")
Back to Journals » OncoTargets and Therapy » Volume 11
Prolyl hydroxylase domain 3 influences the radiotherapy efficacy of pancreatic cancer cells by targeting hypoxia-inducible factor-1α
Authors Tang L, Wu J , Cai S, Huang Y, Zhang X, Fu W, Zhuang Q, Li J
Received 17 September 2018
Accepted for publication 8 November 2018
Published 29 November 2018 Volume 2018:11 Pages 8507—8515
DOI https://doi.org/10.2147/OTT.S187615
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Li-rui Tang,1,* Jun-xin Wu,1,* Shao-li Cai,2 Yun-xia Huang,1 Xue-qing Zhang,1 Wan-kai Fu,1 Qing-yang Zhuang,1 Jin-luan Li1
1Department of Radiation Oncology, Fujian Medical University Cancer Hospital, Fujian Cancer Hospital, Fuzhou, China; 2Key Laboratories of Innate Immune Biology of Fujian Province, Biomedical Research Center of South China, Fujian Normal University, Fuzhou, China
*These authors contributed equally to this work
Purpose: Pancreatic cancer is characterized by a hypoxic microenvironment and resistance to most currently available treatment modalities. Prolyl hydroxylase domain 3 (PHD3) is a rate-limiting enzyme that regulates the degradation of hypoxia-inducible factors (HIFs) and is deregulated in pancreatic cancer cells. Whether such alteration of PHD3 expression contributes to the sustained growth and radioresistance of pancreatic cancer cells remains largely unknown.
Materials and methods: PHD3 was overexpressed in pancreatic cancer Mia-paca2 cells via lentiviral expression. Cell cycle progression and apoptosis were assayed by flow cytometry. HIF-1α, EGFR, and PHD3 protein expression was assessed by Western blotting. Cell survival was determined in a colony formation assay.
Results: PHD3 overexpression suppressed HIF-1α protein expression and EGFR phosphorylation and enhanced the 2 Gy irradiation-mediated reductions in HIF-1α and phosphorylated (p)-EGFR under either normoxic or hypoxic conditions. PHD3 overexpression inhibited the growth and colony formation of Mia-paca2 cells in response to irradiation under either normoxic or hypoxic conditions. PHD3 overexpression exacerbated irradiation-induced apoptosis, with a greater effect under hypoxia than normoxia. Cell cycle distribution analysis demonstrated that PHD3 overexpression resulted in further shortened S phase and lengthened G2/M phase in response to irradiation.
Conclusion: PHD3 expression may contribute to the radiotherapy efficacy of pancreatic cancer cells and serve as a novel biomarker for improving radiotherapy efficacy in pancreatic cancer.
Keywords: pancreatic cancer, PHD3, radiotherapy efficacy, HIF-1α, p-EGFR
Introduction
Pancreatic carcinoma is one of the most lethal solid tumors. Global cancer statistics indicate that ~53,070 new pancreatic cancer cases will occur in the US in 2018, and the 5-year survival rate is only 1.2%–6%.1 Surgical resection is currently recommended as the first-line treatment. However, >80% of pancreatic cancer patients with nonspecific symptoms are diagnosed when the disease is in an advanced stage and the opportunity for radical resection has passed.2 Therefore, radiotherapy and chemotherapy play crucial roles in the treatment of pancreatic cancer.3 Although modern image-guided technology has improved radiotherapy, the efficacy of radiotherapy is still limited by intrinsic and extrinsic radiation resistance in tumor cells.4 To improve the radiation sensitivity of pancreatic cancer cells along with patients’ prognosis, there is an urgent need to elucidate the molecular mechanisms of radiation resistance in pancreatic cancer.
The tumor microenvironment plays a critical role in tumorigenesis and treatment resistance. Intratumoral hypoxia is a widely observed feature of the tumor microenvironment and has been shown to contribute to radiation resistance in numerous solid tumors.5–7 Hypoxia is a prominent biological characteristic of pancreatic cancer, as demonstrated by the lower partial pressure of oxygen in the areas of pancreatic carcinoma compared with that in normal pancreatic tissue.8 Hypoxia inducible factors (HIFs) regulate the expression of the genes required for the adaptation of tumor cells to the hypoxic microenvironment. The HIF family includes HIF-1α, HIF-2α, and HIF-3α. HIF-1α overexpression enhances the proliferation, metabolism, invasion, and metastasis of cancer cells.9,10 Evasion of apoptosis is a hallmark of cancer cells, and it has been shown that HIF-1α can prevent tumor cells from undergoing apoptosis, leading to radioresistance.11 Takasaki et al12 demonstrated that HIF-1α inhibits the intrinsic cell apoptosis pathway. HIF-1α is a downstream factor of the EGFR-mediated pathway, an essential regulatory component for the tumor microenvironment and radioresistance. The abnormal activities of EGFR can promote the formation of precancerous lesions and pancreatic ductal adenocarcinoma.13,14 This type of abnormal change, meanwhile, leads to enhanced proliferation and invasion of pancreatic cancer cells.15 Hypoxia is one of the most important factors in tumor radioresistance. Pancreatic cancer cells adapt to hypoxia and remain viable via HIF-1α-dependent mechanisms.16 Due to the hypoxic conditions in the specific tumor microenvironment of pancreatic cancer, the expression of HIF-1α is significantly higher in these tissues than in other solid tumors.13,16,17 Therefore, HIF-1α may play a pivotal role in the tumorigenesis of pancreatic cancer and related angiogenesis.
