL, Kang ML, et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2012;2(3):568–579. | |
Huang H, Jiang X, Li Z, et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci U S A. 2013;110(29):11994–11999. | |
Sun M, Song CX, Huang H, et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc Natl Acad Sci U S A. 2013;110(24):9920–9925. | |
Gaidzik VI, Paschka P, Späth D, et al. TET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML study group. J Clin Oncol. 2012;30(12):1350–1357. | |
Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. | |
Ko M, Huang Y, Jankowska AM, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468(7325):839–843. | |
Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. | |
Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. | |
Castro-Vega LJ, Buffet A, De Cubas AA, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet. 2014;23(9):2440–2446. | |
Letouzé E, Martinelli C, Loriot C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23(6):739–752. | |
Yang M, Pollard PJ. Succinate: a new epigenetic hacker. Cancer Cell. 2013;23(6):709–711. | |
van Haaften G, Dalgliesh GL, Davies H, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009;41(5):521–523. | |
Dalgliesh GL, Furge K, Greenman C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–363. | |
Yang M, Ge W, Chowdhury R, et al. Asparagine and aspartate hydroxylation of the cytoskeletal ankyrin family is catalyzed by factor-inhibiting hypoxia-inducible factor. J Biol Chem. 2011;286(9):7648–7660. | |
Yang M, Chowdhury R, Ge W, et al. Factor-inhibiting hypoxia-inducible factor (FIH) catalyses the post-translational hydroxylation of histidinyl residues within ankyrin repeat domains. FEBS J. 2011;278(7):1086–1097. | |
Yang M, Hardy AP, Chowdhury R, et al. Substrate selectivity analyses of factor inhibiting hypoxia-inducible factor. Angew Chem Int Ed Engl. 2013;52(6):1700–1704. | |
Zhang N, Fu Z, Linke S, et al. The asparaginyl hydroxylase factor inhibiting HIF-1alpha is an essential regulator of metabolism. Cell Metab. 2010;11(5):364–378. | |
Kuzmanov A, Wielockx B, Rezaei M, Kettelhake A, Breier G. Overexpression of factor inhibiting HIF-1 enhances vessel maturation and tumor growth via platelet-derived growth factor-C. Int J Cancer. 2012;131(5):E603–E613. | |
Khan MN, Bhattacharyya T, Andrikopoulos P, et al. Factor inhibiting HIF (FIH-1) promotes renal cancer cell survival by protecting cells from HIF-1α-mediated apoptosis. Br J Cancer. 2011;104(7):1151–1159. | |
Gerken T, Girard CA, Tung YC, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318(5855):1469–1472. | |
Jia G, Yang CG, Yang S, et al. Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett. October 15, 2008;582(23–24):3313–3319. | |
Jia G, Fu Y, Zhao X, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–887. | |
Berulava T, Ziehe M, Klein-Hitpass L, et al. FTO levels affect RNA modification and the transcriptome. Eur J Hum Genet. 2013;21(3):317–323. | |
Fischer J, Koch L, Emmerling C, et al. Inactivation of the Fto gene protects from obesity. Nature. 2009;458(7240):894–898. | |
Church C, Lee S, Bagg EA, et al. A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS Genet. 2009;5(8):e1000599. | |
Gulati P, Cheung MK, Antrobus R, et al. Role for the obesity-related FTO gene in the cellular sensing of amino acids. Proc Natl Acad Sci U S A. 2013;110(7):2557–2562. | |
Cheung MK, Gulati P, O’Rahilly S, Yeo GS. FTO expression is regulated by availability of essential amino acids. Int J Obes (Lond). 2013;37(5):744–747. | |
Iles MM, Law MH, Stacey SN, et al. A variant in FTO shows association with melanoma risk not due to BMI. Nat Genet. 2013;45(4):428–432. | |
Singleton RS, Liu-Yi P, Formenti F, et al. OGFOD1 catalyzes prolyl hydroxylation of RPS23 and is involved in translation control and stress granule formation. Proc Natl Acad Sci U S A. 2014;111(11):4031–4036. | |
Loenarz C, Sekirnik R, Thalhammer A, et al. Hydroxylation of the eukaryotic ribosomal decoding center affects translational accuracy. Proc Natl Acad Sci U S A. 2014;111(11):4019–4024. | |
Katz MJ, Acevedo JM, Loenarz C, et al. Sudestada1, a Drosophila ribosomal prolyl-hydroxylase required for mRNA translation, cell homeostasis, and organ growth. Proc Natl Acad Sci U S A. 2014;111(11):4025–4030. | |
