")
Back to Journals » Journal of Inflammation Research » Volume 15
Progress of Research into the Interleukin-1 Family in Cardiovascular Disease
Received 23 September 2022
Accepted for publication 30 November 2022
Published 13 December 2022 Volume 2022:15 Pages 6683—6694
DOI https://doi.org/10.2147/JIR.S390915
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Zimin Wu, Cheng Luo, Baoshi Zheng
Department of Cardiovascular Surgery Ward, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi Zhuang Autonomous Region, 530021, People’s Republic of China
Correspondence: Baoshi Zheng, No. 6 Shuangyong Road, Qingxiu District, Nanning, Guangxi Zhuang Autonomous Region, 530021, People’s Republic of China, Tel +86 0771-5355280, Email [email protected]
Abstract: Inflammatory factors, such as the IL-1 family, are generally acknowledged to be involved in systemic diseases and IL-1α and IL-1β, in particular, have been linked to cardiovascular disease with IL-18, IL-33, IL-36, IL-37 and IL-38 yet to be explored. The current review aims to summarize mechanisms of IL-18, IL-33, IL-36, IL-37 and IL-38 in myocardial infarction, hypertension, arrhythmia, valvular disease and aneurysm and to explore the potential for cardiovascular disease treatment strategies and discuss future directions for prevention and treatment.
Keywords: cardiovascular disease, interleukin-1 family, myocardial infarction, hypertension, arrhythmia
Introduction
Cardiovascular disease (CVD) often occurs in middle-aged or older patients1,2 and CVD morbidity and mortality are on the rise along with global aging.3–5 In addition, the population of CVD patients has become younger as obesity, poor lifestyle, dietary habits and substance abuse (cocaine, e-cigarettes) increase in the youthful population.6 Countries in the Asian region, such as China and India, have the highest global burden of CVD due to their large population bases.7,8 Therefore, mechanisms of CVD pathogenesis require urgent attention.
A great deal of research has allowed us to conclude that inflammatory responses are involved in human physiological and pathological processes.9–13 The interleukin-1 family has numerous family members that stimulate inflammation-related genes and contribute to intestinal diseases, tumors, rheumatoid arthritis, liver and kidney diseases, skin diseases and neurological disorders14–19 and have been shown to affect human metabolism, sleep, appetite and mood.20 The interleukin-1 family can be broadly divided into two types, pro-inflammatory, IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β and IL-36γ, and the anti-inflammatory, IL-1Ra, IL-36Ra, IL-37 and IL-38.21 Recent studies have focused on IL-1 and its isoforms, IL-1α, IL-1β and IL-1Ra22–26 but other family members also deserve scrutiny. IL-18 drives myeloid-derived suppressor cell production and suppresses T-cell responses to promote multiple myeloma.27 IL-33 mediates microglia synaptic phagocytosis in central nervous system development.28 Targeted inhibition of IL-36 signaling is used to treat pustular psoriasis29 and both IL-37 and IL-38 suppress allergic disease, with the former binding maternal anti-alopecia homolog 3 and entering the nucleus to affect gene transcription,30 and the latter antagonizing the ERK1/2 and NF-κB pathways and upregulating host defense genes.31 The interleukin-1 family has been linked to CVD and IL-1α creates a pro-thrombotic microenvironment by inducing IL-6 production while IL-1β drives inflammation during atherosclerosis.32 Low-grade chronic inflammation mediated by IL-1 is involved in vascular aging, and increased expression of IL-1Ra has been observed in the elderly.33 IL-1 also affects L-type calcium channels through altered gene expression or IL-1R signaling to trigger heart failure.34 CVD-related mechanisms of IL-1 have been extensively reported and will not be repeated here. Instead, the focus will be on the mechanisms of IL-18, IL-33, IL-36, IL-37 and IL-38 in CVD and development of therapeutic strategies will be discussed.
Regulation of Interleukin-1 Family in Cardiovascular Diseases
Myocardial Infarction
Thrombosis, coronary spasm and atherosclerotic plaque erosion due to acute rupture of coronary atherosclerotic plaque are common causes of coronary heart disease. All may lead to coronary occlusion, distal myocardial blood supply obstruction and myocardial infarction (MI).35,36 Myocardial injury results in the secretion of cytokines by fibroblasts to mediate inflammatory responses.37 Mild inflammatory responses facilitate myocardial repair while excessive responses lead to cardiac adaptive remodeling and systolic dysfunction, precipitating the condition of heart failure.38
MI involves the formation of the leukocyte NLRP3 inflammasome in the infarcted area and the activation of IL-1β and IL-18 in the presence of caspase-1.39 Cardiomyocytes express and activate IL-18 but not IL-1β, even in the presence of high-dose stimulation.39 IL-18 has been shown to be involved in vascular fibrosis and the promotion of atherosclerotic plaques40 and is considered to be a predictor of inflammation risk in MI patients.41 IL-18 acts synergistically with IL-12 and IL-15 to induce IFN-γ secretion, enhance TH1-type immune responses and induce post-MI fibrosis.42,43 IL-18 also triggers release of adhesion molecules, GM-CSF and iNOS, and promotes vascular endothelial cell apoptosis, inhibits cardiac contractile function and induces myocardial fibrotic remodeling.44,45 IL-18 is cross regulated with inducers of extracellular matrix metalloproteinase (EMMPRIN), promoting MMP-9 release, amplifying and exacerbating MI.46 Silencing of EMMPRIN reduced myocardial remodeling by IL-18.43 Moreover, targeted inhibition of the IL-18/TGFβ1/P-SMAD2/3 pathway in a model of MI reduced synthesis of the pro-fibrotic proteins, EDA-Fibronectin, Periostin, Vimentin and α-SMA.47 Similar results were reported for a mouse model of ischemia-reperfusion (I/R). IL-18 blockade reduced infiltration of monocytes and CD4+ T cells in the mouse myocardium, TH17 cell differentiation was inhibited and I/R injury attenuated.48 The conclusion can be drawn from previous studies that elevated post-MI IL-18 exacerbates disease progression. Blockade of the IL-18 signaling pathway appears beneficial but further studies are needed to observe long-term effects.
Many tissues express IL-33. Cardiac fibroblasts are responsible for their synthesis within the heart.49,50 The IL-33 receptor, ST2, has a soluble isoform, sST2, which often acts as a decoy receptor, and the alternative, ST2L to which IL-33 binds and forms an active complex with IL-1R accessory protein.49 Current studies have focused on the IL-33/ST2 axis. IL-33 enriches regulatory T lymphocytes (Tregs) in mouse models of MI and I/R via IL-33/ST2 and increases Sparc expression to promote collagen maturation and maintain cardiac integrity.51,52 Binding of IL-33 to ST2 amplified the group 2 innate lymphocyte (ILC2) population, including IL-13, Areg and BMP-7. IL-13 promotes M2-type macrophage polarization, BMP-7 inhibits TGF-β1 signaling and Areg acts on cardiomyocytes. These factors work together to alleviate post-infarction myocardial fibrosis.53 However, Mia et al found that blockade of the IL-33/ST2 signaling pathway in an MI model attenuated Yap/Taz-induced conversion of cardiac fibroblasts to myofibroblasts, attenuating myocardial fibrosis.54 Thus, IL-33 has the potential to exacerbate myocardial injury. The influences of IL-33 on MI remain controversial and further exploration is required.
IL-36 has four isoforms with different impacts on MI, three agonists, IL-36α, IL-36β and IL-36γ, and one natural inhibitor, IL-36Ra.55,56 IL-36γ upregulates macrophage CD36 expression via the phosphatidylinositol 3-kinase pathway, amplifying the inflammatory response and promoting uptake of oxidized low-density lipoprotein to stimulate foam cell formation and atherogenesis.57 Indeed, targeted inhibition of the IL-36(α/β)/IL-36R pathway attenuated oxidative damage to the vascular endothelium and VCAM-1 and ICAM-1 expression in an I/R mouse.58 IL-36 has hitherto received little attention with respect to MI and isoform functions require characterization.