HIF-1 is degraded by proline hydroxylase domains (PHDs) and factor inhibiting HIF-1, an asparaginyl hydroxylase. The family of PHDs comprises PHD1, PHD2, and PHD3.18,19 In vitro studies suggested that all PHDs can hydroxylate HIFs. However, different hydroxylation sites of HIFs have been identified for different PHDs.20,21 While PHD2 is the main regulator of HIF-1α in normoxia and upon re-oxygenation of hypoxic cells, PHD3 might regulate HIFs in more severe and prolonged hypoxia.22 HIF-1α is recognized by PHDs via Von Hippel-Lindau and is degraded via the ubiquitin-proteasome system.23 PHD1 and PHD2 can hydroxylate the C-terminal hydroxylation site Pro564 and N-terminal hydroxylation site Pro402; however, PHD3 can only hydroxylate the C-terminal hydroxylation site Pro564 and has no activity at the N-terminal site Pro402.18,22 Thus, PHDs may suppress the protein expression of HIFs under hypoxic conditions and enhance the ability of tumor cells to tolerate hypoxic conditions.
Previous studies have shown that PHD3 is a rate-limiting enzyme for the degradation of HIF-1α in the hypoxic microenvironment of pancreatic cancer cells.24,25 However, the effect of PHD3 on the radiotherapy efficacy of pancreatic cancer cells is unknown. In the present study, we investigated the effect of PHD3 overexpression on the radiotherapy efficacy of pancreatic cancer cells and the underlying molecular mechanisms.
Materials and methods
Reagents
DMEM was purchased from Hyclone (Thermo Fisher Scientific, Inc., Waltham, MA, USA). FBS was purchased from Gibco (Thermo Fisher Scientific, Inc.). Annexin V-Fluorescein isothiocyanate (FITC) and propidium iodide (PI) were purchased from eBioscience (Thermo Fisher Scientific, Inc.). Anti-EGFR (EP38Y, ab52894, 1:1,000), anti-p-EGFR (EP774Y, ab40815, 1:1,000), anti-PHD3 (ab30782, 1:1,000) and anti-HIF-1α (mgc3, ab16066, 1:1,000) antibodies were purchased from Abcam (Cambridge, UK).
Cell culture
Human pancreatic adenocarcinoma Mia-paca2 cells were purchased from the Key Laboratory of Stem Cell Biology Shanghai Institute for Biological Sciences Chinese Academy of Sciences, Shanghai, China. Cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin and maintained in a humidified incubator containing 10% CO2, 20% or 2% O2 at 37°C. The cell line was verified by short tandem repeat profiling.
Irradiation
Cells were cultured in 25 cm2 cell culture flasks to logarithmic growth phase and irradiated with a single fraction of 2, 4, 6, or 8 Gy using the Elekta Precise linear accelerator (Elekta Oncology System, Crawley, UK) with high-energy radiation X-ray (6 MV). The irradiation distance was 100 cm, and the radiation field was 15×15 cm. Subsequent experiments were performed after 48 hours of recovery in the incubator.
Lentivirus-mediated overexpression of PHD3
293 T cells were co-transfected with the plasmids of lentivirus vector, pGag/Pol, pRev and pVSV-G, and a plasmid carrying the PHD3 gene. After 48 hours, the medium containing the packaged viruses was harvested. The Mia-paca2 cells in the logarithmic growth phase were seeded into 6-well cell culture plates and cultured with virus supernatant. The medium was changed after 24 hours, and the infected cells were subjected to further experiments.
Colony formation assay
Cells were seeded into 6-well plates at a density of 300 cells/well. After replacement of the medium with fresh medium for 12 hours, the cells were irradiated with a single fraction at different doses (2, 4, 6, or 8 Gy) and cultured for 3 weeks with medium exchange every 3 days. The cells were rinsed with PBS twice, fixed with 4% paraformaldehyde for 15 minutes, and stained with 0.4% crystal violet solution for 30 minutes. The number of clones (>10 cells) was counted under a low power microscope. The planting efficiency (PE) and survival fraction (SF) were calculated as follows: PE = (number of colonies counted/number of cells seeded)×100%; SF = (PE in radiation group/PE in negative control group)×100%. The experiments were repeated for three times with triplicate samples.
Western blotting
Cells were treated with radioimmunoprecipitation assay lysis buffer containing phenylmethane sulfonyl fluoride on ice for 30 minutes, followed by centrifugation at 12,000 rpm and 4°C for 5 minutes. The supernatant was transferred into a new tube, and the protein concentration was determined by bicinchoninic acid protein assay (Beyotime, Shanghai, China). A total of 30 μg of each protein sample was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane. The membrane was blocked by 5% nonfat milk in Tris-buffered saline with 0.05% Tween 20 (TBST) at room temperature for 1 hour, followed by incubation with primary anti-PHD3, anti-HIF-1α, anti-EGFR, anti-p-EGFR, or anti-β-actin antibody overnight at 4°C. After three washes with TBST, the membranes were incubated with mouse or rabbit secondary antibody conjugated with horseradish peroxidase at room temperature for 1 hour. Specific protein bands were visualized by by chemiluminescence (Pierce, Rockford, IL, USA).