Feng T, Yamamoto A, Wilkins SE, et al. Optimal translational termination requires C4 lysyl hydroxylation of eRF1. Mol Cell. 2014;53(4):645–654. | |
Lu Y, Chang Q, Zhang Y, et al. Lung cancer-associated JmjC domain protein mdig suppresses formation of tri-methyl lysine 9 of histone H3. Cell Cycle. 2009;8(13):2101–2109. | |
Sinha KM, Yasuda H, Coombes MM, Dent SY, de Crombrugghe B. Regulation of the osteoblast-specific transcription factor Osterix by NO66, a Jumonji family histone demethylase. EMBO J. 2010;29(1):68–79. | |
Ge W, Wolf A, Feng T, et al. Oxygenase-catalyzed ribosome hydroxylation occurs in prokaryotes and humans. Nat Chem Biol. 2012;8(12):960–962. | |
Ghosh MC, Zhang DL, Jeong SY, et al. Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of HIF2α. Cell Metab. 2013;17(2):271–281. | |
Anderson ER, Taylor M, Xue X, et al. Intestinal HIF2α promotes tissue-iron accumulation in disorders of iron overload with anemia. Proc Natl Acad Sci U S A. 2013;110(50):E4922–E4930. | |
Wilkinson N, Pantopoulos K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2α mRNA translation. Blood. 2013;122(9):1658–1668. | |
Anderson SA, Nizzi CP, Chang YI, et al. The IRP1-HIF-2α axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 2013;17(2):282–290. | |
Takeda K, Cowan A, Fong GH. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation. 2007;116(7):774–781. | |
Takeda K, Aguila HL, Parikh NS, et al. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood. 2008;111(6):3229–3235. | |
Seibler J, Zevnik B, Küter-Luks B, et al. Rapid generation of inducible mouse mutants. Nucleic Acids Res. 2003;31(4):e12. | |
Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244(2):305–318. | |
Ozolins TR, Fisher TS, Nadeau DM, et al. Defects in embryonic development of EGLN1/PHD2 knockdown transgenic mice are associated with induction of Igfbp in the placenta. Biochem Biophys Res Commun. 2009;390(3):372–376. | |
Fisher TS, Lira PD, Stock JL, et al. Analysis of the role of the HIF hydroxylase family members in erythropoiesis. Biochem Biophys Res Commun. 2009;388(4):683–688. | |
Li X, Sutherland S, Takeda K, Fong GH, Lee FS. Integrity of the prolyl hydroxylase domain protein 2:erythropoietin pathway in aging mice. Blood Cells Mol Dis. 2010;45(1):9–19. | |
Schneider M, Van Geyte K, Fraisl P, et al. Loss or silencing of the PHD1 prolyl hydroxylase protects livers of mice against ischemia/reperfusion injury. Gastroenterology. 2010;138(3):1143–1154. | |
Mollenhauer M, Kiss J, Dudda J, et al. Deficiency of the oxygen sensor PHD1 augments liver regeneration after partial hepatectomy. Langenbecks Arch Surg. 2012;397(8):1313–1322. | |
Minamishima YA, Kaelin WG Jr. Reactivation of hepatic EPO synthesis in mice after PHD loss. Science. 2010;329(5990):407. | |
Duan LJ, Takeda K, Fong GH. Prolyl hydroxylase domain protein 2 (PHD2) mediates oxygen-induced retinopathy in neonatal mice. Am J Pathol. 2011;178(4):1881–1890. | |
Rankin EB, Wu C, Khatri R, et al. The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell. 2012;149(1):63–74. | |
Arsenault PR, Pei F, Lee R, et al. A knock-in mouse model of human PHD2 gene-associated erythrocytosis establishes a haploinsufficiency mechanism. J Biol Chem. 2013;288(47):33571–33584. | |
Bishop T, Talbot NP, Turner PJ, et al. Carotid body hyperplasia and enhanced ventilatory responses to hypoxia in mice with heterozygous deficiency of PHD2. J Physiol. 2013;591(Pt 14):3565–3577. | |
Franke K, Kalucka J, Mamlouk S, et al. HIF-1α is a protective factor in conditional PHD2-deficient mice suffering from severe HIF-2α-induced excessive erythropoiesis. Blood. 2013;121(8):1436–1445. | |
Hamm A, Veschini L, Takeda Y, et al. PHD2 regulates arteriogenic macrophages through TIE2 signalling. EMBO Mol Med. 2013;5(6):843–857. | |
Ikeda J, Ichiki T, Matsuura H, et al. Deletion of phd2 in myeloid lineage attenuates hypertensive cardiovascular remodeling. J Am Heart Assoc. 2013;2(3):e000178. | |
Cheng S, Xing W, Pourteymoor S, Mohan S. Conditional Disruption of the Prolyl Hydroxylase Domain-Containing Protein 2 (Phd2) Gene Defines its Key Role in Skeletal Development. J Bone Miner Res. Epub April 17, 2014. | |
Duan LJ, Takeda K, Fong GH. Hematological, hepatic, and retinal phenotypes in mice deficient for prolyl hydroxylase domain proteins in the liver. Am J Pathol. 2014;184(4):1240–1250. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.