IL-37 is a cytokine with anti-inflammatory effects which is elevated in the peripheral blood of MI patients.59 IL-37 amplified Tregs and suppressed TH1 and TH17 cells in PBMC of 129 infarct patients, exerting a protective effect.60 Injection of induced cardiosphere overexpressing IL-37 into an I/R model reduced myocardial infarct size, improved left ventricular function and downregulated pro-inflammatory cytokines.61 Moreover, IL-37 contributed to increased proportions of M2-type macrophages and reduced infiltration by pro-inflammatory macrophages and collagen deposition in the myocardium of an MI model, perhaps due to inhibition of NOTCH1 and NF-κB signaling pathways.62,63 IL-37 also mediated lipid metabolism via the IL-1R8/TLR4/NF-κB signaling pathway to reduce MI.64 In conclusion, IL-37 influences CD4+ T cell typing, promotes macrophage transformation and regulates lipid metabolism during MI. The IL-37 amplification of Tregs is similar to the IL-33/ST2 signaling pathway and research is necessary to demonstrate whether these two cytokines engage in cross-talk.
IL-38 is considered anti-inflammatory in various diseases31,65,66 and inhibited the NLRP3 inflammasome and NF-κB and MAPK signaling pathways downstream of IL-36 to block vascular injury during MI.67 Anti-inflammatory effects also result from IL-38 stimulation of the IL-1RAPL1/JNK/AP1 pathway68 and IL-38 promotes the M1 to M2 phenotypic switching in macrophages.69 The IL-38 receptor has yet to be characterized along with the site of macrophage binding (Figure 1).
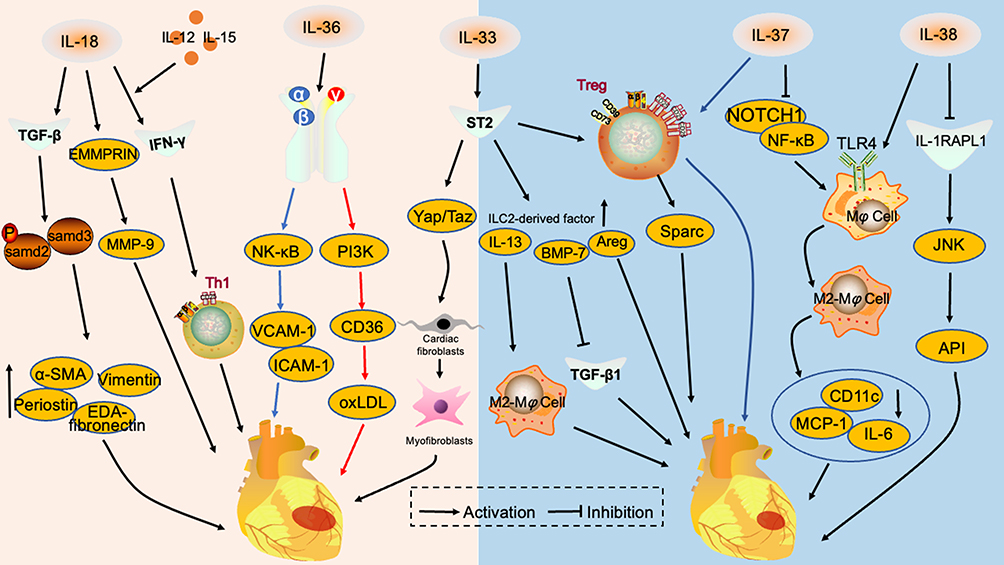 |
Figure 1 Interleukin 1 family in myocardial infarction. IL-18 promotes the expression of pro-fibrotic proteins, EDA-Fibronectin, Periostin, Vimentin and α-SMA, by activating the TGF-β/P-samd2/3 signaling pathway and cross-regulates with EMMRPRIN to promote MMP-9 release. IL-18 acts synergistically with IL-12 and IL-15 to induce IFN-γ release and enhances TH1-type immune responses. These mechanisms aggravate myocardial infarction. IL-36α/β promotes VCAM-1 and ICAM-1 release through NF-κB signaling pathway and aggravates endothelial cell injury. IL-36γ upregulates CD36 through the PI3K signaling pathway, promotes oxLDL secretion and aggravates atheromatous plaque progression. IL-33 stimulates ST2 receptors and activates the Yap/Taz signaling pathway, promoting conversion of cardiac fibroblasts to myofibroblasts. IL-33 binds to ST2 and amplifies the group 2 innate lymphocyte population, including IL-13, Areg and BMP-7. IL-13 promotes M2-type macrophage polarization, BMP-7 inhibits the TGF-β1 signaling pathway and Areg acts directly on cardiomyocytes. IL-37 inhibits NOTCH1 and NF-κΒ signaling pathways, promotes the M2 conversion of macrophages and reduces the secretion of pro-inflammatory factors and chemokines, inhibiting myocardial infarction progression. IL-38 binds to the IL-1RAPL1 receptor to exert anti-inflammatory effects through the JNK/AP1 signaling pathway and alleviate myocardial infarction. Abbreviations: TGF-β, transforming growth factor-β; α-SMA, α-Smooth muscle actin; EMMRPRIN, extracellular matrix metalloproteinase inducer; MMP-9, matrix metalloproteinases-9; IFN-γ, interferon-γ; NF-κB, nuclear factor kappa B; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1; PI3K, phosphatidylinositol 3-kinase; oxLDL, oxidized low-density lipoprotein; ST2, growth ST imulation expressed gene 2; BMP-7, bone morphogenetic protein 7; Sparc, secreted protein acidic and rich in cysteine; TLR4, toll-like receptor 4; MCP-1, monocyte chemoattractant protein-1; IL-1RAPL1, interleukin 1 receptor accessory protein-like 1; Treg, regulatory T cells; JNK, c-jun N-terminal kinase; API, activator protein 1. |
Hypertension
Hypertension is a common and complex clinical syndrome with multiple pathological mechanisms,70 often caused by low-grade chronic inflammation of the kidney and blood vessel walls.71,72
Chronic kidney disease is associated with CVD73 and IL-18, a product of NLRP3 inflammation, has been demonstrated to be involved in the initiation and maintenance of renal inflammation.71,74,75 IL-18 stimulates ICAM-1 and VCAM-1 secretion by vascular endothelial cells to precipitate renal inflammation and hypertension and to mediate leukocyte aggregation, causing damage to the vascular endothelium. Three signaling pathways are involved, IL-18/Src/ERK, IL-18/PI3K/AKT and IL-18/MyD88/TRAF/IRAK/NF-κB.73 However, whether IL-18, like the NLRP3 product, IL-1β, has a role in mediating central system inflammation and causing salt-sensitive hypertension is far from clear.76 IL-18 connects kidney disease and hypertension and elevated IL-18 has been associated with arterial stiffness in patients with metabolic syndrome, weakening the elastic reservoir effect of the artery and increasing late systolic blood pressure.40 IL-18 is a promising therapeutic target in hypertension and its mechanism merits further scrutiny.
Blood pressure correlates with structural changes to the left ventricle and higher blood pressure puts mechanical strain on cardiac myocytes.77 IL-33 was elevated when myocardial endothelial cells were subject to mechanical stretch. It binds to the ST2L receptor, producing an anti-hypertensive, anti-myocardial hypertrophy effect. By contrast, the sST2 receptor competes for IL-33 binding, antagonizing this effect.78 Heterodimerization of ST2L and IL-1RAcP in the presence of IL-33 activates NF-kB and AP-1 to promote hypertension.79 IL-33 restored perivascular adipose tissue (PVAT) activity mediated by eosinophils and vascular anti-constriction to reverse hypertension in a model of obesity.80 Roles of IL-33/ST2 in hypertension remain controversial and the impact of IL-33 on PVAT merits further attention.
Obesity is a common cause of high blood pressure.81–83 IL-36 stimulated the MAPK (p42/p44) pathway, promoting increased intestinal mucus secretion and intestinal commensal A. muciniphila abundance to reduce diet-induced weight gain in mice.84 IL-36 influence on blood pressure was not studied during the above work but the gut microbiome is known to correlate with hypertension.85–88 IL-36 may influence blood pressure by an effect on gut microbes. Nishikawa suggested that IL-36 aggravates acute kidney injury and exacerbates hypertension through upregulation of NF-κB, TNF-α and IL-6 activities.89 The effect ofIL-36 on other organs should be considered in the context of treatment strategies for hypertension.
