")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Progress in Redirecting Antiparasitic Drugs for Cancer Treatment
Authors Huang H, He Q, Guo B , Xu X, Wu Y, Li X
Received 1 March 2021
Accepted for publication 11 May 2021
Published 22 June 2021 Volume 2021:15 Pages 2747—2767
DOI https://doi.org/10.2147/DDDT.S308973
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Haoyang Huang,1 Qing He,2– 5 Binghua Guo,6 Xudong Xu,7 Yinjuan Wu,2– 5 Xuerong Li2– 5
1Department of Clinical Medicine, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, Guangdong, 510080, People’s Republic of China; 2Department of Parasitology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, Guangdong, 510080, People’s Republic of China; 3Key Laboratory for Tropical Diseases Control of Ministry of Education, Sun Yat-sen University, Guangzhou, Guangdong, 510080, People’s Republic of China; 4Provincial Engineering Technology Research Center for Biological Vector Control, Guangzhou, Guangdong, 510080, People’s Republic of China; 5CAEA Center of Excellence on Nuclear Technology Applications for Insect Control, Beijing, 100048, People’s Republic of China; 6School of Ophthalmology and Optometry, Wenzhou Medical University, Wenzhou, Zhejiang, 325035, People’s Republic of China; 7Department of Clinical Medicine, School of Medicine, Sun Yat-sen University, Guangzhou, Guangdong, 510080, People’s Republic of China
Correspondence: Xuerong Li
Department of Parasitology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, Guangdong, 510080, People’s Republic of China
Tel +86 20 87331918
Fax +86 20 87331918
Email [email protected]
Abstract: Drug repurposing is a feasible strategy in developing novel medications. Regarding the cancer field, scientists are continuously making efforts to redirect conventional drugs into cancer treatment. This approach aims at exploring new applications in the existing agents. Antiparasitic medications, including artemisinin derivatives (ARTs), quinine-related compounds, niclosamide, ivermectin, albendazole derivatives, nitazoxanide and pyrimethamine, have been deeply investigated and widely applied in treating various parasitic diseases for a long time. Generally, their pharmacokinetic and pharmacodynamic properties are well understood, while the side effects are roughly acceptable. Scientists noticed that some of these agents have anticancer potentials and explored the underlying mechanisms to achieve drug repurposing. Recent studies show that these agents inhibit cancer progression via multiple interesting ways, inducing ferroptosis induction, autophagy regulation, mitochondrial disturbance, immunoregulation, and metabolic disruption. In this review, we summarize the recent advancement in uncovering antiparasitic drugs’ anticancer properties from the perspective of their pharmacological targets. Instead of paying attention to the previously discovered mechanisms, we focus more on newly emerging ones that are worth noticing. While most investigations are focusing on the mechanisms of their antiparasitic effect, more in vivo exploration in clinical trials in the future is necessary. Moreover, we also paid attention to what limits the clinical application of these agents. For some of these agents like ARTs and niclosamide, drug modification, novel delivery system invention, or drug combination are strongly recommended for future exploration.
Keywords: ferroptosis, autophagy, mitochondria, immunoregulation, glycolysis
Introduction
Cancer has become one of the biggest threats among all sorts of health concerns.1 According to the Global Burden of Disease Study 2017, cancer-related deaths have increased by 25.4% over the past decade.1 As one of the three main cancer treatments, chemotherapy is the major choice for most types of cancers. While scientists have made progress in reducing the death rates using these agents, existing traditional chemoagents generally have some side effects like nephrotoxicity or myelosuppression, which may limit their application in clinical practice.2 Moreover, some cancers may develop resistance to these agents during the treatment, profoundly reducing the therapeutic effect.3 In this way, it is of great significance to include more effective chemo agents that exhibit antitumor properties in current cancer treatment. Novel medication invention is one of the main ways to develop new drugs. However, such an approach is relatively costly and time-consuming under the current drug designed and development model, based on the fact that it would take more than ten years on average before an invented drug is applied clinically, regardless of the low overall success rate.4 Comparably, drug repurposing is more efficient and money-saving for for the corporations, government and the patients, as such a strategy may help reduce the cost of drug invention, finally reduce the price of the medication. With more available preclinical experiments, clinical trials, and practice, it would take less time and lower cost to apply an approved drug in treating cancer.5
Antiparasitic drugs are a group of chemo agents that act against infections caused by parasites, including artemisinin derivatives (ARTs), quinine-related compounds, niclosamide, ivermectin, albendazole derivatives, nitazoxanide and pyrimethamine (Figure 1). These agents exhibit various modes of effect on parasites. Most of the FDA-approved antiparasitic agents have been applied clinically for a long time, and their pharmacokinetic and pharmacodynamic properties have been explored thoroughly. Moreover, their toxicity on normal human cells and possible adverse effects are well understood and mostly acceptable. In this way, scientists thought of the possibility of redirecting antiparasitic agents for treating cancers.
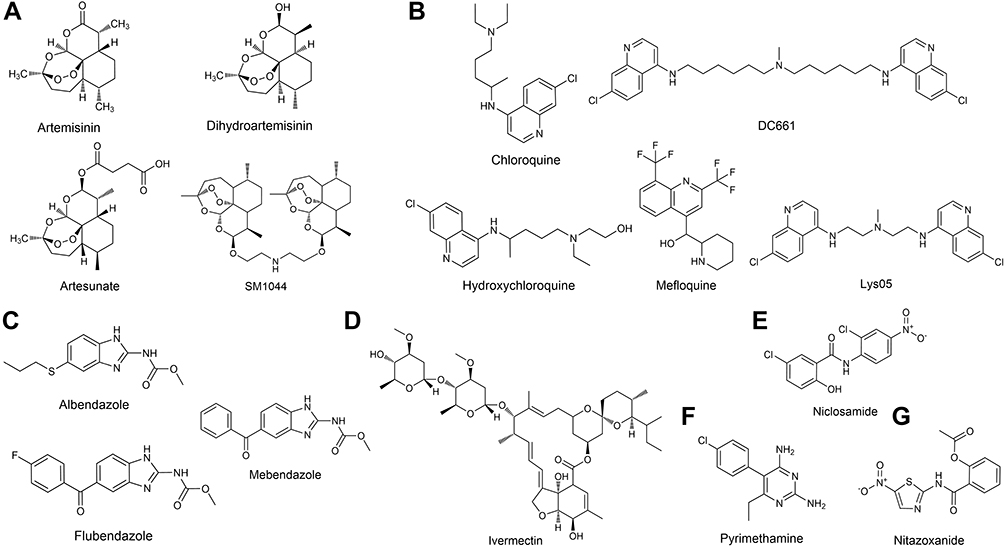 |
Figure 1 Chemical structure of the antiparasitic agents with anticancer properties: Artemisinin derivatives (A); Quinine related compounds (B); Albendazole derivatives (C); Ivermectin (D); Niclosamide (E); Pyrimethamine (F) and Nitazoxanide (G). |
Inspiringly, some of these medications do exhibit antitumor properties. For some of these medications, the mechanism of how they kill cancer cells is similar to how they kill parasites. Moreover, researchers kept exploring and successfully uncovered several novel antitumor mechanisms that target other molecules and biological procedures that have not been discovered before. Multiple phenotypes and metabolisms are involved. Ferroptosis has been regarded as a new approach to induce cancer cell death, and interestingly, ARTs target the process to exert antitumor properties.6 Autophagy acts as a “Double-Edged Sword” in cancer cell metabolism, and its regulation is closely related to cancer cell death.7 Mitochondria is the cancer cells’ energy factory, and its dysfunction contributes a lot to cancer cell death.8 Aerobic glycolysis, known as the “Warburg Effect”, is an essential metabolic pathway and a therapeutic target for cancer cells.9 The tumor microenvironment is attracting scientists’ attention recently, and some of these antiparasitic medications exert immunoregulatory effects, which contribute to their antitumor properties.10 Several signal pathways are also involved in the anticancer potential of the antiparasitic agents. Altogether, these newly discovered anticancer mechanisms profoundly bettered our understanding of these drugs.
In this review, we summarized and discussed the recent progress in redirecting antiparasitic drugs for cancer treatment, specifically from a view of the underlying mechanisms.
Ferroptosis Inducing Effect of Antiparasitic Agents
Scientists have explored the mechanism of how ART and its derivatives kill Plasmodium before. Heme and iron activate ARTs in vivo, and the cleavage of the endoperoxide bridge produces intracellular ROS, which is critical for its antimalarial effect.11,12 Although uncertainty and controversy still exist in its exact antimalarial mechanism, scientists believe that both iron and ROS play an essential role in its antiparasitic effect.