Cell apoptosis and cycle analysis
After treatment, the cells were processed to obtain a single-cell suspension and washed with ice cold PBS two times, followed by incubation with 100 μL binding buffer containing 5 μL PI and 5 μL Annexin V-FITC at room temperature in the dark for 15 minutes. After addition of 400 μL binding buffer, the cells were subjected to cell apoptosis and cycle distribution analysis by flow cytometry. Experiments were repeated three times.
Statistical analysis
All data were analyzed using SPSS software (version 24.0; IBM, Armonk, NY, USA). Results are expressed as the mean ± standard error. Comparisons between groups were examined by Student’s t-test. P<0.05 was considered statistically significant.
Results
PHD3 overexpression enhances irradiation-induced downregulation of EGFR/HIF-1α signaling
To assess whether PHD3 regulates the activity of the EGFR/HIF-1α pathway, we performed Western blotting to evaluate the effect of lentivirus-mediated PHD3 overexpression on the expression of EGFR, p-EGFR, and HIF-1α after 2 Gy irradiation under either normoxic or hypoxic conditions. Compared with those in the negative control group, PHD3 protein levels were increased by lentivirus-mediated overexpression. Under normoxic conditions and treatment with 2 Gy radiation (Figure 1A), PHD3 overexpression had no significant effect on the total EGFR expression (P>0.05) but obviously decreased the expression of p-EGFR and HIF-1α (P<0.05; Figure 1C). We next cultured normal and PHD3-overexpressing cells under hypoxic conditions (Figure 1B), followed by treatment with 2 Gy radiation. Compared with those in irradiated normal cells, the expression levels of HIF-1α and p-EGFR were significantly suppressed in PHD3-overexpressing cells (P<0.05) with no significant difference in EGFR expression (P>0.05; Figure 1D). Interestingly, we also found that radiation slightly promoted the expression level of PHD3, which was accompanied by decreased expression of HIF-1α and p-EGFR in Mia-paca2 cells under either normoxic or hypoxic conditions. Furthermore, PHD3 overexpression alone resulted in significant decreases in HIF-1α and p-EGFR expression under either normoxic or hypoxic conditions. These results suggest that activation of the EGFR/HIF-1α pathway is associated with PHD3 expression in Mia-paca2 cells under normoxic and hypoxic conditions.
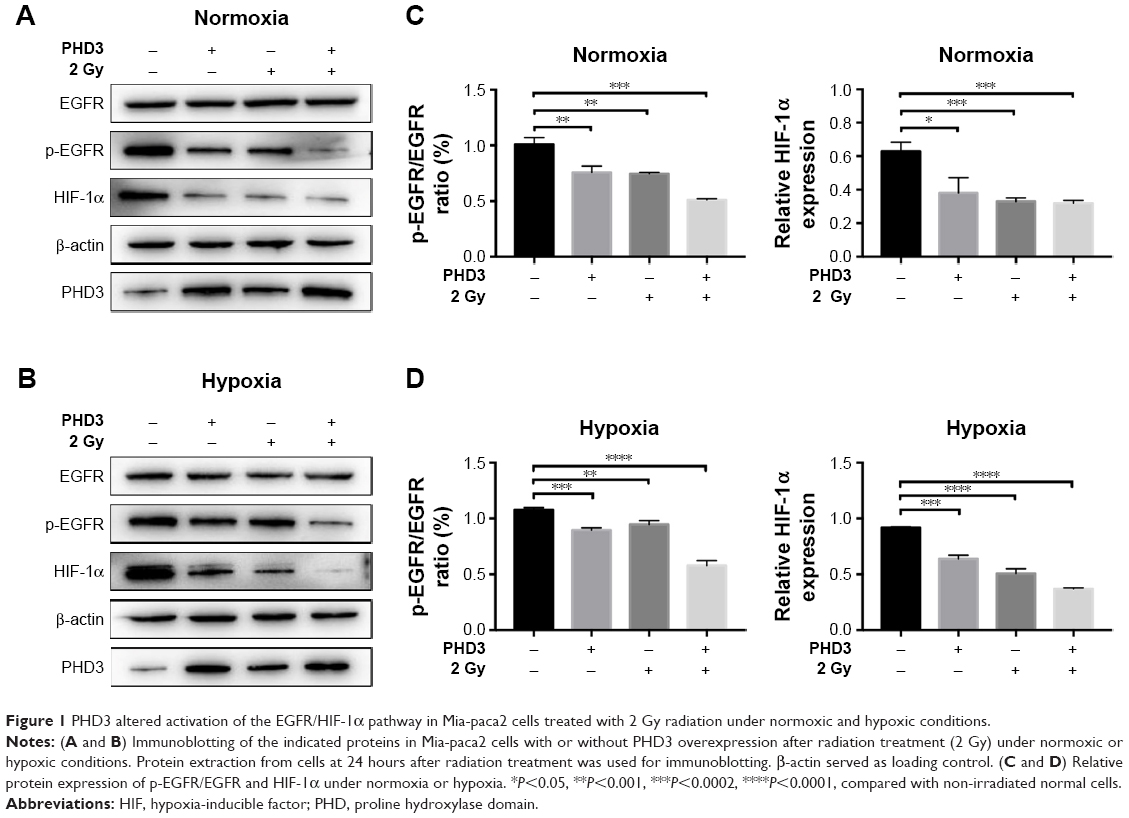 | Figure 1 PHD3 altered activation of the EGFR/HIF-1α pathway in Mia-paca2 cells treated with 2 Gy radiation under normoxic and hypoxic conditions. |
PHD3 overexpression suppresses the colony-forming capability of Mia-paca2 cells exposed to high radiation under hypoxia
To investigate whether increased expression of PHD3 affects the colony-forming capability of Mia-paca2 cells exposed to radiation treatment, we performed colony formation assays. The SFs of normal and PHD3-overexpressing cells treated with 2, 4, or 6 Gy radiation were significantly lower than those of non-irradiated cells under normoxic or hypoxic conditions (Figure 2A and B). However, when cells were treated with 8 Gy radiation under normoxic conditions, there was no significant difference of the SF between normal and PHD3-overexpressing cells; in contrast, PHD3 overexpression significantly enhanced the loss of cell viability under the hypoxic conditions (Figure 2B). These results indicate that PHD3 overexpression could significantly suppress the colony-forming abilities of Mia-paca2 cells treated with a low dose of irradiation under either normoxic or hypoxic conditions. PHD3 overexpression did not alter the proliferation of Mia-paca2 cells treated with a high dose of irradiation under normoxic conditions, but did suppress their viability under hypoxic conditions.