IL-37 has been shown to be associated with hypertension,90 regulating immune cell differentiation and inhibiting inflammatory factor release.91 IL-37 may reduce vascular endothelial damage during hypertension by mediating NO bioavailability while reducing NADPHO-associated products.92 Mice injected with recombinant IL-37 had reduced secretion of the inflammatory factors, IL-1β, CXCL-1 and TNFα, in epididymal adipose tissue and improved systemic insulin resistance status. A similar effect was observed in human adipocytes and AMPK and the mTOR signaling pathway have been implicated.93 The protective effect of IL-37 in hypertension appears largely due to inhibition of inflammatory factor release.
There is a paucity of reports demonstrating direct effects of IL-38 on hypertension but its anti-inflammatory and hyperlipidemia modulating effects may be involved.31,94 IL-38 inhibited human adipocyte differentiation and reduced secretion of inflammatory cytokines, IL-1β and MCP-1,95 and IL-38 reduction of joint inflammation involved the NF-κB signaling pathway also known to be involved in hypertension.96,97 Macrophages, smooth muscle cells and vascular endothelial cells also release IL-38 under apoptotic conditions and affect the circulatory Bcl-2/Bax/Caspase-3 signaling pathway, attenuating hyperlipidemia.68,94 No direct relationship between IL-38 and hypertension has yet been shown but this molecule does seem to ameliorate the disorder (Figure 2).
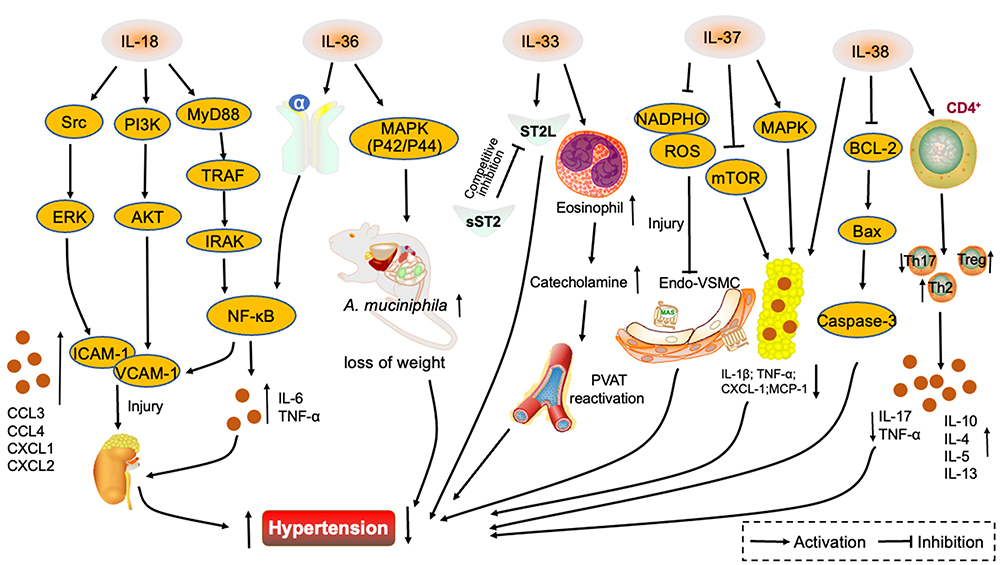 |
Figure 2 Interleukin 1 family in hypertension. IL-18 promotes VCAM-1 and ICAM-1 secretion through three signaling pathways, Src/ERK, PI3K/AKT and MyD88/TRAF/IRAK/NF-κB. Renal vascular endothelial injury is exacerbated and hypertension promoted. IL-36α stimulates the NF-κB signaling pathway, IL-6 and TNF-α are secreted, renal vasculature damaged and hypertension aggravated. IL-36 activates the MAPK signaling pathway, upregulates A. muciniphila abundance, regulates lipid metabolism and lowers blood pressure. Competitive binding of IL-33 by sST2 receptors attenuates the protective effect of IL-33/ST2L on blood pressure. IL-33 also promotes an increase in eosinophils, catecholamine secretion, perivascular adipose tissue activity and results in the attenuation hypertension. IL-37 attenuates the damage of NADPHO metabolites on vascular endothelium, while activating the MAPK signaling pathway and inhibiting the mTOR pathway, attenuating inflammatory factor secretion in adipose tissue and alleviating hypertension. IL-38 reduces inflammatory factor secretion in adipocytes and inhibits the BCL-2/Bax/Caspase-3 signaling pathway while binding to CD4+ T cells, decreasing Th17 ratio and upregulating Th2 and Treg ratios and controlling blood pressure. Abbreviations: Src, tyrosine protein kinase; ERK, extracellular regulated protein kinases; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; MyD88, myeloid differentiation factor 88; TRAF, tumor necrosis factor receptor-associated factor; IRAK, interleukin-1 receptor-associated kinase; NF-κB, nuclear factor kappa B; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1; CCL3, chemokines-3; CCL4, chemokines-4; CXCL1, chemokine (C-X-C motif) ligand 1; CXCL2, chemokine (C-X-C motif) ligand 2; TNF-α, tumor necrosis factor-α; MAPK, mitogen-activated protein kinase; A. muciniphila, Akkermansia muciniphila; ST2L, the membrane-bound isoform; sST2, soluble protein; PVAT, perivascular adipose tissue; NADPHO, nicotinamide adenine dinucleotide phosphate oxidase; ROS, reactive oxygen species; mTOR, mammalian target of rapamycin; BCL-2, B cell lymphoma protein-2; Bax, apoptosis proteins; Caspase-3, cysteine aspartate protease 3; Treg, regulatory T cells; MCP-1, monocyte chemoattractant protein-1. |
Arrhythmias
Arrhythmias occur alone or as a complication of various types of heart disease and in severe cases increase the risk of death.98 Inflammatory leukocytes may cause arrhythmias by switching phenotypes or interfering with conduction between cardiomyocytes.99
Increased IL-18 in the peripheral circulation has been associated with atrial fibrillation and could be considered a potential therapeutic target.100,101 Indeed, the QT interval in sickle cell cardiomyopathy patients was strongly correlated with plasma IL-18 and IL-18 increased susceptibility to ventricular arrhythmias, perhaps through the NF-κB signaling pathway.102 Inhibition of IL-18 via a binding protein improved cardiac diastolic function and targeting this molecule may be beneficial for patients at risk of sudden cardiac death.
IL-37 level correlated with the type of atrial fibrillation (AF), levels being lower in patients with paroxysmal and persistent AF than in those with permanent AF.103 IL-37 treatment decreased IL-6 and CRP secretion in in vitro experiments.103 Thus, IL-37 appears to have a protective effect in AF, perhaps through inhibition of the NF-κB signaling pathway.
Targeting the inflammatory response may be an appropriate therapeutic foundation for the treatment of atrial fibrillation.
Valvular Heart Disease
Valvular heart disease is common in industrialized countries, and the incidence rate in the United States is approximately 2.5%.104 Surgery is the best option for patients with end-stage disease.
IL-18 is expressed in the aortic valve and correlates with aortic stenosis.105 IL-18 induced the conversion of valvular interstitial cells to myofibroblasts and induced osteopontin (OPN) secretion via NF-κB to accelerate valve calcification.106 HO-1 and FPN were also activated through the p38 MAPK/ERK1/2 signaling pathway, promoting phagocytosis and degradation of erythrocytes by M1-type macrophages, exacerbating valvular calcification.107
IL-33 and sST2 were found to be elevated in peripheral blood from patients with non-rheumatic aortic disease and α-SMA, OPN and ST2 were co-expressed, demonstrating that IL-33 induces phenotypic transformation of valvular mesenchymal cells through the NF-κB and p38 MAPK pathways and exacerbates aortic lesions.108 Moreover, IL-33 binding to ST2 receptors activated valvular interstitial cells and promoted mesenchymal transformation of valve endothelial cells, contributing to mitral mucinous tumor degeneration.109
IL-37 was found to be expressed in both aortic disease and mitral valve disease but with lower levels at the site of aortic stenosis.110 Recombinant IL-37 attenuated the osteogenic response in valves by inhibiting the NF-κB and ERK1/2 signaling pathways and reducing bone morphogenetic protein-2 and alkaline phosphatase release.111
IL-18 and IL-33 appear to facilitate valvular calcific disease via specific protein activating effects while IL-37 was protective. However, the paucity of relevant literature and the lack of mature animal models means that further research is needed to consolidate these conclusions.