Interestingly, the antitumor effect of ARTs shares at least to some degree a similar mechanism with its antiparasitic effect (Table 1). In the early 21st century, scientists observed that ARTs’ anticancer effect correlates with the existence of iron and ROS production.13,14 However, the big picture of how these two factors exert their tumor inhibitory function remains fuzzy for a long time. In 2012, Dixon et al firstly define the concept of “Ferroptosis” as “Iron-dependent nonapoptotic cell death”, which dramatically deepens understanding of iron-related cell death.15 Generally, ferroptosis refers to an iron-dependent and reactive oxygen species (ROS)-producing cell death procedure. Later, researchers recognized that ferroptosis induction is exactly how ARTs kill cancer cells.16–19
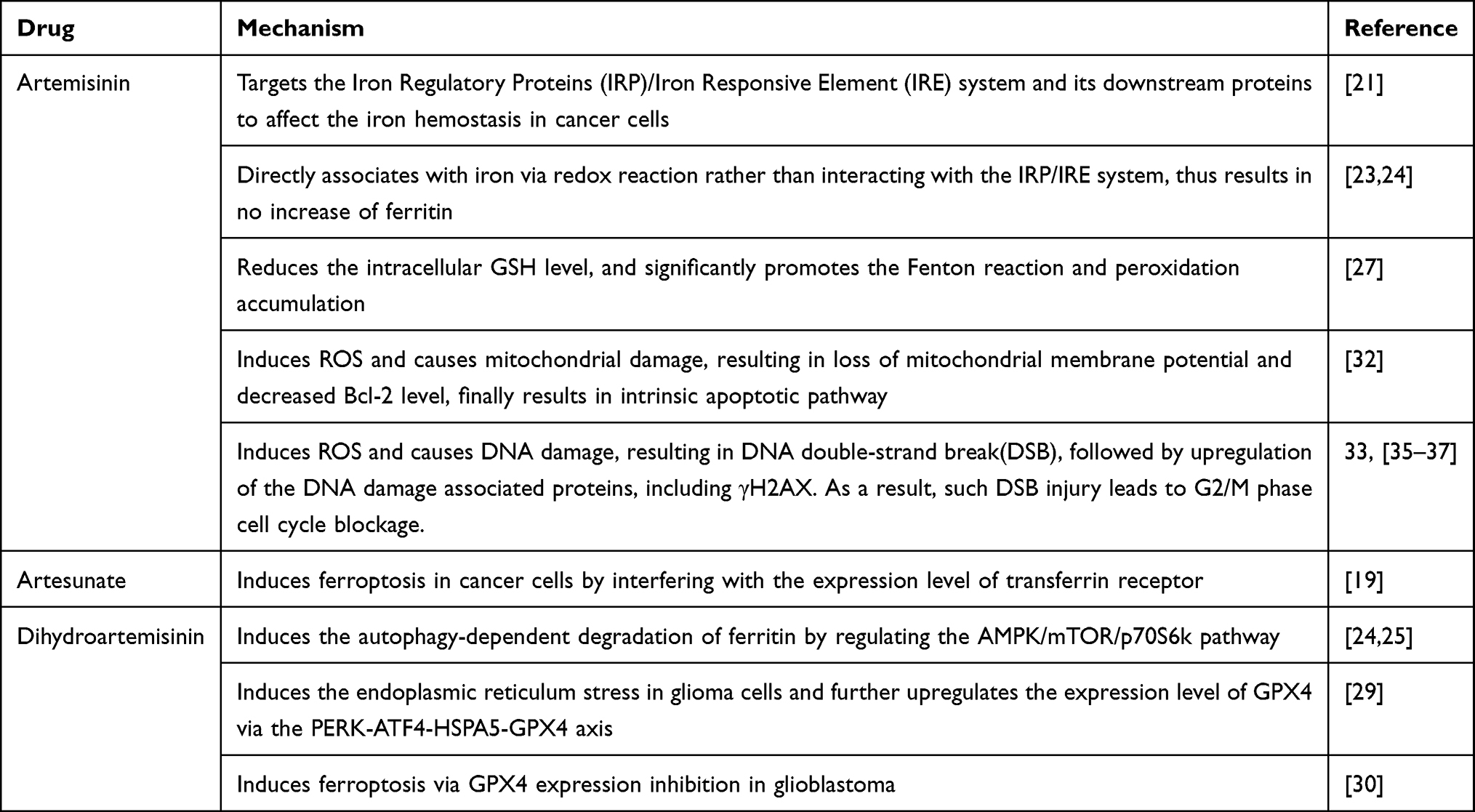 |
Table 1 Ferroptosis Inducing Effect of Antiparasitic Agents |
ARTs function as multi-target ferroptosis inducers in cancer cells (Figure 2). One of the mechanisms is by stepping on the “accelerator” of the ferroptosis procedure. The transferrin receptor is the source of intracellular iron and thus is the upstream component in the ferroptosis pathway. Artesunate (ARS) and many other ARTs upregulate the expression level of the transferrin receptor, thus connecting the ferroptosis with ARS-induced cancer cell inhibition.19,20 The IRP/IRE (Iron Regulatory Proteins/Iron Responsive Element) system is probably the most critical regulator maintaining the intracellular iron hemostasis.21 It regulates the expression of a series of iron concentration-regulating proteins at the post-transcription level.20 ARTs target this system and its downstream proteins to disturb the iron hemostasis in cancer cells. In normal situations, the IRP recognizes the free iron and reduces its binding capacity with IRE.20 This results in upregulation of the ferritin expression. As the iron storage protein, ferritin helps maintain the normal intracellular iron concentration.22 ARTs can directly associate with iron via Redox reaction.23 However, Chen’s study revealed that these associated irons fail to interact with the IRP/IRE system, resulting in no increase of ferritin.24 However, the ART-associated irons’ superoxidative reaction-catalyzing ability remains similar to free irons. In this way, free irons escape from ferritin’s regulation and accumulate in the cell while preserving its ability to promote ferroptosis.24 Interestingly, scientists found another pathway that ARTs can influence ferritin level. Dihydroartisinin (DHA) induces autophagy-dependent ferritin degradation by regulating the AMPK/mTOR/p70S6k pathway. Again, this results in increased intracellular iron concentration.24,25
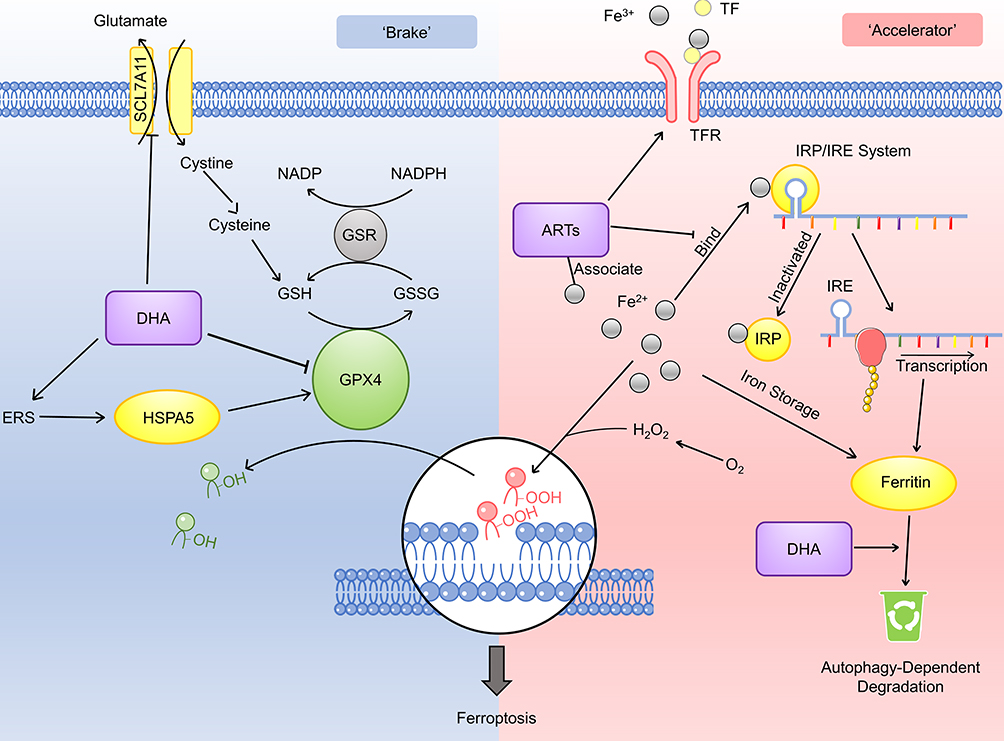 |
Figure 2 Mechanism of the ferroptosis regulating effect of the antiparasitic agents. The inhibitory regulation of ferroptosis (the “brake”) is shown on the left side and the promotive regulation (the “accelerator”) is shown on the right side. DHA downregulates the PRIM2/SLC7A11 axis and further suppresses the GSH production. DHA also downregulates the GPX4 expression and releases the brake of ferroptosis. However, there is also evidence suggesting that DHA may upregulate GPX4 via upregulating the endoplasmic reticulum stress (ERS) and the HSPA5 expression. ARTs upregulate the expression of TFR, which would subsequently increase the intracellular iron level. ARTs associate with Fe2+ and prevent its combination with the IRP/IRE system, thus inhibit the expression level of ferritin and increase the intracellular iron level. DHA can also promote the degradation of ferritin. |
ARTs may also regulate the negative-regulatory factors (the “brake”) of ferroptosis. As an anti-oxidative substance, glutathione (GSH) reduces the oxidative injury caused by ROS, therefore reduces the injury caused by ferroptosis.26 However, the administration of the artesunate or DHA reduces the intracellular GSH level.27,28 Further exploration suggests that DHA downregulates the PRIM2/SLC7A11 axis.28 As a key cysteine concentration regulator, SLC7A11 regulates the GSH biosynthesis. In this way, DHA suppresses the intracellular GSH level. In a common ferroptosis pathway, glutathione peroxidase 4 (GPX4) functions as an inhibitory factor via facilitating the transition from GSH to oxidized glutathione (GS-SG).16 Scientists have demonstrated that DHA induces ferroptosis via downregulating GPX4 expression.29 In Chen’s study, however, they showed the other aspect of DHA’s effect on GPX4 level. Their investigation demonstrated that DHA exerts endoplasmic reticulum stress in glioma cells. Subsequently, HSPA5 (heat shock protein family A (Hsp70) member 5) is upregulated, further increasing GPX4 expression.30 In this way, the cells neutralized the DHA-induced ROS stress and escaped from ferroptosis. Not surprisingly, inhibition on the PERK/ATF4/HSPA5 pathway further strengthens the ferroptosis-inducing effect of DHA.29 To further release the brake of the ferroptosis pathway, Chen et al combined DHA with a potent GPX4 inhibitor RSL3.24 Such a combination synergistically promotes the ferroptosis procedure and strengthens the inhibitory effect on various cancer cell lines. In the future, animal experiments would be needed to evaluate the anticancer property of such combination in vivo. At the same time, it would be meaningful to try combining DHA with other GPX4 inhibitors, such as ML-120 or ML-162, in treating cancer cells. Taken together, we can summarize that ARTs are multi-target ferroptosis promoters, both targeting the “accelerator” and the “brake” in this procedure.