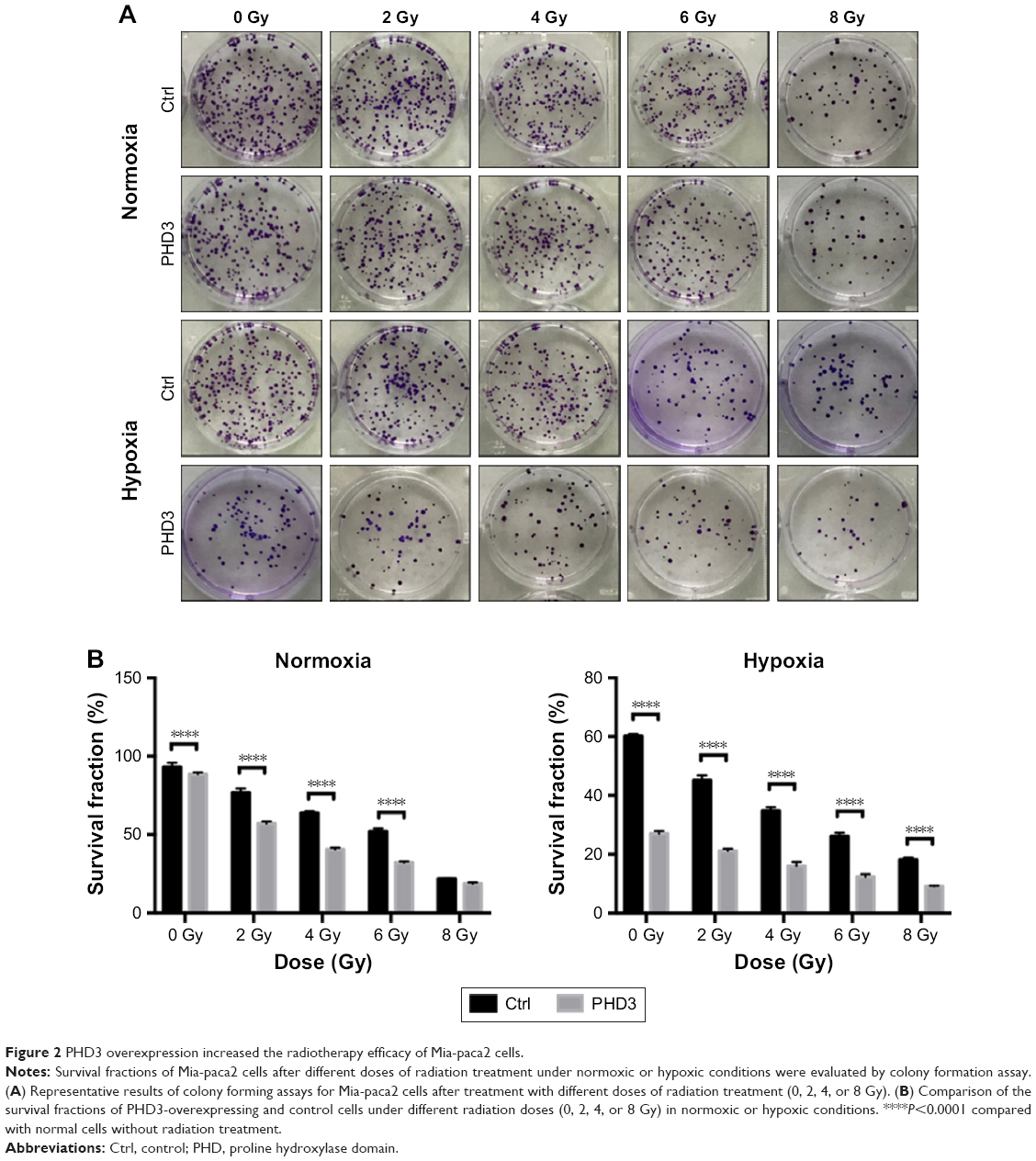 | Figure 2 PHD3 overexpression increased the radiotherapy efficacy of Mia-paca2 cells. |
PHD3 overexpression enhances irradiation-mediated apoptosis of Mia-paca2 cells under hypoxia
To examine whether PHD3 is involved in cell apoptosis via regulation of the EGFR/HIF-1α pathway in Mia-paca2 cells, we cultured normal and PHD3-overexpressing cells under normoxic or hypoxic conditions with or without exposure to 2 Gy irradiation. Cell apoptosis was analyzed by Annexin V-FITC/PI staining coupled with flow cytometry. Under the normoxic conditions, the apoptosis rate of PHD3-overexpressing cells was significantly higher than that of the normal cells. Irradiation (2 Gy) significantly increased the apoptosis rate of normal cells, and PHD3 overexpression further increased the apoptosis rate of the cells irradiated with 2 Gy (Figure 3A). When the cells were treated under hypoxic conditions, the apoptosis rate was increased in cells overexpressing PHD3 after exposure to 2 Gy radiation compared with that of cells that received the same treatment under normoxic conditions (Figure 3B). These results demonstrated that PHD3 overexpression increased radiation-induced apoptosis in Mia-paca2 cells, with a greater effect in the hypoxic condition.