Aneurysms
Aneurysms, whether thoracic or abdominal aortic, are extremely dangerous and aortic dissection is a fatal hazard.112 The pathophysiological basis concerns inflammatory cascade and extracellular matrix breakdown due to arterial injury.113
IL-18 has recently been found that to enhance OPN expression in the intima, leading to the release of matrix metalloproteinases from macrophages and decreased collagen and elastin which exacerbates the progression of abdominal aortic aneurysms.114 Obesity is a pre-disposing factor for cardiovascular disease, including aneurysms, and changes in PVAT may form part of the underlying mechanism.115,116 Adipocytes and PVAT were found to bind IL-18r and Na-Cl cotransport protein (NCC) at the sites of abdominal aortic aneurysm lesions which enhanced IL-18 binding to macrophages, arterial smooth muscle cells and vascular endothelial cells, exacerbating endothelial injury and promoting disease progression. IL-18r and NCC were found to act synergistically and also participated independently in the pathophysiology of abdominal aortic aneurysms.117
Animal experiments have shown beneficial effects of IL-33.118 Activation of the IL-33/ST2 signaling pathway amplifies Tregs to inhibit inflammatory factors and chemokines such as IL-6 and MCP-1 in smooth muscle cells while M2 macrophage conversion is promoted. However, Tregs are crucial for this mechanism and in their absence, the protective effect was lost (Figure 3).
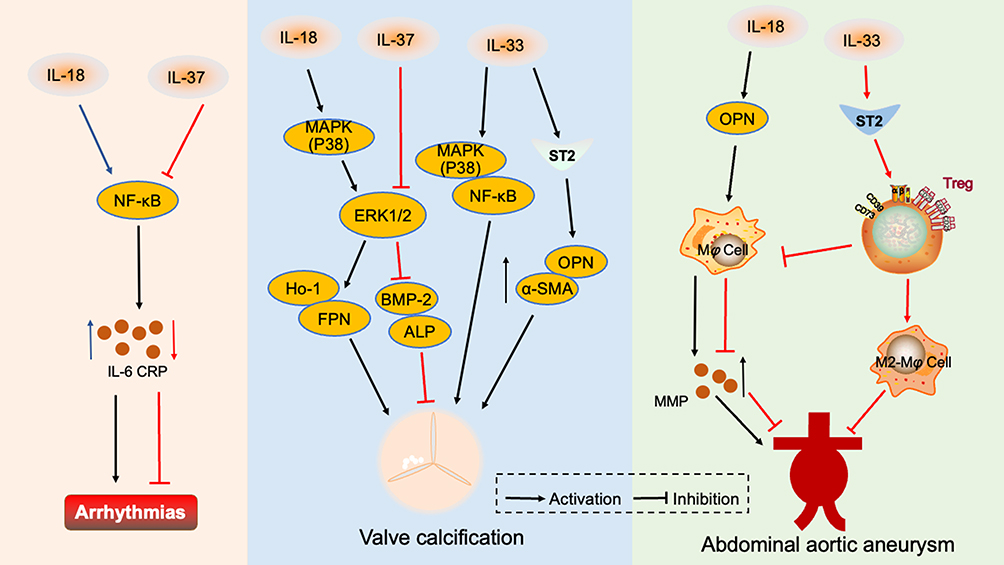 |
Figure 3 Interleukin 1 family in other cardiovascular diseases. IL-18 activates the NF-κB signaling pathway, causing the release of inflammatory factors and inducing arrhythmia. However, IL-37 reduces the incidence of arrhythmia by inhibiting the NF-κB pathway. IL-18 activates the MAPK(p38)/ERK1/2 signaling pathway, promotes HO-1 and FPN secretion, and accelerates valve calcification. IL-33 mediates the MAPK(p38)/ NF-κB signaling pathway and promotes OPN and α-SMA secretion through IL-33/ST2, which aggravates valve calcification. IL-37 inhibits ERK1/2 and reduces BMP-2 and ALP release, thus reducing valve calcification. IL-18 promotes the secretion of OPN which causes macrophages to secrete MMP and aggravates abdominal aortic aneurysm. IL-33 binds to the ST2 receptor and enriches Treg, to exert an inhibitory effect on abdominal aortic aneurysm by inhibiting macrophage MMP secretion and promoting macrophage conversion to the M2 phenotype. Abbreviations: NF-κB, nuclear factor kappa B; CRP, c-reactive protein; MAPK, mitogen-activated protein kinase; ERK1/2, extracellular regulated protein kinase 1/2; HO-1, heme oxygenase-1; FPN, ferroportin; ST2, growth ST imulation expressed gene 2; OPN, osteopontin; α-SMA, α-Smooth muscle actin; BMP-2, bone morphogenetic protein 2; ALP, alkaline phosphatase; MMP, matrix metalloproteinases; Treg, regulatory T cells. |
IL-18 appears to promote aneurysm development and the relationship between IL-18 and PVAT could provide new treatment options. IL-33 is a protective factor despite its aggravating effect on CVD. IL33/ST2 pathway specificity in aneurysmal awaits further clarification.
Prospect and Future
IL-18 promotes several cardiac diseases through multiple pathways and statins, eg, atorvastatin, commonly prescribed for coronary heart disease have lipid-lowering effects and inhibit the NLRP3 inflammasome to reduce IL-18 secretion.119 Other drugs such as remifentanil and baicalin120,121 may also inhibit IL-18. In addition, NCC inhibitors such as hydrochlorothiazide, have potential value in limiting the progression of abdominal aortic aneurysms. Interleukin receptors may thus be a suitable focus for therapeutic research and interactions between IL-18 and other cytokines require attention. IL-18 acts synergistically with IL-1 to exacerbate systemic acute infectious inflammation122 and a synergistic interaction between IL-22 and IL-18 in the prevention and treatment of rotavirus infection was also found by Zhang.123 However, synergistic effects of IL-18 with other inflammatory factors have been little reported and should be considered when designing drugs.
While focusing on the therapeutic potential of IL-33 for CVD its harmful effects should also be kept in mind. IL-33 induced eosinophilic pericarditis through the IL-33 receptor124 and the beneficial effects of IL-33 in CVDs, such as hypertension, are attenuated by its sST2 decoy receptor. Promising effects of IL-33 injections have been reported for hypertension but the resulting splenomegaly does significantly limit its therapeutic potential.80 Several IL-33 blocking strategies signaling are feasible with anti-IL-33 mAb, anti-ST2 or sST2 being under development for asthma and COPD.125,126 In addition, the hybrid factor, IL-233, a complex of IL-2 and IL-33, enhances the protective effect of Tregs and ILC2 in AKI and prevents I/R injury.127
IL-36 inhibitors are currently used for psoriasis29 and effect on blood pressure, via modulation of metabolic patterns and gut microbiota, and on vascular endothelial damage, demonstrate the therapeutic potential for CVD. Elevated urinary IL-36α and enhanced IL-36α staining in renal biopsy samples are found in AKI89 and a contrasting increase in IL-36γ in P. aeruginosa infection-mediated lung injury.128 IL-36β enhances disease progression in a mouse model of colitis by promoting the Th2 response while decreasing the Foxp3+ Tregs response.129 Different isoforms of IL-36 mediate disease in different ways and attention should be paid to its subtypes.
IL-37 and lL-38 suppress the secretion of inflammatory factors, inhibit macrophage polarization and transport and modulate immunity. Recombinant IL-37 enhances mesenchymal stem cells for the treatment of SLE.130 Probiotics and prebiotics increase IL-38 gene expression and reduce airway inflammation to control asthma.131 Positive effects of IL-37 and IL-38 have been shown in animal models of CVD but have been little studied in hypertension, valvular disease, aneurysms and arrhythmias and deserve further investigation.