The correlation between ferroptosis and cancer immunity regulation is attracting scientists’ attention.38 On one hand, cancer cells that undergo ferroptosis may release signals like may AA (Arachidonic acid) metabolites that affect immune cells and regulate local immune reaction. Notably, such ferroptosis acts like a double-edged sword, meaning that it can be immunosuppressive or immunopromotive depending on the signal that a ferroptotic cell secretes.38 On the other hand, ferroptosis also happens in immune cells (eg macrophage-1/2) under pharmaceutical induction, and will interfere with their function and anticancer properties.38,39 Regarding the ARTs’ research field, there are investigations focusing on how ARTs affect cancer immunity. However, there are currently no available papers focusing on whether their immunoregulating effect correlates with ferroptosis. In the future, it would be interesting to study how ART-induced ferroptosis influences the tumor microenvironment, will it cause immunosuppression or immunopromotion? Likewise, will immune cells undergo ferroptosis under the treatment of ARTs and what would be the downstream effects?
Autophagy Regulating Effect of Antiparasitic Agents
Autophagy functions as a double-edged sword in cancer cells and increasingly attracts scientists’ attention since its discovery.40,41 It refers to a highly conservative process that degrades cytoplasmic components to survive critical stress.42 Collectively, there are mainly five steps in the autophagic process: (i) initiation; (ii) Cargo initiation; (iii) Autophagosome maturation; (iv) Fusion between autophagosome and lysosome, (v) Formation of autolysosome and nutrient recycling.42 We observed an interesting phenomenon that most antiparasitic agents target autophagy procedure, either as inducer or inhibitor, and contribute to their anticancer property (Table 2). In this way, we will discuss the autophagy-regulating effect of the antiparasitic agents in detail (Figure 3).
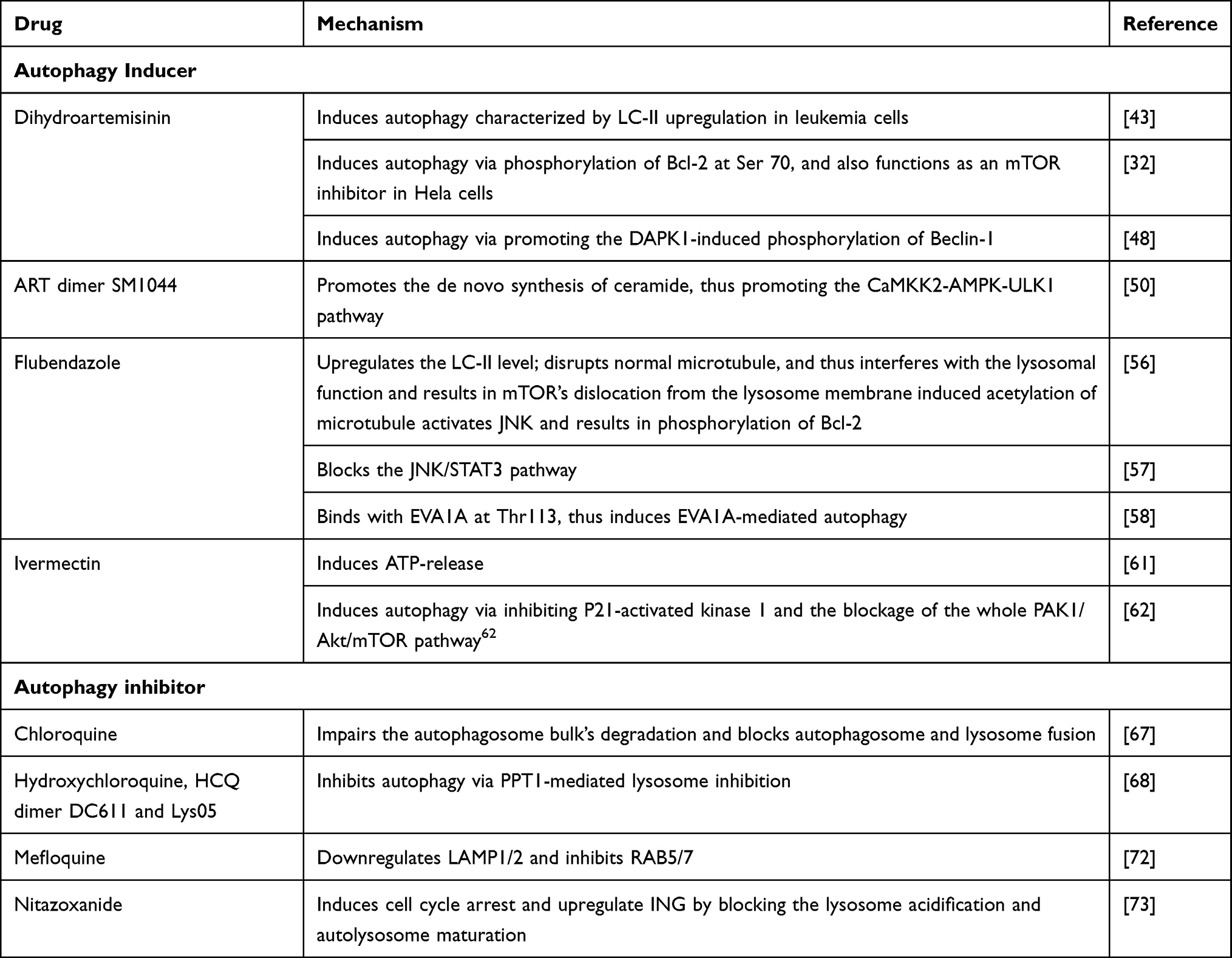 |
Table 2 Autophagy Regulating Effect of Antiparasitic Agents |
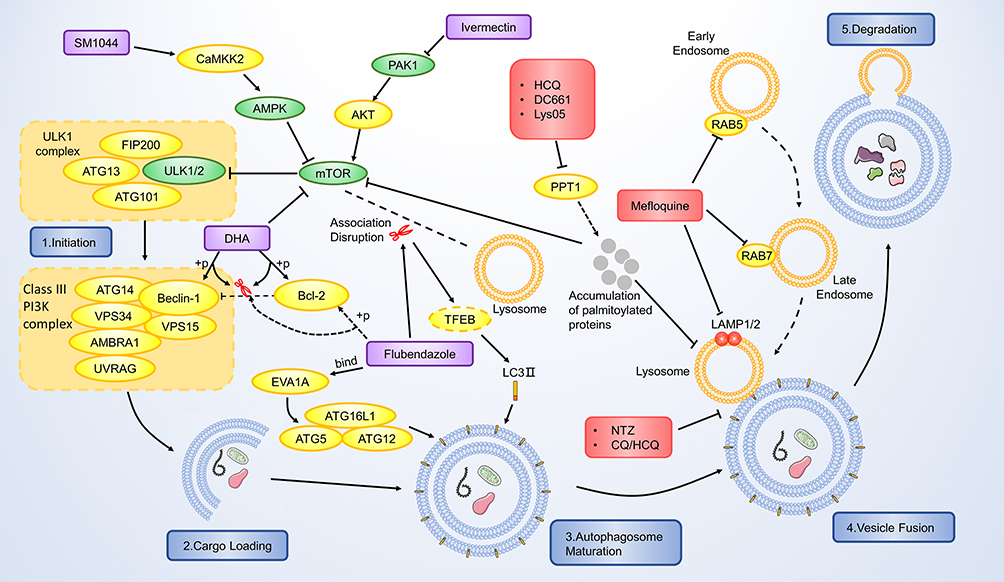 |
Figure 3 Mechanism of the autophagy regulating effect of the antiparasitic agents. The autophagy-promotive agents are shown in purple boxes and autophagy-inhibitory agents are shown in red boxes. mTOR is one of the major inhibitory elements that targets the initiation of the autophagic procedure. SM1044, DHA and ivermectin inhibit mTOR and subsequently activate the autophagic process. Flubendazole disassociates mTOR from the lysosome, leading to nuclear translocation of TFEB and finally upregulates LC3. It can also phosphorylate Bcl-2 via JNK-1 activation and release its inhibition on Beclin-1, which is part of the Class III PI3K complex. Flubendazole also binds with EVA1A and further induces EVA1A mediated autophagy. HCQ, DC661 and Lys05 inhibit the PPT1 and result in accumulation of palmitoylated proteins, which would concurrently inhibit lysosome and mTOR. Nitazoxanide and CQ/HCQ inhibit the fusion between autophagosome and lysosome. Mefloquine targets RAB5/7 and LAMP1/2 to inhibit the lysosome maturation. |
Antiparasitic Agents as Autophagy Inducer
Wang et al were the first to link the ARTs’ antitumor effect with autophagy. In 2012, they found that DHA induces autophagy characterized by microtubule-associated protein light chain 3-II (LC3-II) upregulation in leukemia cells.43 From then on, research focusing on this field started to show up, and the past several years witnessed a significant advancement in understanding ARTs-related autophagy. The autophagy induction effect was validated in various cancers, including breast cancer, colorectal cancer, ovarian cancer, leukemia, and glioma.44 Scientists revealed multiple mechanisms involved in ARTs-induced autophagy, in which the Beclin-1/Bcl-2 complex is one of their main targets. The importance of Bcl-2 and Beclin-1 in autophagy has been demonstrated clearly.45,46 Beclin-1 plays a central role in autophagy as it regulates the formation and maturation of autophagosomes. Moreover, the dissociation of Beclin-1 from Bcl-2 is critical for its biological activity. Wang et al revealed that DHA induces autophagy via phosphorylation of Bcl-2 at Ser 70.32 According to the previous report, phosphorylation of Bcl-2 at this site would dissociate Beclin-1, thus triggering the autophagic pathway.47 Similarly, in cholangiocarcinoma, DHA was reported to induce autophagy via promoting the Death Associated Protein Kinase 1(DAPK1)-induced phosphorylation of Beclin-1, again detaches Beclin-1 from Beclin-1/Bcl-2 complex.48 ARTs also regulate some autophagy-related pathways. The mammalian target of the rapamycin (mTOR) pathway plays an inhibitory role in autophagy, and targeting the mTOR pathway to induce autophagy has become a cancer treatment therapy.49 Wang et al reported that DHA functions as an mTOR inhibitor in Hela cells, thus inducing autophagy.32 Interestingly, a novel ART derivative, SM1044, also shows the autophagy-inducing property in a different manner.50 It promotes the de novo synthesis of ceramide, promoting the CaMKK2-AMPK-ULK1 pathway, and finally induces autophagy.