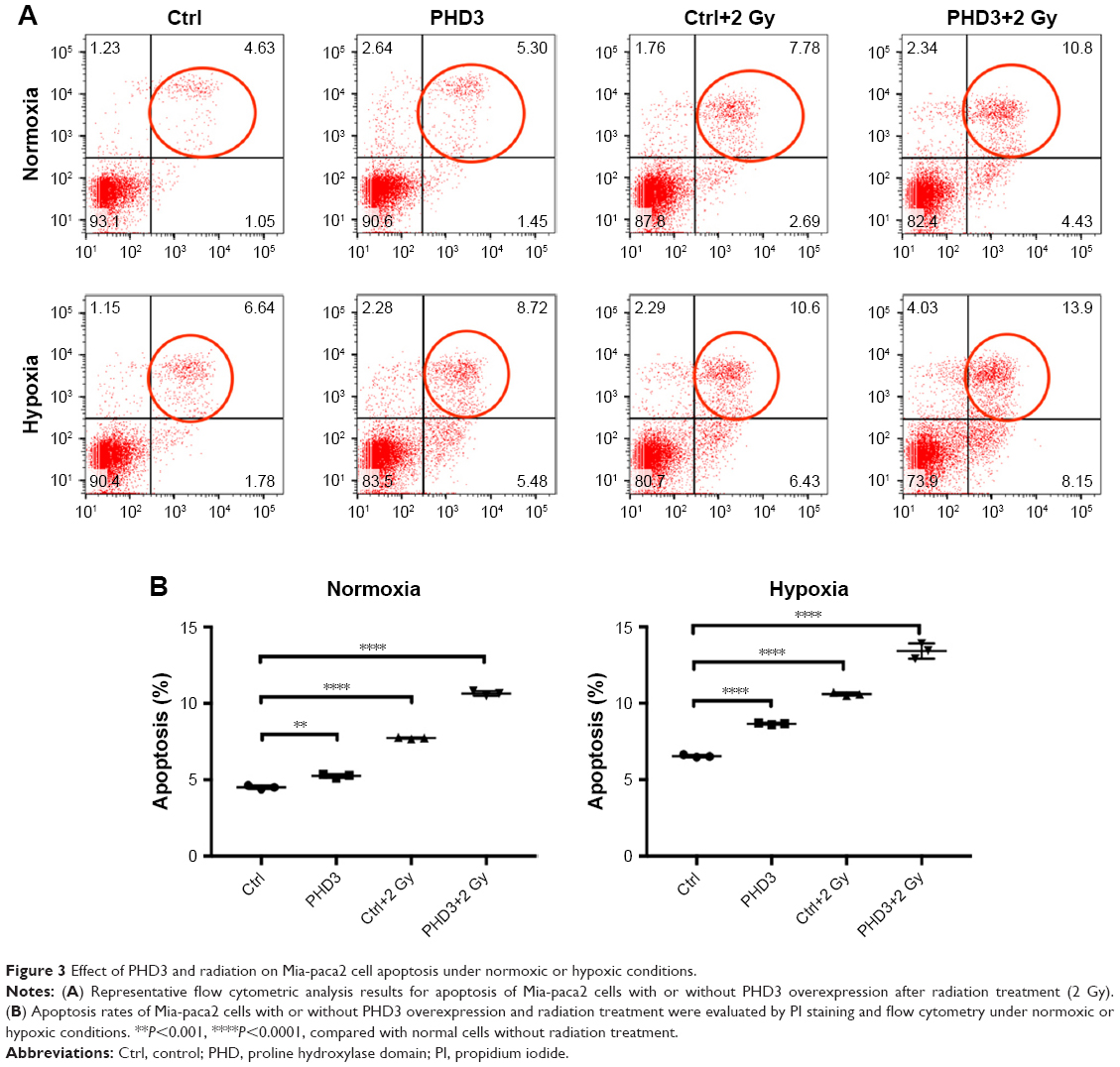 | Figure 3 Effect of PHD3 and radiation on Mia-paca2 cell apoptosis under normoxic or hypoxic conditions. |
PHD3 overexpression increases irradiation-mediated cell cycle arrest at G2/M phase in Mia-paca2 cells
To evaluate the role of the PHD3 in the cell cycle progression of Mia-paca2 cells, we cultured normal and PHD3-overexpressing cells under either normoxic or hypoxic conditions with or without 2 Gy irradiation. We performed cell cycle distribution analysis by PI staining and flow cytometry. Under normoxic and hypoxic conditions, PHD3 overexpression or irradiation treatment caused a significant increase in the percentage of cells in the G2/M phase compared with the percentage of untreated normal cells in this phase (Figure 4A). Moreover, the percentage of cells in the G2/M phase was markedly increased in PHD3-overexpressing cells treated with 2 Gy radiation compared that that in the first three groups (Figure 4B). Therefore, overexpression of PHD3 led to cell cycle arrest at the G2/M phase, which was enhanced by radiation treatment.