Conclusion
The interleukin-1 cytokines, IL-18, IL-33, IL-36, IL-37 and IL-38, have been linked to CVD. IL-18 and IL-36 promote the development of MI, hypertension, calcific valve disease and aneurysm by inducing release of inflammatory factors, chemokines, adhesion factors, related proteins and promoting lipid deposition. By contrast, IL-37 and IL-38 inhibit the above pathways and exert a protective effect. IL-36 increases the abundance of intestinal flora and reduces hypertension. Controversy surrounds the protective and promotional role of CVD by IL-33. Further elucidation of mechanisms, regulatory relationships and effector targets is needed for therapeutic development.
Acknowledgments
The authors would like to express their gratitude to EditSprings (https://www.editsprings.cn) for the expert linguistic services provided. The authors are grateful to Sisi Mo for her help with the study design and manuscript preparation in this paper.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (8206020365).
Disclosure
The authors declare no conflict of interest.
References
1. Liberale L, Montecucco F, Tardif JC, Libby P, Camici GG. Inflamm-ageing: the role of inflammation in age-dependent cardiovascular disease. Eur Heart J. 2020;41(31):2974–2982. doi:10.1093/eurheartj/ehz961
2. Sidney S, Go AS, Jaffe MG, Solomon MD, Ambrosy AP, Rana JS. Association between aging of the US population and heart disease mortality from 2011 to 2017. JAMA Cardiol. 2019;4(12):1280–1286. doi:10.1001/jamacardio.2019.4187
3. Roth GA, Mensah GA, Johnson CO, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J Am Coll Cardiol. 2020;76(25):2982–3021. doi:10.1016/j.jacc.2020.11.010
4. Paneni F, Diaz Cañestro C, Libby P, Lüscher TF, Camici GG. The aging cardiovascular system: understanding it at the cellular and clinical levels. J Am Coll Cardiol. 2017;69(15):1952–1967. doi:10.1016/j.jacc.2017.01.064
5. Liberale L, Badimon L, Montecucco F, Lüscher TF, Libby P, Camici GG. Inflammation, aging, and cardiovascular disease: JACC review topic of the week. J Am Coll Cardiol. 2022;79(8):837–847. doi:10.1016/j.jacc.2021.12.017
6. Andersson C, Vasan RS. Epidemiology of cardiovascular disease in young individuals. Nat Rev Cardiol. 2018;15(4):230–240. doi:10.1038/nrcardio.2017.154
7. Zhao D, Liu J, Wang M, Zhang X, Zhou M. Epidemiology of cardiovascular disease in China: current features and implications. Nat Rev Cardiol. 2019;16(4):203–212. doi:10.1038/s41569-018-0119-4
8. Prabhakaran D, Jeemon P, Roy A. Cardiovascular diseases in India: current epidemiology and future directions. Circulation. 2016;133(16):1605–1620. doi:10.1161/circulationaha.114.008729
9. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. 2020;17(6):327–340. doi:10.1038/s41569-019-0326-7
10. Soehnlein O, Libby P. Targeting inflammation in atherosclerosis — from experimental insights to the clinic. Nat Rev Drug Discov. 2021;20(8):589–610. doi:10.1038/s41573-021-00198-1
11. Stark K, Massberg S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol. 2021;18(9):666–682. doi:10.1038/s41569-021-00552-1
12. Ye J, Que B, Huang Y, et al. Interleukin-12p35 knockout promotes macrophage differentiation, aggravates vascular dysfunction, and elevates blood pressure in angiotensin II-infused mice. Cardiovasc Res. 2019;115(6):1102–1113. doi:10.1093/cvr/cvy263
13. Zhang M, Gao J, Zhao X, et al. p38α in macrophages aggravates arterial endothelium injury by releasing IL-6 through phosphorylating megakaryocytic leukemia 1. Redox Biol. 2021;38:101775. doi:10.1016/j.redox.2020.101775
14. Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10(2):89–102. doi:10.1038/nri2691
15. Kanneganti T-D, Phimister EG. Inflammatory Bowel Disease and the NLRP3 inflammasome. N Engl J Med. 2017;377(7):694–696. doi:10.1056/NEJMcibr1706536
16. Dmitrieva-Posocco O, Dzutsev A, Posocco DF, et al. Cell-type-specific responses to interleukin-1 control microbial invasion and tumor-elicited inflammation in colorectal cancer. Immunity. 2019;50(1):166–180.e7. doi:10.1016/j.immuni.2018.11.015
17. Afsar B, Covic A, Ortiz A, Afsar RE, Kanbay M. The future of IL-1 targeting in kidney disease. Drugs. 2018;78(11):1073–1083. doi:10.1007/s40265-018-0942-2
18. Wong R, Lénárt N, Hill L, et al. Interleukin-1 mediates ischaemic brain injury via distinct actions on endothelial cells and cholinergic neurons. Brain Behav Immun. 2019;76:126–138. doi:10.1016/j.bbi.2018.11.012
19. Libby P, Kobold S. Inflammation: a common contributor to cancer, aging, and cardiovascular diseases—expanding the concept of cardio-oncology. Cardiovasc Res. 2019;115(5):824–829. doi:10.1093/cvr/cvz058
20. Chan AH, Schroder K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J Exp Med. 2020;217(1):e20190314. doi:10.1084/jem.20190314
21. Akdis M, Burgler S, Crameri R, et al. Interleukins, from 1 to 37, and interferon-γ: receptors, functions, and roles in diseases. J Allergy Clin Immunol. 2011;127(3):701–21.e1–70. doi:10.1016/j.jaci.2010.11.050
22. Garlanda C, Mantovani A. Interleukin-1 in tumor progression, therapy, and prevention. Cancer Cell. 2021;39(8):1023–1027. doi:10.1016/j.ccell.2021.04.011
23. Migliorini P, Italiani P, Pratesi F, Puxeddu I, Boraschi D. The IL-1 family cytokines and receptors in autoimmune diseases. Autoimmun Rev. 2020;19(9):102617. doi:10.1016/j.autrev.2020.102617
24. Zhu AS, Mustafa T, Connell JP, Grande-Allen KJ. Tumor necrosis factor alpha and interleukin 1 beta suppress myofibroblast activation via nuclear factor kappa B signaling in 3D-cultured mitral valve interstitial cells. Acta Biomater. 2021;127:159–168. doi:10.1016/j.actbio.2021.03.075
25. Schunk SJ, Triem S, Schmit D, et al. Interleukin-1α is a central regulator of leukocyte-endothelial adhesion in myocardial infarction and in chronic kidney disease. Circulation. 2021;144(11):893–908. doi:10.1161/circulationaha.121.053547
26. Xiong S, Zhang L, Richner JM, Class J, Rehman J, Malik AB. Interleukin-1RA mitigates SARS-CoV-2-induced inflammatory lung vascular leakage and mortality in humanized K18-hACE-2 mice. Arterioscler Thromb Vasc Biol. 2021;41(11):2773–2785. doi:10.1161/atvbaha.121.316925
27. Nakamura K, Kassem S, Cleynen A, et al. Dysregulated IL-18 is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell. 2018;33(4):634–648.e5. doi:10.1016/j.ccell.2018.02.007
28. Vainchtein ID, Chin G, Cho FS, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359(6381):1269–1273. doi:10.1126/science.aal3589
29. Bachelez H, Choon SE, Marrakchi S, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. 2019;380(10):981–983. doi:10.1056/NEJMc1811317
30. Huang Z, Xie L, Li H, et al. Insight into interleukin-37: the potential therapeutic target in allergic diseases. Cytokine Growth Factor Rev. 2019;49:32–41. doi:10.1016/j.cytogfr.2019.10.003
31. Sun X, Hou T, Cheung E, et al. Anti-inflammatory mechanisms of the novel cytokine interleukin-38 in allergic asthma. Cell Mol Immunol. 2020;17(6):631–646. doi:10.1038/s41423-019-0300-7
32. Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res. 2020;126(9):1260–1280. doi:10.1161/circresaha.120.315937
33. Gkaliagkousi E, Lazaridis A, Dogan S, et al. Theories and molecular basis of vascular aging: a review of the literature from vascagenet group on pathophysiological mechanisms of vascular aging. Int J Mol Sci. 2022;23(15):8672. doi:10.3390/ijms23158672
34. Buckley LF, Abbate A. Interleukin-1 blockade in cardiovascular diseases: a clinical update. Eur Heart J. 2018;39(22):2063–2069. doi:10.1093/eurheartj/ehy128
35. Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction (2018). Circulation. 2018;138(20):e618–e651. doi:10.1161/cir.0000000000000617
36. Nagele P, Simplified A. Proposal to redefine acute myocardial infarction versus acute myocardial injury. Circulation. 2020;141(18):1431–1433. doi:10.1161/circulationaha.119.044996
37. Ruparelia N, Chai JT, Fisher EA, Choudhury RP. Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nat Rev Cardiol. 2017;14(3):133–144. doi:10.1038/nrcardio.2016.185
38. Sun K, Li YY, Jin J. A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Signal Transduct Target Ther. 2021;6(1):79. doi:10.1038/s41392-020-00455-6
39. Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15(4):203–214. doi:10.1038/nrcardio.2017.161
40. Mozos I, Malainer C, Horbańczuk J, et al. Inflammatory markers for arterial stiffness in cardiovascular diseases. Front Immunol. 2017;8:1058. doi:10.3389/fimmu.2017.01058
41. Ridker PM, MacFadyen JG, Thuren T, Libby P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1β inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur Heart J. 2020;41(23):2153–2163. doi:10.1093/eurheartj/ehz542
42. Giral H, Franke V, Moobed M, et al. Rapid inflammasome activation is attenuated in post-myocardial infarction monocytes. Front Immunol. 2022;13:857455. doi:10.3389/fimmu.2022.857455
43. Su Z, Lin R, Chen Y, et al. Knockdown of EMMPRIN improves adverse remodeling mediated by IL-18 in the post-infarcted heart. Am J Transl Res. 2015;7(10):1908–1916.