Autophagy induced by ARTs also correlates with the other pharmaceutical effects. The relationship between autophagy and ferroptosis is worth discussing. On the one hand, Du’s study suggested that DHA-induced autophagy accelerates the degradation of ferritin, and autophagy related 7 (ATG7)-knockdown abolishes cancer cells from DHA-induced cell death.25 Further study revealed that DHA induces the lysosomal degradation of ferritin, as we have discussed before. On the other hand, Chen et al demonstrated that at low concentrations, DHA-induced ferroptosis is independent of autophagy.24 Moreover, although knockdown of autophagy related 5 (ATG5) delayed DHA-induced ferroptosis, DHA can still sensitize cancer cells to erastin-induced ferroptosis in such condition, suggesting the DHA can induce autophagy in an autophagy-independent manner.24 Collectively, these data suggest that DHA-induced autophagy contributes to the process of ferroptosis. However, the autophagy process is probably unnecessary for DHA-induced ferroptosis, as several autophagy-independent pathways result in ferroptosis. Autophagy also correlates with ARTs-induced cell cycle arrest. Previous studies revealed that DHA/ARS-induced cell cycle arrest is dependent on autophagy, and inhibition of autophagy attenuates such cell cycle blockage.51,52 Interestingly, DHA downregulates the telomere shelterin component TBP-related factor 2(TRF2) via autophagy-mediated degradation, thus impairing the normal cell cycle.51 Regarding apoptosis, the autophagy process seems to play a protective role in ART-induced apoptosis. Jiang et al reported that autophagy inhibition augments ARS-induced apoptosis.53 Similarly, Ganguli’s study suggests that autophagy inhibition showed a synergistic effect with ART by promoting apoptosis.54 It is conceivable that ARTs emerge as stress for cancer cells, as it may induce apoptosis. Autophagy may help cancer cells survive those critical situations, thus exhibiting a cytoprotective effect. Taken together, as a “Double-edged Sword”, ARTs-induced autophagy mainly functions as a cancer killer. It will significantly help scientists better understand the pharmaceutical effect of ARTs if more investigations focus on how autophagy cross-talk with the other mechanisms of ARTs.
Recent years witnessed a considerable breakthrough in understanding autophagy-related mechanisms of benzimidazole derivatives. In 2015, Zhang et al screened out flubendazole as an autophagy-related 4B (Atg4B) docking molecule via the in-silico analysis, suggesting this agent’s potential target.55 Almost simultaneously, Chauhan et al identified flubendazole as an autophagy inducer through experiments.56 It significantly upregulates the LC3-II level, suggesting that it augments the autophagic process.56 As flubendazole disrupts normal microtubule, it interferes with the lysosomal function and results in mTOR’s dislocation from the lysosome membrane.56 The downregulation of the mTOR pathway leads to the dephosphorylation and nuclear translocation of the transcription factor EB (TFEB), which is a critical transcript factor in lysosomal biogenesis and autophagy. Moreover, flubendazole-induced acetylation of microtubule activates JNK and results in phosphorylation of Bcl-2.56 As a result, Beclin-1 is disassociated from Bcl-2 and again induces autophagy. Lin et al further revealed that flubendazole also blocks the JNK/STAT3 pathway, which is the upstream negatively regulating pathway of the autophagic process, resulting in autophagy induction.57 Zhen et al revealed that flubendazole exerts autophagy inducing effect in an EVA1A-related manner.58 EVA1A has been reported to induce autophagosome formation via interacting with ATG16L1.59 Their analysis revealed that flubendazole probably binds with EVA1A at Thr113, thus induces EVA1A-mediated autophagy. While most studies suggest that flubendazole/mebendazole generated autophagy function as an anticancer mechanism in cancer cells, controversy still exists. Sung et al demonstrated that while mebendazole functions as an antiangiogenic agent via inhibiting the proliferation of endothelial cells, mebendazole-induced autophagy is observed, and inhibiting such autophagy enhances its anti-proliferative effect.60 In this way, mefloquine-induced autophagy seems to be cytoprotective. It would be interesting to validate the overall effect of mebendazole/flubendazole-induced autophagy on both carcinogenesis and angiogenesis in a mouse model, thus concluding whether it is a beneficial mechanism or potential therapeutic target.
Scientists discovered the significance of autophagy in the ivermectin-induced anticancer effect in 2015. Draganov et al demonstrated that ivermectin induces ATP-release and further induces autophagy in breast cancer cells.61 Later, Dou et al revealed that ivermectin inhibits cancer cell proliferation in an autophagy-mediated manner.62 Such ivermectin-induced autophagy correlates with inhibition of P21-activated kinase 1 and the whole PAK1/Akt/mTOR pathway blockage.62 Similarly, Liu et al demonstrated a similar mechanism in glioma cells.63 Mitophagy, an emerging concept, refers to selective autophagic degradation of mitochondria.64 Zachari et al reported that ivermectin induces mitophagy in embryonic fibroblasts.65 It would be interesting to validate whether ivermectin induces such mitophagy in cancer cells in future exploration.
Antiparasitic Agents as Autophagy Inhibitor
While autophagy induction promotes cancer cell death, inhibiting autophagy may also contribute to anticancer property. The autophagy inhibitory effect of chloroquine (CQ) and its derivatives has attracted scientists’ interest for a long time. While more and more studies focused on its application in various cancer types, the detailed mechanism of how CQs induces autophagy remained insufficiently understood. The past several years witnessed several inspiring progress in understanding the underlying mechanisms. It has been known that CQ, HCQ and Lys05 mainly target autophagy by blocking autophagosome and lysosome fusion.66 Mauthe et al revealed that CQ induces autophagy inhibition differently from BafA1, a commonly used autophagy inhibitor.67 It impairs the autophagosome bulk’s degradation, while the lysosomal acidity is not interfered with.67 Interestingly, Rebecca et al demonstrated a different autophagy-inducing mechanism of CQ derivatives.68,69 They identified palmitoyl-protein thioesterase 1 (PPT1) as a common target of CQ derivatives including hydroxychloroquine (HCQ), DC661, and Lys05.68,69 PPT1 functions as a thioester bond breaker of the palmitoylated proteins. It promotes tumor proliferation growth, and targeting PPT1 inhibits tumor progression. Their studies revealed that inhibition on PPT1 leads to accumulation of palmitoylated proteins, which would disrupt the lysosome catabolism. Inspiringly, their investigations revealed that PPT1 inhibition also results in direct inhibition of mTORC1, which is similar to the pharmaceutical mechanism of the autophagy inducers. While the upstream of autophagy is activated, the downstream events (lysosome function) is totally blocked. In this way, PPT1 inhibitors would harvest the anticancer effect of both autophagy inhibitors and autophagy inducers. To some degree, this is similar to co-administration of the autophagy inducer and autophagy inhibitor, which have shown synergistic anticancer effect, however with one single agent.70 Comparing the final anticancer effect between PPT1 inhibition and coadministration of autophagy inducer and inhibitor would be interesting. Current publications are still insufficient to evaluate the other downstream effects of PPT1 inhibition, and how cancer cells would response to such concurrent inhibition of autophagy and mTORC1 remains fuzzy. It is worthy to pay more attention on PPT1 inhibitors and to see if it could be applied in clinical practice. Mefloquine (MQ), another CQ derivative, was also found to induce mitochondrial autophagy in esophageal squamous carcinoma cells.71 Takeda investigated deeper and revealed that MQ inhibits the expression of Lysosomal Associated Membrane Protein 1/2 (LAMP1/2), the lysosome markers that are essential for the normal function of the lysosome and the autophagy process.72 Further investigation revealed that it targets the lysosomal function via inhibiting RAB5/7 expression, which is critical in lysosome formation.72
Nitazoxanide (NTZ) is another worth noticing autophagy inhibitor. Wang et al revealed that NTZ induces cell cycle arrest by upregulating ING (Inhibitor of Growth 1) in glioblastoma, which is mediated by inhibiting its autophagic degradation.73 Further exploration demonstrated that NTZ blocks the lysosome acidification and autolysosome maturation, thus reducing the late-stage autophagic flux. In the future, validating such autophagy inhibitory effect of NTZ in other cancer cells and exploring the other downstream effect of such autophagic inhibition would be of great significance.
Generally, the autophagy inhibitory effect has two main significance. First, based on the fact that autophagy can help cancer cells suffer intolerant situations, autophagy inhibition decreases cancer cells.74,75 Once the cancer cells fail to perform the autophagy procedure fully, apoptosis is often triggered and causing cell death.74,75 Second, autophagy inhibition profoundly decreases cancer cells’ surviving ability under other anticancer agents’ treatment, thus enhancing their pharmacologic effect. In recent years, scientists moved on to investigate more downstream effects of autophagy inhibition. While most researches focused on the direct impact of CQ-induced autophagy on cancer cells, New et al paid more attention to its impact on cancer-associated fibroblasts (CAF).76 They observed a relatively high level of basal autophagy in CAF cells and demonstrated that the autophagy-dependent secretion of CAF promotes the progression of HNSCC. Targeting CAF autophagy with CQ inhibits proliferation, migration, and invasion of cancer cells, suggesting its therapeutic effect. Interestingly, in another study, Zhao et al revealed that CAF might promote cancer cell stemness via inducing autophagy.77 In this way, CQ may inhibit the CAF-induced stemness via inhibiting CAF-induced autophagy.
Application of Autophagy Regulators: Future Directions
Autophagy functions as a double-edged sword in cancer cells.78,79 On one hand, mutation-induced autophagy reduction helps cancer cells accumulate ROS and abnormal organelles, contributing to malignant transformation during tumorigenesis.74 On the other hand, strengthened autophagy help cancer cells survive under stress condition (eg hypoxia or medication) in the later stage of cancer progression.74 Moreover, while autophagy enhances tumor immunity from some aspects (eg autophagy enhances MHCI/II process and presentation, which are critical in malignancy recognition), it attenuates the immune response from other aspects (eg autophagy degrades NK-derived granzyme B so that NK-mediated cell death is attenuated).80 In this way, the proper application of autophagy regulators would be of great importance.