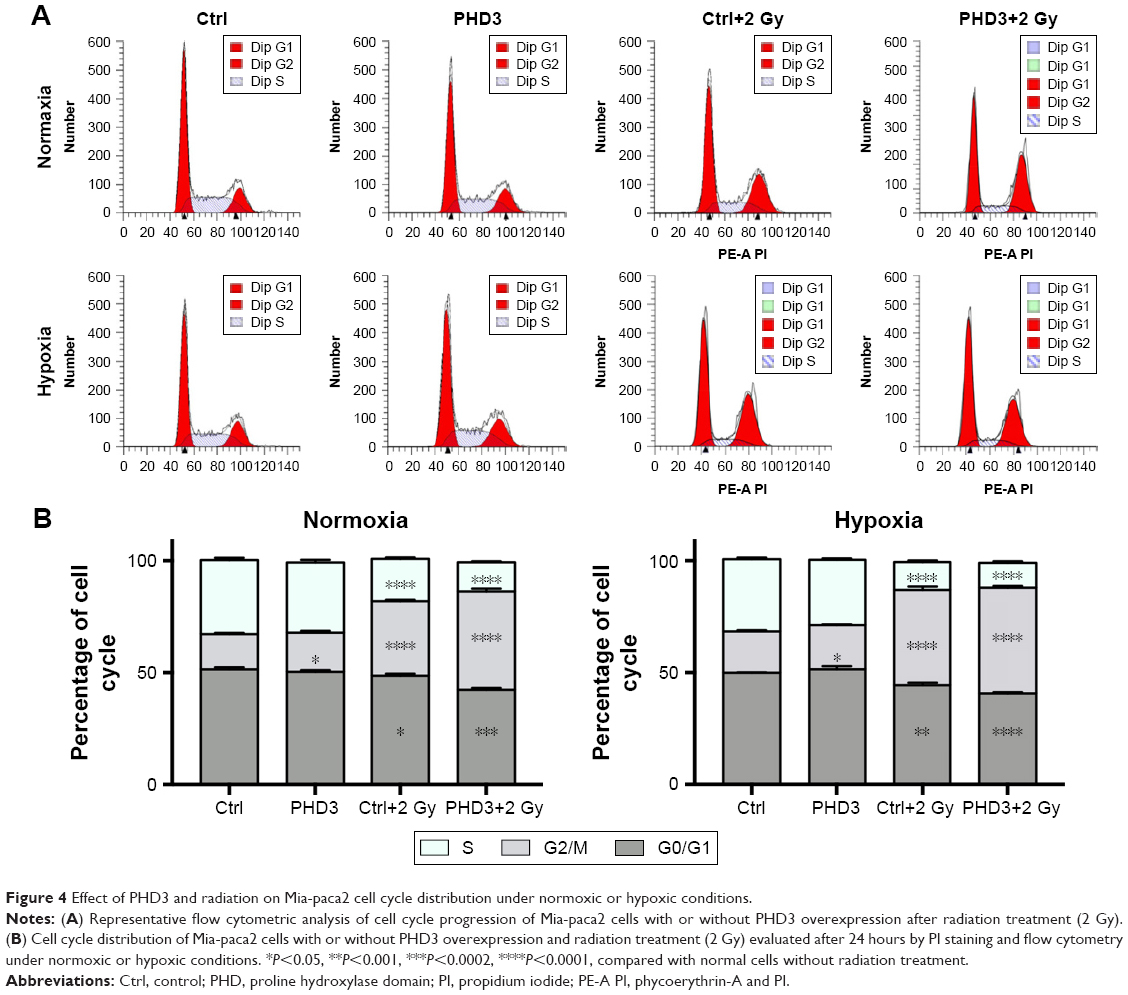 | Figure 4 Effect of PHD3 and radiation on Mia-paca2 cell cycle distribution under normoxic or hypoxic conditions. |
Discussion
The rates of local control in the majority of clinical trials of radiotherapy for pancreatic cancer range from 49%–100%.26 The tumor microenvironment has been shown to restrain the efficacy of radiotherapy in pancreatic cancer.4 Previous studies revealed that intratumoral hypoxia has invasive and metastatic properties, which are associated with resistance to radiotherapy.5–7 In the present study, we found that changing the biological characteristics of cells by suppressing the hypoxia tolerance of tumor cells could enhance the radiotherapy efficacy of pancreatic cancer cells.
HIF-1α is an essential transcription factor that mediates hypoxic-adaptive responses in cells and maintains cancer cell clonogenicity in hypoxic conditions.27,28 HIF-1α is a downstream factor of the EGFR-mediated signaling pathway, an essential regulatory component of the tumor microenvironment and radioresistance.12 There exists a positive feedback loop between HIF-1α and EGFR signaling. HIF-1α downregulation can further stimulate the EGFR/extracellular-regulated protein kinase (ERK) pathway, thereby potentially decreasing the cascade amplification of the EGFR/MAP kinase–ERK kinase/ERK/HIF-1α loop resulting from hypoxic conditions.29 Here, our data revealed that PHD3 regulated the activity of the EGFR/HIF-1α pathway as demonstrated by decreased expression of HIF-1α and p-EGFR upon overexpression of PHD3 under normoxic or hypoxic conditions. Interestingly, we observed significantly more robust inhibition of HIF-1α and p-EGFR in PHD3-overexpressing cells treated with 2 Gy radiation. Therefore, it is possible that upregulation of PHD3 enhances the radiation-induced inhibition of the EGFR/HIF-1α pathway.