44. Maffia P, Grassia G, Di Meglio P, et al. Neutralization of interleukin-18 inhibits neointimal formation in a rat model of vascular injury. Circulation. 2006;114(5):430–437. doi:10.1161/circulationaha.105.602714
45. Kaplanski G. Interleukin-18: biological properties and role in disease pathogenesis. Immunol Rev. 2018;281(1):138–153. doi:10.1111/imr.12616
46. Xie SL, Chen YY, Zhang HF, et al. Interleukin 18 and extracellular matrix metalloproteinase inducer cross-regulation: implications in acute myocardial infarction. Transl Res. 2015;165(3):387–395. doi:10.1016/j.trsl.2014.09.001
47. Zhao J, Chen Y, Chen Q, et al. Curcumin ameliorates cardiac fibrosis by regulating macrophage-fibroblast crosstalk via IL18-P-SMAD2/3 signaling pathway inhibition. Front Pharmacol. 2021;12:784041. doi:10.3389/fphar.2021.784041
48. Gu H, Xie M, Xu L, Zheng X, Yang Y, Lv X. The protective role of interleukin-18 binding protein in a murine model of cardiac ischemia/reperfusion injury. Transpl Int. 2015;28(12):1436–1444. doi:10.1111/tri.12683
49. Miller AM, Liew FY. The IL-33/ST2 pathway--A new therapeutic target in cardiovascular disease. Pharmacol Ther. 2011;131(2):179–186. doi:10.1016/j.pharmthera.2011.02.005
50. Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest. 2007;117(6):1538–1549. doi:10.1172/jci30634
51. Xia N, Lu Y, Gu M, et al. A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction. Circulation. 2020;142(20):1956–1973. doi:10.1161/circulationaha.120.046789
52. Cheng X, Liao YH, Ge H, et al. TH1/TH2 functional imbalance after acute myocardial infarction: coronary arterial inflammation or myocardial inflammation. J Clin Immunol. 2005;25(3):246–253. doi:10.1007/s10875-005-4088-0
53. Chen WY, Wu YH, Tsai TH, et al. Group 2 innate lymphoid cells contribute to IL-33-mediated alleviation of cardiac fibrosis. Theranostics. 2021;11(6):2594–2611. doi:10.7150/thno.51648
54. Mia MM, Cibi DM, Ghani S, et al. Loss of Yap/Taz in cardiac fibroblasts attenuates adverse remodelling and improves cardiac function. Cardiovasc Res. 2022;118(7):1785–1804. doi:10.1093/cvr/cvab205
55. Melton E, Qiu H. Interleukin-36 cytokine/receptor signaling: a new target for tissue fibrosis. Int J Mol Sci. 2020;21(18):6458. doi:10.3390/ijms21186458
56. Gabay C, Towne JE. Regulation and function of interleukin-36 cytokines in homeostasis and pathological conditions. J Leukoc Biol. 2015;97(4):645–652. doi:10.1189/jlb.3RI1014-495R
57. Zhang M, Liu J, Gao R, et al. Interleukin-36γ aggravates macrophage foam cell formation and atherosclerosis progression in ApoE knockout mice. Cytokine. 2021;146:155630. doi:10.1016/j.cyto.2021.155630
58. El-Awaisi J, Kavanagh DP, Rink MR, Weston CJ, Drury NE, Kalia N. Targeting IL-36 improves age-related coronary microcirculatory dysfunction and attenuates myocardial ischemia/reperfusion injury in mice. JCI Insight. 2022;7(5):e155236. doi:10.1172/jci.insight.155236
59. McCurdy S, Yap J, Irei J, Lozano J, Boisvert WA. IL-37-a putative therapeutic agent in cardiovascular diseases. Qjm. 2022;115(11):719–725. doi:10.1093/qjmed/hcab011
60. Mao X, Zhu R, Zhang F, et al. IL-37 plays a beneficial role in patients with acute coronary syndrome. Mediators Inflamm. 2019;2019:9515346. doi:10.1155/2019/9515346
61. Li J, Zhang WJ, Yao H, Li TM. Therapeutic effects of interleukin-37 and induced cardiosphere on treating myocardial ischemia-reperfusion injury. Int Immunopharmacol. 2020;88:106719. doi:10.1016/j.intimp.2020.106719
62. Wang YM, Zhang JJ, Wu BW, et al. IL-37 improves mice myocardial infarction via inhibiting YAP-NLRP3 signaling mediated macrophage programming. Eur J Pharmacol. 2022;934:175293. doi:10.1016/j.ejphar.2022.175293
63. Su Z, Tao X. Current understanding of IL-37 in human health and disease. Front Immunol. 2021;12:696605. doi:10.3389/fimmu.2021.696605
64. Zhu R, Zhang F, Pan C, Yu K, Zhong Y, Zeng Q. Role of IL-37- and IL-37-treated dendritic cells in acute coronary syndrome. Oxid Med Cell Longev. 2021;2021:6454177. doi:10.1155/2021/6454177
65. Tsilioni I, Pantazopoulos H, Conti P, Leeman SE, Theoharides TC. IL-38 inhibits microglial inflammatory mediators and is decreased in amygdala of children with autism spectrum disorder. Proc Natl Acad Sci U S A. 2020;117(28):16475–16480. doi:10.1073/pnas.2004666117
66. Gao X, Chan PKS, Lui GCY, et al. Interleukin-38 ameliorates poly(I:C) induced lung inflammation: therapeutic implications in respiratory viral infections. Cell Death Dis. 2021;12(1):53. doi:10.1038/s41419-020-03283-2
67. van de Veerdonk FL, Stoeckman AK, Wu G, et al. IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc Natl Acad Sci U S A. 2012;109(8):3001–3005. doi:10.1073/pnas.1121534109
68. Mora J, Schlemmer A, Wittig I, et al. Interleukin-38 is released from apoptotic cells to limit inflammatory macrophage responses. J Mol Cell Biol. 2016;8(5):426–438. doi:10.1093/jmcb/mjw006
69. Li Z, Ding Y, Peng Y, et al. Effects of IL-38 on macrophages and myocardial ischemic injury. Front Immunol. 2022;13:894002. doi:10.3389/fimmu.2022.894002
70. Madhur MS, Elijovich F, Alexander MR, et al. Hypertension: do inflammation and immunity hold the key to solving this epidemic? Circ Res. 2021;128(7):908–933. doi:10.1161/circresaha.121.318052
71. Thomas JM, Ling YH, Huuskes B, et al. IL-18 (Interleukin-18) produced by renal tubular epithelial cells promotes renal inflammation and injury during deoxycorticosterone/salt-induced hypertension in mice. Hypertension. 2021;78(5):1296–1309. doi:10.1161/hypertensionaha.120.16437
72. Krishnan SM, Sobey CG, Latz E, Mansell A, Drummond GR. IL-1β and IL-18: inflammatory markers or mediators of hypertension? Br J Pharmacol. 2014;171(24):5589–5602. doi:10.1111/bph.12876
73. Thomas JM, Huuskes BM, Sobey CG, Drummond GR, Vinh A. The IL-18/IL-18R1 signalling axis: diagnostic and therapeutic potential in hypertension and chronic kidney disease. Pharmacol Ther. 2022;239:108191. doi:10.1016/j.pharmthera.2022.108191