There have been many preclinical studies and clinical trials focusing on autophagy inhibitors (eg CQ and HCQ) in the cancer field (discussed later in section 3). In most of these trials, autophagy inhibitors are applied combined with other medications to reduce chemo-resistance. This is a good strategy but not all we can do. Although both autophagy enhancement and inhibition may bring benefits to cancer reduction, it should be highlighted that a basal level autophagy flux is essential for cancer cell survival.81 From this aspect, a complete inhibition on autophagy may be a practical approach to kill cancer cells, regardless of what role autophagy is functioning. Combining CQ, HCQ, or MQ with other autophagy inhibitors like GSK690693 or Autophinib may be a potent combination in cancer inhibition. Regarding autophagy inducers, we believe that future exploration should focus more on its cancer immunity regulatory effect. Although autophagy is considered to have a complicated effect on immune response, it improves the effectiveness of antigen-presenting cell (APC) and cytotoxic T lymphocytes (CTL), which are the major players in cancer immunity. Similarly, although autophagy upregulates many cytokines in the tumor microenvironment, the majority are immunoreactive ones.80 In this way, it is worth trying the combination of autophagy inducers with immunotherapies, like CAR-T (chimeric antigen receptor T-cell) therapy or other adoptive cellular immunotherapies.
Mitochondria Disrupting Effect of Antiparasitic Agents
Mitochondrial damage is another worth noting mechanism involved in the antiparasitic agents’ anticancer property (Table 3). Generally, targeting mitochondria may lead to two results. First, direct injury on mitochondria will trigger the intrinsic apoptosis pathway, leading to cell death. Second, as the energy factory, mitochondria dysfunction will significantly disrupt normal cell metabolism, leading to energy insufficiency and reducing viability.
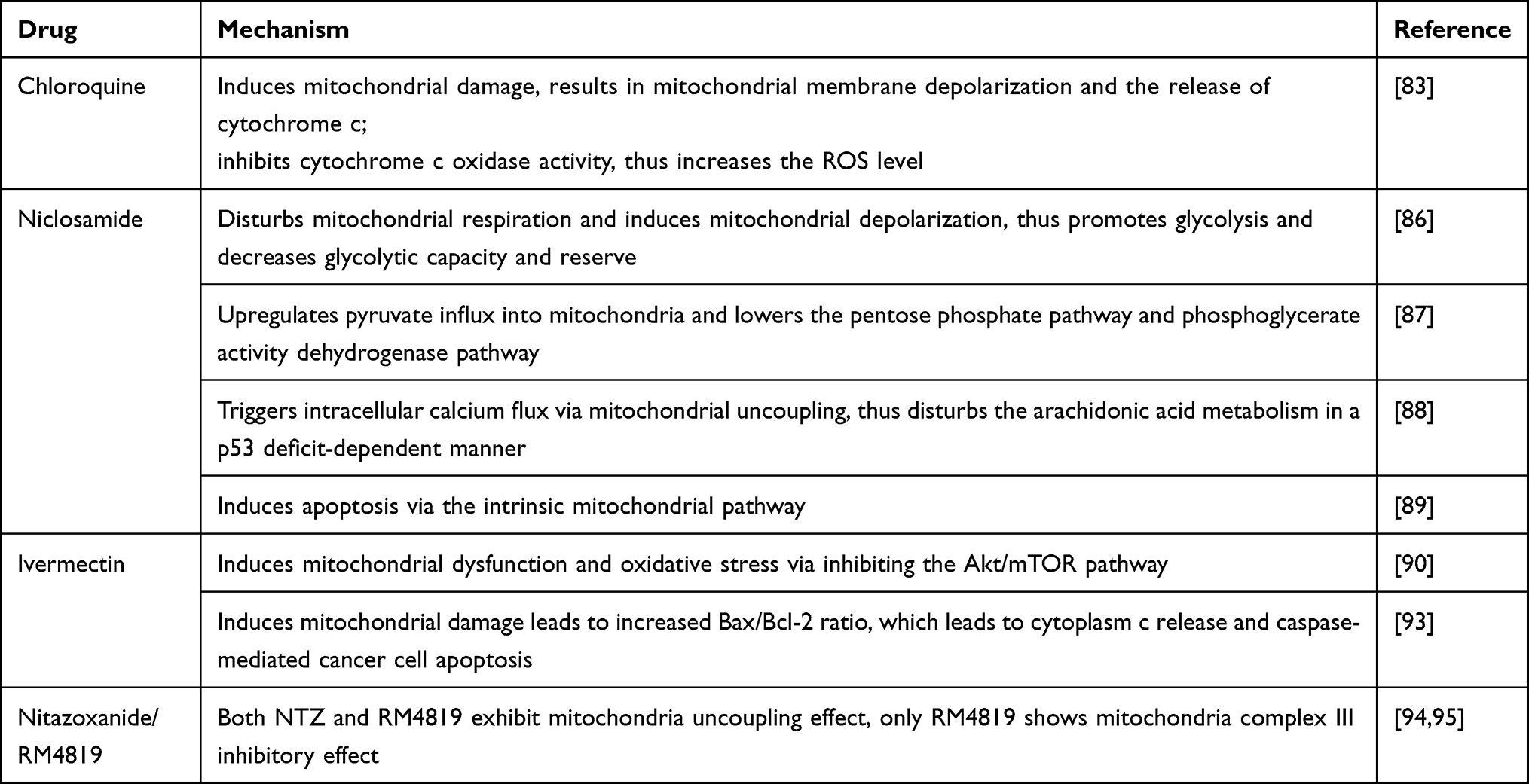 |
Table 3 Mitochondria Disrupting Effect of Antiparasitic Agents |
Recent studies continue to deepen our understanding of the mechanism in CQs.82,83 Liang et al revealed that CQ targets the invasiveness and migrating ability of cancer stem-like cells of triple-negative breast cancer (TNBC) by inducing mitochondrial damage, characteristic of abnormal mitochondrial structure.83 Such mitochondrial damage leads to mitochondrial membrane depolarization and the release of cytochrome c.83 CQ also inhibits cytochrome c oxidase activity, which transfers the electron from cytochrome c to oxygen, leading to an increased level of ROS.83 In this way, the CQ-induced ROS results in DNA double-strand damage characteristic of γ-H2AX accumulation. Besides inducing DNA damage, CQ also impairs the DNA repair ability of TNBC cancer cells, thus further worsening its DNA-related injury.83 Both autophagy inhibition, mitochondrial damage, and downstream ROS production may contribute to cancer cell death. Notably, Vessoni et al pointed out that, although CQ induces all these pathologic processes, only the mitochondrial membrane potential loss is responsible for the cancer cell sensitivity to CQ.84 Their experiments suggest that although CQ does induce purine oxidation, no DNA double-strand damage occurs when the loss of MMP is already detectable, suggesting that the DNA injury happens after the loss of MMP and cell death induced by CQ.84 Combining these two studies, we can conclude that both ROS generation and DNA double-strand damage are the downstream events of CQ-induced mitochondrial damage, while the CQ-related cancer cell death relies more on the loss of MMP. Mefloquine exhibits a similar mechanism in inducing mitochondrial damage followed by ROS generation.82 Altogether, these studies strongly suggest that the mitochondria-targeted strategy plays a critical role in CQs’ anticancer property.
As an anti-helminth agent, niclosamide functions mainly as a mitochondrial uncoupler that inhibits oxidative phosphorylation and reduces ATP production at the therapeutic dose.85 However, it can also inhibit human mitochondrial function at a higher dose in vitro. Based on such a pharmacologic mechanism, scientists thought of redirecting it as an anticancer agent. In recent years, many studies focus on its inhibitory effect on oxidative phosphorylation in cancer cells mitochondria. Such inhibition on mitochondria interferes with cellular metabolism. Shangguan et al validated that niclosamide inhibits ovarian cancer cells both in vitro and in vivo.86 They showed that niclosamide disturbs mitochondrial respiration and induces mitochondrial depolarization in ovarian carcinoma cells.86 In this way, niclosamide promotes glycolysis and decreases glycolytic capacity and reserve, suggesting that it disrupts normal aerobic glycolysis. Alasdair et al demonstrated that mitochondrial uncoupling upregulates pyruvate influx into mitochondria and lowers the pentose phosphate pathway and phosphoglycerate activity dehydrogenase pathway.87 Furthermore, it inhibits colon cancer cells’ proliferation in vitro and suppresses the hepatic metastasis in vivo experiments. Most interestingly, Kumar et al revealed a novel antitumor mechanism of niclosamide in p53-defective cancers.88 Their study demonstrated that niclosamide triggers intracellular calcium flux via mitochondrial uncoupling, thus disturbing the arachidonic acid metabolism in a p53 deficit-dependent manner. Moreover, their experiment revealed that an increased arachidonic acid level is responsible for the niclosamide-induced cytochrome c release and apoptosis. Yu et al showed that niclosamide induces apoptosis in thyroid cancer cells in vitro and inhibits tumor growth in vivo.89 Further, they observed mitochondrial membrane potential change and activation of Bax and inhibition of Bcl-2 in niclosamide-treated cells, suggesting that the intrinsic mitochondrial pathway mediates niclosamide-induced apoptosis. Altogether, these studies revealed that niclosamide targets mitochondria as its primary anticancer mechanism.