It has been reported that PHD3 can limit the activation of EGFR during tumorigenesis.30,31 In addition, previous studies have shown that activation of irradiation-mediated intracellular signaling pathways can enhance tumor necrosis factor alpha (TNF-α) expression in nucleus pulposus cells, which, in turn, further upregulates the expression level of PHD3 in a nuclear factor-kappa B (NF-κB)-dependent manner.32,33 Intriguingly, we found that 2 Gy irradiation increased the expression of PHD3 in Mia-paca2 cells, which was consistent with the findings of previous studies.32,33 Therefore, irradiation-mediated suppression of EGFR signaling may at least partially mediated through the TNF-α/NF-κB regulated expression of PHD3, which warrants further verification.
Under hypoxic conditions, HIF-1α strengthens glycolysis to provide the necessary energy for the growth and proliferation of pancreatic cancer cells.34,35 Our data demonstrated that, under the normoxic conditions, cell proliferation was decreased after radiation treatment, whereas proliferation was most significantly decreased upon the overexpression of PHD3. Both of these effects were more pronounced under hypoxic conditions. Therefore, under the hypoxic conditions, PHD3 overexpression in combination with radiation significantly inhibits Mia-paca2 cell proliferation.
Cells in G2 phase are more sensitive to radiation treatment, whereas those in S phase are resistant to radiation.36,37 HIF-1α is highly expressed under hypoxic conditions and inhibits the pancreatic cancer cells from shifting to G2 phase, thereby influencing their radiotherapy efficacy and apoptosis under hypoxic conditions.38 Our results demonstrated that overexpression of PHD3 combined with 2 Gy irradiation could significantly enhance the apoptosis rate under hypoxic conditions. Further cell cycle distribution analyses showed that PHD3 overexpression with 2 Gy radiation treatment markedly increased the percentage of cells in the G2/M phase. These results suggest that PHD3 overexpression combined with 2 Gy irradiation is capable of reversing the evasion of apoptosis and arresting pancreatic cancer cells in radiosensitive phases of the cell cycle.
Conclusion
PHD3 overexpression enhanced the radiation-induced inhibition of Mia-paca2 cell proliferation, apoptosis, and cell cycle arrest at the G2/M phase by suppressing the EGFR/HIF-1α signaling axis. Our findings may provide a basis for PHD3 overexpression having an adjuvant effect with radiotherapy. However, future studies are required to comprehensively investigate the regulatory mechanism of PHD3 in order to provide a novel target for pancreatic cancer radiotherapy.
Acknowledgments
This study was supported by the Fujian Province Natural Science Foundation (no. 2016J01437 and 2017J01260), the Fujian Medical Innovation Project (no. 2015-CX-8), the Peking University Cancer Hospital & Institute, Key Laboratory of Carcinogenesis and Translational Research, Ministry of Education/Beijing (2017 Open Project-9), the Key Clinical Specialty Discipline Construction Program of Fujian, China, the National Clinical Key Specialty Construction Program, and the Joint Funds for the Innovation of Science and Technology, Fujian Province (no. 2017Y9074).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. | ||
Wason MS, Colon J, das S, et al. Sensitization of pancreatic cancer cells to radiation by cerium oxide nanoparticle-induced ROS production. Nanomedicine. 2013;9(4):558–569. | ||
Ben-Josef E, Lawrence TS, Zalupski MM. Radiotherapy for locally advanced pancreatic cancer. J Clin Oncol. 2007;25(30):4861. | ||
Fasih A, Elbaz HA, Hüttemann M, Konski AA, Zielske SP. Radiosensitization of pancreatic cancer cells by metformin through the AMPK pathway. Radiat Res. 2014;182(1):50–59. | ||
Rogers JL, Bayeh L, Scheuermann TH, et al. Development of inhibitors of the PAS-B domain of the HIF-2α transcription factor. J Med Chem. 2013;56(4):1739–1747. | ||
Stegeman H, Span PN, Kaanders JH, Bussink J. Improving chemoradiation efficacy by PI3-K/AKT inhibition. Cancer Treat Rev. 2014;40(10):1182–1191. | ||
Taiakina D, dal Pra A, Bristow RG. Intratumoral hypoxia as the genesis of genetic instability and clinical prognosis in prostate cancer. Adv Exp Med Biol. 2014;772:189–204. | ||
Koong AC, Mehta VK, Le QT, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys. 2000;48(4):919–922. | ||
Tong WW, Tong GH, Chen XX, Zheng HC, Wang YZ. HIF2α is associated with poor prognosis and affects the expression levels of survivin and cyclin D1 in gastric carcinoma. Int J Oncol. 2015;46(1):233–242. | ||
Putra AC, Eguchi H, Lee KL, et al. The A Allele at rs13419896 of EPAS1 is associated with enhanced expression and poor prognosis for non-small cell lung cancer. PLoS One. 2015;10(8):e0134496. | ||