74. Parikh CR, Abraham E, Ancukiewicz M, Edelstein CL. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J Am Soc Nephrol. 2005;16(10):3046–3052. doi:10.1681/asn.2005030236
75. Akcay A, Nguyen Q, Edelstein CL. Mediators of inflammation in acute kidney injury. Mediators Inflamm. 2009;2009:137072. doi:10.1155/2009/137072
76. Liu D, Zeng X, Li X, Mehta JL, Wang X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. 2018;113(1):5. doi:10.1007/s00395-017-0663-9
77. Ghali R, Altara R, Louch WE, et al. IL-33 (Interleukin 33)/sST2 axis in hypertension and heart failure. Hypertension. 2018;72(4):818–828. doi:10.1161/hypertensionaha.118.11157
78. Chen WY, Hong J, Gannon J, Kakkar R, Lee RT. Myocardial pressure overload induces systemic inflammation through endothelial cell IL-33. Proc Natl Acad Sci U S A. 2015;112(23):7249–7254. doi:10.1073/pnas.1424236112
79. Yin X, Cao H, Wei Y, Li HH. Alteration of the IL-33-sST2 pathway in hypertensive patients and a mouse model. Hypertens Res. 2019;42(11):1664–1671. doi:10.1038/s41440-019-0291-x
80. Saxton SN, Whitley AS, Potter RJ, Withers SB, Grencis R, Heagerty AM. Interleukin-33 rescues perivascular adipose tissue anticontractile function in obesity. Am J Physiol Heart Circ Physiol. 2020;319(6):H1387–h1397. doi:10.1152/ajpheart.00491.2020
81. Mouton AJ, Li X, Hall ME, Hall JE. Obesity, hypertension, and cardiac dysfunction: novel roles of immunometabolism in macrophage activation and inflammation. Circ Res. 2020;126(6):789–806. doi:10.1161/circresaha.119.312321
82. Hall JE, Do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity, kidney dysfunction and hypertension: mechanistic links. Nat Rev Nephrol. 2019;15(6):367–385. doi:10.1038/s41581-019-0145-4
83. Hu T, Wu Q, Yao Q, Jiang K, Yu J, Tang Q. Short-chain fatty acid metabolism and multiple effects on cardiovascular diseases. Ageing Res Rev. 2022;81:101706. doi:10.1016/j.arr.2022.101706
84. Giannoudaki E, Hernandez-Santana YE, Mulfaul K, et al. Interleukin-36 cytokines alter the intestinal microbiome and can protect against obesity and metabolic dysfunction. Nat Commun. 2019;10(1):4003. doi:10.1038/s41467-019-11944-w
85. Verhaar BJH, Collard D, Prodan A, et al. Associations between gut microbiota, faecal short-chain fatty acids, and blood pressure across ethnic groups: the HELIUS study. Eur Heart J. 2020;41(44):4259–4267. doi:10.1093/eurheartj/ehaa704
86. Yang T, Santisteban MM, Rodriguez V, et al. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65(6):1331–1340. doi:10.1161/hypertensionaha.115.05315
87. Adnan S, Nelson JW, Ajami NJ, et al. Alterations in the gut microbiota can elicit hypertension in rats. Physiol Genomics. 2017;49(2):96–104. doi:10.1152/physiolgenomics.00081.2016
88. Kaye DM, Shihata WA, Jama HA, et al. Deficiency of prebiotic fiber and insufficient signaling through gut metabolite-sensing receptors leads to cardiovascular disease. Circulation. 2020;141(17):1393–1403. doi:10.1161/circulationaha.119.043081
89. Nishikawa H, Taniguchi Y, Matsumoto T, et al. Knockout of the interleukin-36 receptor protects against renal ischemia-reperfusion injury by reduction of proinflammatory cytokines. Kidney Int. 2018;93(3):599–614. doi:10.1016/j.kint.2017.09.017
90. Satiroglu O, Gürlek B, Durakoglugil ME, et al. The role of serum interleukin-37 levels, inflammation and blood pressure in patients with preeclampsia. Clin Exp Hypertens. 2020;42(7):669–674. doi:10.1080/10641963.2020.1772813
91. Ye J, Wang Y, Wang Z, et al. Circulating IL-37 levels are elevated in patients with hypertension. Exp Ther Med. 2021;21(6):558. doi:10.3892/etm.2021.9990
92. Ballak DB, Brunt VE, Sapinsley ZJ, et al. Short-term interleukin-37 treatment improves vascular endothelial function, endurance exercise capacity, and whole-body glucose metabolism in old mice. Aging Cell. 2020;19(1):e13074. doi:10.1111/acel.13074
93. Ballak DB, Li S, Cavalli G, et al. Interleukin-37 treatment of mice with metabolic syndrome improves insulin sensitivity and reduces pro-inflammatory cytokine production in adipose tissue. J Biol Chem. 2018;293(37):14224–14236. doi:10.1074/jbc.RA118.003698
94. Lai M, Peng H, Wu X, Chen X, Wang B, Su X. IL-38 in modulating hyperlipidemia and its related cardiovascular diseases. Int Immunopharmacol. 2022;108:108876. doi:10.1016/j.intimp.2022.108876
95. Li Y, Chen S, Sun J, Yu Y, Li M. Interleukin-38 inhibits adipogenesis and inflammatory cytokine production in 3T3-L1 preadipocytes. Cell Biol Int. 2020;44(11):2357–2362. doi:10.1002/cbin.11428
96. Luo P, Zhao T, He H. IL-38-mediated NLRP3/caspase-1 inhibition is a disease-modifying treatment for TMJ inflammation. Ann N Y Acad Sci. 2022;1508(1):92–104. doi:10.1111/nyas.14704
97. Su Q, Yu XJ, Wang XM, et al. Na(+)/K(+)-ATPase Alpha 2 isoform elicits rac1-dependent oxidative stress and TLR4-induced inflammation in the hypothalamic paraventricular nucleus in high salt-induced hypertension. Antioxidants. 2022;11(2):288. doi:10.3390/antiox11020288
98. Byrne R, Constant O, Smyth Y, et al. Multiple source surveillance incidence and aetiology of out-of-hospital sudden cardiac death in a rural population in the West of Ireland. Eur Heart J. 2008;29(11):1418–1423. doi:10.1093/eurheartj/ehn155
99. Grune J, Yamazoe M, Nahrendorf M. Electroimmunology and cardiac arrhythmia. Nat Rev Cardiol. 2021;18(8):547–564. doi:10.1038/s41569-021-00520-9
100. Luan Y, Guo Y, Li S, et al. Interleukin-18 among atrial fibrillation patients in the absence of structural heart disease. Europace. 2010;12(12):1713–1718. doi:10.1093/europace/euq321
101. Vm M, Al S, Aa A, et al. Circulating interleukin-18: association with IL-8, IL-10 and VEGF serum levels in patients with and without heart rhythm disorders. Int J Cardiol. 2016;215:105–109. doi:10.1016/j.ijcard.2016.04.002
102. Gupta A, Fei Y-D, Kim TY, et al. IL-18 mediates sickle cell cardiomyopathy and ventricular arrhythmias. Blood. 2021;137(9):1208–1218. doi:10.1182/blood.2020005944
103. Li W, Li S, Li X, Jiang S, Han B. Interleukin-37 elevation in patients with atrial fibrillation. Clin Cardiol. 2017;40(2):66–72. doi:10.1002/clc.22630