Scientists did not discover the inhibitory effect of ivermectin on mitochondria in cancer cells until 2016. In 2016, Liu et al revealed that ivermectin induces proliferation inhibition and apoptosis in both glioblastoma and endothelial cells, thus exhibit antitumor effect and anti-angiogenesis effect.90 Furthermore, they showed that it induces mitochondrial dysfunction and oxidative stress in glioblastoma and endothelial cells, which correlates with the inhibition of the Akt/mTOR pathway.90 Scientists revealed similar phenotypes, including mitochondrial dysfunction and oxidative stress induction in ivermectin-treated chronic myeloid leukemia cells and in renal cell carcinoma correspondingly.91,92 Interestingly, in Wang’s study, ivermectin exhibits renal cancer cell inhibitory effect while sparing the normal kidney cells, suggesting it potent in further clinical treatment.92 In Zhang’s experiment, they demonstrated that ivermectin-induced mitochondrial damage leads to increased Bax/Bcl-2 ratio, which leads to cytoplasm c release and caspase-mediated cancer cell apoptosis.93 Altogether, these studies demonstrated that ivermectin targets mitochondrion and induces its dysfunction, leading to its antitumor effect.
Nitazoxanide (NTZ) and its bromo-derivative RM4819 also target mitochondria to inhibit cancer cells.94,95 Tantawy et al demonstrated that NTZ influences the Bcl-2 family and results in mitochondria dysfunction characteristic of cytochrome-c release.94 In another study, Ripani et al revealed that while both NTZ and RM4819 exhibit mitochondria uncoupling effect, only RM4819 shows mitochondria complex III inhibitory effect.95 Due to such pharmaceutical target difference, RM4819 is far more effective than NTZ in disrupting mitochondria function. Their experiment further revealed that these 2 agents have minimal effect on the normal enteral epithelium. Altogether, these results suggest that NTZ and RM4819 could be potent anticancer agents, and further in vivo exploration may be needed.
Immunoregulating Effect of Antiparasitic Agents
Interestingly, several antiparasitic drugs exhibit immunoregulatory effects as anticancer mechanisms (Table 4). Immunoregulatory effects include directly affecting some specific immune cells and regulating the cancer microenvironment as a whole. Immunoregulation may further enhance the antitumor effect of these agents, which is worth discussing.
 |
Table 4 Immuoregulating Effect of Antiparasitic Agents |
In the past five years, scientists dramatically deepened the understanding of ARTs immunoregulatory effects. Many of these studies focus on how ARTs affect immune cells. Cao et al demonstrated that ART upregulates the proportion of CD4+ IFN-γ+ T cells and Granzyme B + cytotoxic T lymphocyte while abolishing the immunosuppressive effect of Treg cells and MDSCs (myeloid-derived suppressor cells).96 In another study, Qian et al revealed that ARS enhances the cytotoxicity of γδ T cells via upregulating the expression of granzyme B in γδ T cells.97 However, current research on how ARS affects γδ-T cell immunity is far not sufficient. In the future, we may focus on the detailed mechanism of how ARS upregulates the granzyme B level, and if other mechanisms exist in ARS-induced γδ-T cell enhancement. Considering further application, since ARS enhances γδ T cell function, combining ARS with γδ-T related immunotherapies (like CAR γδ-T therapy) would be worth of trying.98 NK cells are also targets of ARTs. Houh et al revealed that ART enhances the cytotoxic effect of NK cells and stimulates the granule exocytosis.99 Although it does not interfere with the NK activating receptors’ expression level, it modulates their downstream proteins, including Vav-1 and ERK1/2.99 Similarly, Lu et al reported that ART sensitizes cancer cells to NK cell-induced cytolysis manifested by enhancing conjugation between NK cells and tumor cells.100 Further exploration revealed that ART enriched apoptotic genes in cancer cells, making them more susceptible to NK cell-mediated lysis. In the future, combining ART with NK cell immunotherapies (adoptive NK transfer, CAR-NK, etc.) in animal experiments would be interesting. Besides directly acting on immune cells, ART also regulates the immunoreactive mediators’ expression level (T-bet, IFN-γ and TNF-α).96,97 While cancer cells may secrete immunosuppressive mediators to inhibit T cells’ cytotoxicity, both ART and ARS help cancer cells regain sensitivity to γδ T cells via inhibiting TGF-b secretion.97,101 These results suggested that ARTs may help construct a more stressful microenvironment for cancer cells, and its potential application in cancer-related immune regulation is worth of exploring in the future.
Several pieces of research focused on CQs’ immunoregulatory effect. Autophagy inhibition, the original impact of CQs, may contribute to such an immunoregulatory effect. A recent study revealed that CQ-induced autophagy inhibition enhances cancer cells’ immunogenicity, thus enhancing dendritic cell (DC) maturation and T cell response.102 Their experiment showed that combining 5-FU with CQ significantly induces carcinoembryonic antigen (CEA) family protein expression, which leads to enhanced immunogenicity and promotes DC maturation. As it is known that apoptotic cells are usually weak in immunogenicity, such a difference in immunogenicity probably correlates with autophagy-inhibition induced apoptosis, as it is different from physiological apoptosis. Although to some degree, there is insufficient evidence proving that such enhancement in immunogenicity directly correlates with CQ-induced autophagy blockage, their study is still inspirational, and further investigations should focus more on the exact mechanism of how such CQ-induced immunogenicity enhancement is mediated. The immunoregulatory effect of CQ can also be autophagy inhibition-independent. Han et al revealed that, although HCQ does inhibit autophagy of CML cancer cells, it sensitizes CML cell to Vγ9Vδ2 T cell-mediated lysis in an autophagy-independent way.103 Further exploration demonstrated that HCQ induces UL16 Binding Protein 4(ULBP4), the NKG2D ligand, on the CML cells’ surface. The enhanced connection between ULBP4 and NKG2D augments the recognition capability of Vγ9Vδ2 T cells on CML cells. Interestingly, HCQ does not directly alter the production or degradation of ULBP4. Instead, it facilitates the translocation of ULBP4 from the cytoplasm to the cell membrane.103 In another study, Zhang et al demonstrated that CQ promotes cancer cell killer CD8+T cell and downregulates immunosuppressive cells, including tumor-associated macrophages, myeloid-derived suppressor cells, and Tregs in breast cancer in vivo.104 Notably, it suppresses TGF-b production of the cancer cells in vitro and in vivo, suggesting that it functions as a cancer microenvironment regulator.104 While most studies revealed that CQ enhances tumor-related immune response, Masuelli et al demonstrated a different result, suggesting that CQ may be an immune inhibitor.105 Their study revealed that while CQ enhances curcumin (CUR) in vitro and nude mice, it reduces the antitumor effect of curcumin in immunocompetent mice. Further investigation suggests that CQ downregulates the immune response via upregulating the FoxP3 positive Treg cells and reducing the T cytotoxic cells. Moreover, a higher expression level of HIF-1a in the CQ+CUR group is observed. The authors hypothesized that the immunosuppressive effect of CQ probably correlates with its damage-associated molecular pattern (DAMP)-inducing effect or the immune-suppressive cytokines released due to higher HIF-1a. Currently available results are still insufficient to conclude whether CQ/HCQ functions as an immune enhancer or inhibitor in the cancer microenvironment. Future studies should focus more on the exact mechanism of how CQ boosts or inhibits immune response in immunocompetent mice models via analyzing the expression level of DAMP molecules, including HIF-1a, the cytokine levels, and the response of different types of immune cells to determine the overall effect of CQs on the cancer-related immune response.
In 2011, Takakura et al firstly demonstrated that pyrimethamine (Pyr) functions as a STAT3 inhibitor in polycystic kidney disease models.106 However, such STAT3 inhibitory effect had not been studied in the cancer field until 2018. In this year, Khan et al validated the STAT3 inhibitory effect of Pyr in breast cancer.107 Such inhibition on STAT3 suppresses the proliferation and invasion of breast cancer cells in vitro. Interestingly, Pyr also exhibits an immunoregulatory effect via inhibiting STAT3. Their experiments demonstrated that administration of Pyr suppresses Treg cells as well as TH-17 associated immune response, while enhancing the cytotoxicity of CD8+ T cells via promoting exocytosis. It has been fully demonstrated that STAT3 plays a critical role in carcinogenesis in various types of cancers. Future studies should explore the STAT3 inhibitory effect of Pyr in other cancers, especially in which STAT3 plays an important role in immunoregulation.
Metabolic Disrupting Effect of Antiparasitic Agents
Cancer cell metabolism is characterized by aerobic glycolysis, known as the Warburg effect. Disrupting glycolysis can significantly disturb the normal metabolism of cancer cells. The past several years witnessed progress in understanding the glycolysis disrupting effect of antiparasitic agents, and we will discuss them in detail.
Artemisinins function as glycolysis inhibitors. In 2015, Mi et al revealed that DHA downregulates the glycolysis metabolism level characteristic with decreased ATP and lactate levels in non-small cell lung carcinoma (NSCLC).108 Further exploration suggests that it inhibits the mTOR pathway and downregulates the expression level of GLUT1, resulting in reduced glucose uptake and decreased glycolysis metabolism.108 Similarly, in esophageal carcinoma, Li et al observed that DHA suppresses glycolysis via inhibiting the expression level of pyruvate kinase M2.109 In prostate cancer cell line LNCaP, DHA reduces the glucose uptake, ATP level, and lactate level.110 Western blotting revealed that the expression of a series of glycolysis-related proteins, including GLUT1, hexokinase 2, platelet-type fructose phosphate kinase, pyruvate kinase M2, and lactate dehydrogenase are decreased.110 Furthermore, they demonstrated that DHA suppresses the expression of HIF-1a, which is the upstream regulator of glycolysis, via regulating the PI3K/Akt/mTOR pathway.110 Similarly, scientists also observed the glycolysis inhibitory effect of ARS in B cell lymphoma.111 Benzimidazole derivatives also exhibit a glycolysis-disrupting effect. Zhou et al reported that albendazole inhibits the expression of HIF-1a in NSCLC cells.112 Such suppression on HIF-1a results in glycolysis inhibition.112 Under the hypoxia condition, albendazole inhibits glycolytic enzymes, including HK, PK, and LDH, while reducing lactate.112 Altogether, this experiment suggests that albendazole may disrupt hypoxia-induced glycolysis via suppressing the expression level of HIF-1a. Fenbendazole, another benzimidazole derivative, also disturbs the glycolytic metabolism. It inhibits the glucose uptake and the activity of hexokinase 2, thus significantly attenuating glycolysis. The in-silico analysis revealed that fenbendazole probably interferes with hexokinase II by mimicking the structure of glucose or G6P, thus binding to the enzyme’s active site and disrupting its normal function.113
Disturbance of glycolysis would profoundly interfere with the survival ability of cancer cells in hypoxia conditions. Notably, although radiotherapy or TACE (transcatheter arterial chemoembolization) are both potent therapy against the tumor, they are not effective enough for the cancer cells that are hypoxia-resistant. In this way, combining these agents with radiotherapy or TACE should be considered. Several studies combine ARTs or benzimidazole derivatives with radiotherapy and showed that these agents do exhibit radiosensitizing effects.114,115 In this way, it would be interesting to carry on clinical trials that combine radiotherapy with these agents. Regarding TACE, no animal experiment is available and future investigations would be needed to explore whether such strategy is effective.