Krakstad C, Chekenya M. Survival signalling and apoptosis resistance in glioblastomas: opportunities for targeted therapeutics. Mol Cancer. 2010;9:135. | ||
Takasaki C, Kobayashi M, Ishibashi H, Akashi T, Okubo K. Expression of hypoxia-inducible factor-1α affects tumor proliferation and antiapoptosis in surgically resected lung cancer. Mol Clin Oncol. 2016;5(2):295–300. | ||
Ardito CM, Grüner BM, Takeuchi KK, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22(3):304–317. | ||
Zhao S, Wang Y, Cao L, Ouellette MM, Freeman JW. Expression of oncogenic K-ras and loss of Smad4 cooperate to induce the expression of EGFR and to promote invasion of immortalized human pancreas ductal cells. Int J Cancer. 2010;127(9):2076–2087. | ||
Siveke JT, Einwächter H, Sipos B, Lubeseder-Martellato C, Klöppel G, Schmid RM. Concomitant pancreatic activation of Kras(G12D) and Tgfa results in cystic papillary neoplasms reminiscent of human IPMN. Cancer Cell. 2007;12(3):266–279. | ||
Joshi S, Kumar S, Ponnusamy MP, Batra SK. Hypoxia-induced oxidative stress promotes MUC4 degradation via autophagy to enhance pancreatic cancer cells survival. Oncogene. 2016;35(45):5882–5892. | ||
Wang G, Wang JJ, Fu XL, Guang R, To ST. Advances in the targeting of HIF-1α and future therapeutic strategies for glioblastoma multiforme (Review). Oncol Rep. 2017;37(2):657–670. | ||
Kim SY, Yang EG. Recent advances in developing inhibitors for hypoxia-inducible factor prolyl hydroxylases and their therapeutic implications. Molecules. 2015;20(11):20551–20568. | ||
Taylor MS. Characterization and comparative analysis of the EGLN gene family. Gene. 2001;275(1):125–132. | ||
García-Heredia JM, Felipe-Abrio B, Cano DA, Carnero A. Genetic modification of hypoxia signaling in animal models and its effect on cancer. Clin Transl Oncol. 2015;17(2):90–102. | ||
Huang J, Zhao Q, Mooney SM, Lee FS. Sequence determinants in hypoxia-inducible factor-1alpha for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J Biol Chem. 2002;277(42):39792–39800. | ||
Takei A, Ekström M, Mammadzada P, et al. Gene transfer of prolyl hydroxylase domain 2 inhibits hypoxia-inducible angiogenesis in a model of choroidal neovascularization. Sci Rep. 2017;7:42546. | ||
Jokilehto T, Jaakkola PM. The role of HIF prolyl hydroxylases in tumour growth. J Cell Mol Med. 2010;14(4):758–770. | ||
Bruick RK, Mcknight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–1340. | ||
Epstein AC, Gleadle JM, Mcneill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54. | ||
de Geus SWL, Eskander MF, Kasumova GG, et al. Stereotactic body radiotherapy for unresected pancreatic cancer: a nationwide review. Cancer. 2017;123(21):4158–4167. | ||
Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33(4):207–214. | ||
Dai ZJ, Gao J, Ma XB, et al. Up-regulation of hypoxia inducible factor-1α by cobalt chloride correlates with proliferation and apoptosis in PC-2 cells. J Exp Clin Cancer Res. 2012;31:28. | ||
Wang G, Li Y, Yang Z, Xu W, Yang Y, Tan X. ROS mediated EGFR/MEK/ERK/HIF-1α loop regulates glucose metabolism in pancreatic cancer. Biochem Biophys Res Commun. 2018;500(4):873–878. | ||
Garvalov BK, Foss F, Henze AT, et al. PHD3 regulates EGFR internalization and signalling in tumours. Nat Commun. 2014;5:5577. | ||
Dopeso H, Jiao HK, Cuesta AM, et al. PHD3 controls lung cancer metastasis and resistance to EGFR inhibitors through TGFα. Cancer Res. 2018;78(7):1805–1819. | ||
van Valen F, Kentrup-Lardong V, Truckenbrod B, Rübe C, Winkelmann W, Jürgens WW. Regulation of the release of tumour necrosis factor (TNF) alpha and soluble TNF receptor by gamma irradiation and interferon gamma in Ewing’s sarcoma/peripheral primitive neuroectodermal tumour cells. J Cancer Res Clin Oncol. 1997;123(5):245–252. | ||
Fujita N, Gogate SS, Chiba K, Toyama Y, Shapiro IM, Risbud MV. Prolyl hydroxylase 3 (PHD3) modulates catabolic effects of tumor necrosis factor-α (TNF-α) on cells of the nucleus pulposus through co-activation of nuclear factor κB (NF-κB)/p65 signaling. J Biol Chem. 2012;287(47):39942–39953. | ||
Semenza GL. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J Bioenerg Biomembr. 2007;39(3):231–234. | ||
Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol. 2009;19(1):12–16. | ||
Toyoshima M. Analysis of p53 dependent damage response in sperm-irradiated mouse embryos. J Radiat Res. 2009;50(1):11–17. | ||
Zheng L, Tang W, Wei F, et al. Radiation-inducible protein RbAp48 contributes to radiosensitivity of cervical cancer cells. Gynecol Oncol. 2013;130(3):601–608. | ||
Hubbi ME, Gilkes DM, Hu H, Kshitiz, Ahmed I, Semenza GL. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1α to promote cell-cycle progression. Proc Natl Acad Sci U S A. 2014;111(32):E3325–E3334. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.