104. Iung B, Vahanian A. Epidemiology of valvular heart disease in the adult. Nat Rev Cardiol. 2011;8(3):162–172. doi:10.1038/nrcardio.2010.202
105. Bartoli-Leonard F, Zimmer J, Aikawa E. Innate and adaptive immunity: the understudied driving force of heart valve disease. Cardiovasc Res. 2021;117(13):2506–2524. doi:10.1093/cvr/cvab273
106. Zhou J, Zhu J, Jiang L, Zhang B, Zhu D, Wu Y. Interleukin 18 promotes myofibroblast activation of valvular interstitial cells. Int J Cardiol. 2016;221:998–1003. doi:10.1016/j.ijcard.2016.07.036
107. Xu R, Zhu D, Guo J, Wang C. IL-18 promotes erythrophagocytosis and erythrocyte degradation by m1 macrophages in a calcific microenvironment. Can J Cardiol. 2021;37(9):1460–1471. doi:10.1016/j.cjca.2021.04.007
108. He Y-B, Guo J-H, Wang C, Zhu D, Lu L-M. IL-33 promotes the progression of nonrheumatic aortic valve stenosis via inducing differential phenotypic transition in valvular interstitial cells. J Cardiol. 2020;75(2):124–133. doi:10.1016/j.jjcc.2019.06.011
109. Garcia-Pena A, Ibarrola J, Navarro A, et al. Activation of the Interleukin-33/ST2 pathway exerts deleterious effects in myxomatous mitral valve disease. Int J Mol Sci. 2021;22(5):2310. doi:10.3390/ijms22052310
110. Kapelouzou A, Kontogiannis C, Tsilimigras DI, et al. Differential expression patterns of Toll Like Receptors and Interleukin-37 between calcific aortic and mitral valve cusps in humans. Cytokine. 2019;116:150–160. doi:10.1016/j.cyto.2019.01.009
111. Zeng Q, Song R, Fullerton DA, et al. Interleukin-37 suppresses the osteogenic responses of human aortic valve interstitial cells in vitro and alleviates valve lesions in mice. Proc Natl Acad Sci U S A. 2017;114(7):1631–1636. doi:10.1073/pnas.1619667114
112. Lu H, Rateri DL, Bruemmer D, Cassis LA, Daugherty A. Involvement of the renin–angiotensin system in abdominal and thoracic aortic aneurysms. Clin Sci. 2012;123(9):531–543. doi:10.1042/cs20120097
113. Anagnostakos J, Lal BK. Abdominal aortic aneurysms. Prog Cardiovasc Dis. 2021;65:34–43. doi:10.1016/j.pcad.2021.03.009
114. Suehiro C, Suzuki J, Hamaguchi M, et al. Deletion of interleukin-18 attenuates abdominal aortic aneurysm formation. Atherosclerosis. 2019;289:14–20. doi:10.1016/j.atherosclerosis.2019.08.003
115. Eckstein -H-H, Maegdefessel L. Linking obesity with abdominal aortic aneurysm development. Eur Heart J. 2020;41(26):2469–2471. doi:10.1093/eurheartj/ehz882
116. Zhang Z-B, Ruan -C-C, Lin J-R, et al. Perivascular adipose Tissue–Derived PDGF-D contributes to aortic aneurysm formation during obesity. Diabetes. 2018;67(8):1549–1560. doi:10.2337/db18-0098
117. Liu C-L, Ren J, Wang Y, et al. Adipocytes promote interleukin-18 binding to its receptors during abdominal aortic aneurysm formation in mice. Eur Heart J. 2020;41(26):2456–2468. doi:10.1093/eurheartj/ehz856
118. Li J, Xia N, Wen S, et al. IL (Interleukin)-33 suppresses abdominal aortic aneurysm by enhancing regulatory T-cell expansion and activity. Arterioscler Thromb Vasc Biol. 2019;39(3):446–458. doi:10.1161/atvbaha.118.312023
119. Koushki K, Shahbaz SK, Mashayekhi K, et al. Anti-inflammatory action of statins in cardiovascular disease: the role of inflammasome and toll-like receptor pathways. Clin Rev Allergy Immunol. 2021;60(2):175–199. doi:10.1007/s12016-020-08791-9
120. Zhao J, Wang Z, Yuan Z, Lv S, Su Q. Baicalin ameliorates atherosclerosis by inhibiting NLRP3 inflammasome in apolipoprotein E-deficient mice. Diab Vasc Dis Res. 2020;17(6):1479164120977441. doi:10.1177/1479164120977441
121. Ni X-Q, Hu Z-Y. Remifentanil improves myocardial ischemia-reperfusion injury in rats through inhibiting IL-18 signaling pathway. Eur Rev Med Pharmacol Sci. 2020;24(7):3915–3922. doi:10.26355/eurrev_202004_20858
122. Vanden Berghe T, Demon D, Bogaert P, et al. Simultaneous targeting of IL-1 and IL-18 is required for protection against inflammatory and septic shock. Am J Respir Crit Care Med. 2014;189(3):282–291. doi:10.1164/rccm.201308-1535OC
123. Zhang Z, Zou J, Shi Z, et al. IL-22–induced cell extrusion and IL-18–induced cell death prevent and cure rotavirus infection. Sci Immunol. 2020;5(52):eabd2876. doi:10.1126/sciimmunol.abd2876
124. Abston ED, Barin JG, Cihakova D, et al. IL-33 independently induces eosinophilic pericarditis and cardiac dilation: ST2 improves cardiac function. Circ Heart Fail. 2012;5(3):366–375. doi:10.1161/circheartfailure.111.963769
125. Kim RY, Oliver BG, Wark PAB, Hansbro PM, Donovan C. COPD exacerbations: targeting IL-33 as a new therapy. Lancet Respir Med. 2021;9(11):1213–1214. doi:10.1016/s2213-2600(21)00182-x
126. Wechsler ME, Ruddy MK, Pavord ID, et al. Efficacy and safety of itepekimab in patients with moderate-to-severe asthma. N Engl J Med. 2021;385(18):1656–1668. doi:10.1056/NEJMoa2024257
127. Stremska ME, Jose S, Sabapathy V, et al. IL233, A novel IL-2 and IL-33 hybrid cytokine, ameliorates renal injury. J Am Soc Nephrol. 2017;28(9):2681–2693. doi:10.1681/asn.2016121272
128. Aoyagi T, Newstead MW, Zeng X, et al. Interleukin-36γ and IL-36 receptor signaling mediate impaired host immunity and lung injury in cytotoxic Pseudomonas aeruginosa pulmonary infection: role of prostaglandin E2. PLoS Pathog. 2017;13(11):e1006737. doi:10.1371/journal.ppat.1006737
129. Zhu J, Xu Y, Li Z, Liu S, Fu W, Wei Y. Interleukin-36β exacerbates DSS-induce acute colitis via inhibiting Foxp3+ regulatory T cell response and increasing Th2 cell response. Int Immunopharmacol. 2022;108:108762. doi:10.1016/j.intimp.2022.108762
130. Xu J, Chen J, Li W, et al. Additive therapeutic effects of mesenchymal stem cells and IL-37 for systemic lupus erythematosus. J Am Soc Nephrol. 2020;31(1):54–65. doi:10.1681/asn.2019050545
131. Wu Z, Mehrabi Nasab E, Arora P, Athari SS. Study effect of probiotics and prebiotics on treatment of OVA-LPS-induced of allergic asthma inflammation and pneumonia by regulating the TLR4/NF-kB signaling pathway. J Transl Med. 2022;20(1):130. doi:10.1186/s12967-022-03337-3
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.