Pyrimethamine (Pyr) functions as a plasmodium dihydrofolate reductase (pDHFR) inhibitor in fighting against malaria.116 It inhibits the transformation from dihydrofolate to tetrahydrofolate, resulting in nucleic acid synthesis dysfunction. Interestingly, a recent study revealed that pDHFR has a similar structure with human dihydrofolate reductase (hDHFR), and pyrimethamine does exert an inhibitory effect on hDHFR by directly binding to it, thus inhibiting tumor cell proliferation and metastasis.117 Moreover, researchers demonstrated that compared with methotrexate, another hDHFR inhibitor, Pyr additionally inhibits epithelial–mesenchymal transition, metastasis, and invasiveness of cancer cells. Further, they revealed that thymidine phosphorylase is another target of Pyr, which can explain such a difference in the inhibitory effect on cancer cells. Since it may interfere with DNA synthesis and metabolism, considering combining it with radiotherapy is reasonable. An in vitro experiment suggested that administration of Pyr on glioblastoma cells before chemo-radiotherapy would enhance their anticancer effect.118 In the future, more exploration in animal experiment and in more cancer cell types would be necessary.
Dark Side: Limitations and Future Directions
Although in vitro and animal experiments suggest that these agents are potent cancer killers, limitations that slowed their pace into clinical application emerge at the clinical trial stage. We analyze the results of the clinical trials and explore the limitations of these agents. Finally, we manage to provide possible solutions to overcome these limitations and give our opinions on the future directions of these agents. The data of the clinical trials are obtained from www.clinicaltrials.gov.
Artemisinins
We summarized the ongoing and completed clinical trials which apply ARTs in cancer treatment. (Table S1) Two completed Phase I trials published their results. Considering the medication tolerability, 18 mg/kg given on days 1 and 8 of a 21-day cycle or 200 mg/d orally are both tolerable dosage regimens.119,120 The major observed DL-AE (Dose Limited-Adverse Event) is anemia, which is also commonly seen in malaria ARS therapy and is acceptable and well tolerated. However, as previous animal studies aroused concerns about the potential ototoxicity of ARS, intermittent vertigo, ongoing tinnitus, or subclinical hearing loss is observed in several patients (4/23).119–121 Thus, future clinical trials should continue to pay attention to the possible ototoxicity, especially in long-term surveillance. In these two clinical trials, drug efficacy is kind of disappointing. No complete or partial remission was observed. A possible reason lies in its pharmacokinetic property. Pharmacokinetic analysis suggested that the concentration of ARS rapidly declined in the first 20 mins (T1/2=7–8 mins), although its active metabolite DHA exists much longer (T1/2=0.5–3.5 h). This suggests that future investigations in vitro should focus more on DHA instead of ARS. Besides, considering modifying the chemical structure of ARS that may slow down its transformation into DHA would be another way out. In the clinical trials, the plasma concentration of DHA (1.46 µM) is far not high enough to reach the IC50 concentration of the preclinical trials (8–100 µM) in the acceptable dosage (18 mg/kg given on days 1 and 8 of a 21-day cycle).119 These pharmacokinetics data suggested that future in vivo or in vitro studies should focus on DHA’s anticancer effect at a low dose, and we would better consider it as a combination therapy medication. Currently, there are no completed Phase II/III clinical trials, and the ongoing ones focus on colorectal carcinoma. Previous in vitro and in vivo experiments suggested that ARS has an anticancer effect on various cancers, including head and neck carcinoma, breast cancer and lymphoma, etc.27,111,122 Thus, it would be meaningful to carry on clinical trials of ARS on these cancer types after the pharmacokinetic issue is solved.
Benzimidazole Derivatives
We may first discuss the pharmaceutical properties of benzimidazole derivatives according to several previous clinical trials, although they are not correlated with cancer. In 2010, a clinical trial explored the maximum tolerated dose of albendazole and revealed that up to 2400 mg/d is tolerable.123 Moreover, the pharmacokinetic analysis showed that ABZ is rapidly metabolized into albendazole sulfoxide (ABZ-SO), the major active metabolite of ABZ. ABZ-SO. The mean maximum plasma concentration of ABZ-SO reaches 2.7 ± 2.1 (0.80–4.7) μg/mL, which is much higher compared with its IC50 value in cell experiments.124,125 Currently, there are no ongoing or finished clinical trials applying ABZ to treat cancers. Based on the promising antitumor effect of ABZ in vitro and in vivo experiments, carrying out more clinical trials applying ABZ to treat cancers is necessary. Moreover, the pharmacokinetic properties of ABZ suggested that future in vitro studies should focus more on the antitumor characteristics of ABZ-SO since ABZ remains only a short time in the plasma and ABZ-SO is the major active metabolite. Regarding MBZ, Guerini et al have summarized the pharmacokinetic properties of MBZ in detail.126 The maximum plasma concentration of MBZ under the tolerated dose is higher than the IC50 value in vitro experiments. There are several clinical trials applying mebendazole in treating cancers (Table S2). However, most of them are at the “Recruiting” stage and currently there are no available results. A terminated clinical trial suggested that MBZ may not be effective enough to inhibit cancer progression in GI cancer patients. However, no detailed results are available. The ongoing trials focus more on brain tumors and it is worth waiting for further results.
Ivermectin
Currently, there are no available clinical trials that apply ivermectin in treating cancers. Several recently published reviews focused on the antitumor pharmacokinetic properties of ivermectin.127,128 The pharmacodynamic evaluations revealed that the highest approachable plasma concentration of ivermectin in humans is higher than the average effective concentration in animal experiments. Moreover, several recent clinical trials which use ivermectin to treat other diseases again confirm that its side effect and toxicity are relatively acceptable.129,130 In constant with the previous authors’ conclusion, we strongly suggest carrying out more clinical trials using ivermectin to treat cancers.
Chloroquine Derivatives
For chloroquine derivatives, there are more clinical trials compared with other antiparasitic agents (Tables S3 and S4). CQ’s anticancer potential in brain tumors is attracting scientists’ attention (NCT02496741, NCT01894633, NCT01727531, NCT02378532, NCT02432417, NCT00224978). It is a pity that not all of these trials have published data. A clinical trial (n=30) published in 2006 suggested that additional CQ to chemotherapy may improve the mid-term survival of glioblastoma multiforme (GBM) patients.131 Another completed clinical trial (NCT02378532) which started in 2015 combines chloroquine with chemoradiation in GBM patients. Unfortunately, no published result is available. An ongoing clinical trial (NCT02432417) combines CQ with temozolomide and radiotherapy in GBM patients.132 Previous studies suggested that upregulation of EGFRVIII would help cancer cells survive stress conditions by inducing autophagy in GBM cancer cells.132 In the future, whether CQ-induced autophagy-inhibition may improve the overall survival of GBM patients, especially those with EGFRVIII-positive subtype, should be investigated. The result of this clinical trial is worth waiting for.
Niclosamide
The available clinical trials are summarized in Table S5. As mitochondrial uncoupler, it has a natural potent to harm other cells like cardiac or muscular cells. Whether such cytotoxic effect would limit its clinical application in treating cancer is attracting scientists’ attention. A recent study focused on this problem and investigated how cardiac cells would react to mitochondrial uncouplers including niclosamide.133 Gao et al demonstrated that while high-level niclosamide does exhibit cardiotoxicity, it exerts a cardio-protective effect by activating the STAT3 pathway at lower concentration (so-called “mild mitochondrial uncoupling”).133 According to previous preclinical experiments, the minimum clinically meaning dose is about 163.5 ng/mL (combined with enzalutamide), which equals approximately 0.5 μM. At such a concentration, niclosamide induces about 5% cardiac cell death and a 20% loss of membrane potential (MMP), which may disturb the normal cardiac function. Cardiac cell protection or targeted delivery to cancer cells could be the way out. No matter which approach is selected, cardiotoxicity monitoring should be included in clinical trials. Currently, there is no available study focusing on whether niclosamide would affect muscle cells, and investigations on this issue would be necessary. Another barrier to the clinical application of niclosamide is whether it can achieve targeted concentration in vivo. A published clinical trial suggested that at the maximum tolerated dose (500 mg TID), only 1/3 of the patients reach 163.5 ng/mL.134 Scientist thought of improving the delivery system and several nanobased-agents were invented, which may improve its bioavailability.135 In the future, scientists may explore these agents in clinical trials.
Pyrimethamine
Only 2 ongoing clinical trials are available on www.clinicaltrials.gov. (Table S6). In one of the trials, the researchers have published some of the results.136 Their investigation suggested that in the maximum dose cohort (50 mg/d), the plasma concentration of Pyr reached 6.17 μM, which has not reached the target concentration (10 μM) for maximum STAT3 inhibition. However, there is no dose-related toxicity observed at such dose, suggesting that a higher dosage may be tolerable. Researchers observed the STAT3 inhibition in CLL cells in some of the treated patients. Interestingly, their research suggested that it may be possible to predict whether the CLL cells would respond to Pyr in vivo by treating the CLL cells in vitro and evaluate the post-treatment STAT3 expression change. Moreover, a rebound of STAT3 expression at the progression stage of the treatment. In this way, it is worthy to consider combining Pyr with other STAT3 inhibitors in future exploration.
Nitazoxanide
Currently, there is only one clinical trial applying nitazoxanide (NTZ) in cancer treatment (NCT02366884), and the result is still unavailable. There are also insufficient in vitro and in vivo experiments to evaluate the detailed anticancer mechanism of nitazoxanide. Based on previous studies, colorectal cancer cells seem to be more sensitive to nitazoxanide (For HCT116 cell line: IC50=11.2 μM) compare with glioblastoma (IC 50: around 400 μM).73,94 According to the reported clinical trials, the maximum plasma concentration of NTZ at 1g/d dosage is 10.2 μg/mL (equals 33.2 μM).136 Moreover, a previous study suggested that up to 4g/d NTZ is tolerated in humans.136 In this way, future exploration may focus on evaluating the highest tolerated dose of NTZ in human and analysis its pharmacokinetic property.
Conclusion
Drug redirection is increasingly attracting the attention of both researchers and pharmaceutical companies. Repositioning anti-parasitic agents to kill cancer cells could be a feasible way to develop cancer medications, and investigations on the detailed mechanisms and related clinical trials are continuing to accumulating. We summarized the recent advance in exploring the antiparasitic drugs’ anticancer properties. Most of these agents target multiple biological procedures and exhibit inspiring anticancer potential. Their pharmaceutical effects also provide ideas for novel drug design and development. However, there is also a dark side in this field. Several limitations that may block their final clinical application still exist and is worthy of noticing. For most of these agents, whether the target plasma concentration is achievable under the tolerated dose is the major issue. Drug combination or pharmacokinetic property improvement by modifying the chemical structure or applying the delivery systems may be the way out. Future in vitro or animal experiments should also focus more on the pharmaceutical properties of these agents at the range of achievable plasma concentration. Hopefully, some of these anti-parasitic agents may emerge as clinically applicable anticancer medications in the future.
Abbreviations
ARTs, artemisinin derivatives; ART, artemisinin; ROS, reactive oxygen species; IRP, iron regulatory proteins; IRE, iron responsive element; ARS, artesunate; DHA, dihydroartemisinin; GSH, glutathione; GPX4, glutathione peroxidase 4; GS-SG, oxidized glutathione; Bcl-2, B-cell lymphoma 2; DSB, double-strand break; LC3-II, Microtubule-associated protein light chain 3-II; DAPK1, death-associated protein kinase 1; mTOR, mammalian target of the rapamycin; ATG7, autophagy related 7; ATG5, autophagy related 5; TRF2, TBP-related factor 2; Atg4b, autophagy-related 4B; TFEB, transcription factor EB; JNK, Jun N-terminal protein kinase; JNK/STAT3, pathway; EVA1A, Eva-1 homolog A; ATG16L1, Autophagy Related 16 Like 1; CQ, chloroquine; PPT-1, palmitoyl-protein thioesterase 1; HCQ, hydroxychloroquine; MQ, mefloquine; LAMP1/2, lysosomal-associated membrane protein 1/2; CAF, cancer-associated fibroblasts; HNSCC, head and neck squamous cell carcinoma; TNBC, triple-negative breast cancer; MMP, matrix metalloproteinase; ATP, adenosine triphosphate; Bax, BCL (B Cell Lymphoma)-Associated X; MDSCs, myeloid-derived suppressor cells; ERK1/2, extracellular signal-regulated kinases 1 and 2; DC, dendritic cell; 5-FU, 5-Fluorouracil; IFN-γ, interferon-γ; TNF-α, tumor necrosis factor-α; CEA, carcinoembryonic antigen; CML, chronic myeloid leukemia; ULBP4, UL16 Binding Protein 4; NKG2D, receptor natural killer group 2D; CUR, curcumin; FoxP3, forkhead box P3; HIF-1a, hypoxia-inducible factor 1a; DAMP, damage-associated molecular pattern; TGF-b, transforming growth factor-β; NSCLC, non-small cell lung carcinoma; GLUT1, glucose transporter type 1; LNCaP, lymph node carcinoma of the prostate; G6P, glucose-6-phosphate.
Consent for Publication
The authors consent that the details of any images, videos, recordings, etc. can be published, and all the authors have been shown the article contents to be published.
Acknowledgments
We appreciate Ms. Nicole S. Li of the University of Illinois for her help in improving the language of the manuscript. This work was supported by National Key R&D Program of China (No.2020YFC1200100), the Guangdong Natural Science Foundation (No. 2019A1515010583) and the National Natural Science Foundation of China (No.81641094) to XL, and Fundamental Research Funds for the Central Universities (No. 20ykpy158) to YW.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Roth GA, Abate D, Abate KH, et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1736–1788. doi: 10.1016/S0140-6736(18)32203-7
2. Mun EJ, Babiker HM, Weinberg U, Kirson ED, Von Hoff DD. Tumor-treating fields: a fourth modality in cancer treatment. Clin Cancer Res. 2018;24(2):266–275. doi:10.1158/1078-0432.CCR-17-1117
3. Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575(7782):299–309. doi:10.1038/s41586-019-1730-1
4. Sertkaya A, Wong HH, Jessup A, Beleche T. Key cost drivers of pharmaceutical clinical trials in the United States. Clin Trials. 2016;13(2):117–126. doi:10.1177/1740774515625964
5. Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18(1):41–58. doi:10.1038/nrd.2018.168
6. Mou Y, Wang J, Wu J, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12(1):34. doi:10.1186/s13045-019-0720-y
7. White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res. 2015;21(22):5037–5046. doi:10.1158/1078-0432.CCR-15-0490
8. Deng P, Haynes CM. Mitochondrial dysfunction in cancer: potential roles of ATF5 and the mitochondrial UPR. Semin Cancer Biol. 2017;47:43–49. doi:10.1016/j.semcancer.2017.05.002
9. Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41(3):211–218. doi:10.1016/j.tibs.2015.12.001
10. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–68. doi:10.1016/j.canlet.2016.01.043
11. Gunjan S, Sharma T, Yadav K, et al. Artemisinin derivatives and synthetic trioxane trigger apoptotic cell death in asexual stages of plasmodium. Front Cell Infect Microbiol. 2018;8:256. doi:10.3389/fcimb.2018.00256
12. Klonis N, Creek DJ, Tilley L. Iron and heme metabolism in Plasmodium falciparum and the mechanism of action of artemisinins. Curr Opin Microbiol. 2013;16(6):722–727. doi:10.1016/j.mib.2013.07.005
13. Kim SJ, Kim MS, Lee JW, et al. Dihydroartemisinin enhances radiosensitivity of human glioma cells in vitro. J Cancer Res Clin Oncol. 2006;132(2):129–135. doi:10.1007/s00432-005-0052-x
14. Efferth T, Benakis A, Romero MR, et al. Enhancement of cytotoxicity of artemisinins toward cancer cells by ferrous iron. Free Radical Biol Med. 2004;37(7):998–1009. doi:10.1016/j.freeradbiomed.2004.06.023
15. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
16. Lu B, Chen XB, Ying MD, He QJ, Cao J, Yang B. The role of ferroptosis in cancer development and treatment response. Front Pharmacol. 2017;8:992. doi:10.3389/fphar.2017.00992
17. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
18. Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radical Biol Med. 2019;133:130–143. doi:10.1016/j.freeradbiomed.2018.09.043
19. Ooko E, Saeed ME, Kadioglu O, et al. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine. 2015;22(11):1045–1054. doi:10.1016/j.phymed.2015.08.002
20. Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2(5):517–532. doi:10.18632/oncoscience.160
21. Muckenthaler MU, Galy B, Hentze MW. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr. 2008;28(1):197–213. doi:10.1146/annurev.nutr.28.061807.155521
22. Manz DH, Blanchette NL, Paul BT, Torti FM, Torti SV. Iron and cancer: recent insights. Ann N Y Acad Sci. 2016;1368(1):149–161. doi:10.1111/nyas.13008
23. Sibmooh N, Udomsangpetch R, Kujoa A, Chantharaksri U, Mankhetkorn S. Redox reaction of artemisinin with ferrous and ferric ions in aqueous buffer. Chem Pharm Bull (Tokyo). 2001;49(12):1541–1546. doi:10.1248/cpb.49.1541
24. Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27(1):242–254. doi:10.1038/s41418-019-0352-3
25. Du J, Wang T, Li Y, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radical Biol Med. 2019;131:356–369. doi:10.1016/j.freeradbiomed.2018.12.011
26. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radical Biol Med. 2020;152:175–185. doi:10.1016/j.freeradbiomed.2020.02.027
27. Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254–262. doi:10.1016/j.redox.2016.12.010
28. Yuan B, Liao F, Shi ZZ, et al. Dihydroartemisinin inhibits the proliferation, colony formation and induces ferroptosis of lung cancer cells by inhibiting PRIM2/SLC7A11 axis. Onco Targets Ther. 2020;13:10829–10840. doi:10.2147/OTT.S248492
29. Chen Y, Mi Y, Zhang X, et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res. 2019;38(1):402. doi:10.1186/s13046-019-1413-7
30. Yi R, Wang H, Deng C, et al. Dihydroartemisinin initiates ferroptosis in glioblastoma through GPX4 inhibition. Biosci Rep. 2020;40(6). doi:10.1042/BSR20193314
31. Kumar B, Kalvala A, Chu S, et al. Antileukemic activity and cellular effects of the antimalarial agent artesunate in acute myeloid leukemia. Leukemia Res. 2017;59:124–135. doi:10.1016/j.leukres.2017.05.007
32. Wang L, Li J, Shi X, et al. Antimalarial dihydroartemisinin triggers autophagy within HeLa cells of human cervical cancer through Bcl-2 phosphorylation at Ser70. Phytomedicine. 2019;52:147–156. doi:10.1016/j.phymed.2018.09.221
33. Wen L, Liu L, Wen L, Yu T, Wei F. Artesunate promotes G2/M cell cycle arrest in MCF7 breast cancer cells through ATM activation. Breast Cancer. 2018;25(6):681–686. doi:10.1007/s12282-018-0873-5
34. Greenshields AL, Shepherd TG, Hoskin DW. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol Carcinog. 2017;56(1):75–93. doi:10.1002/mc.22474
35. Greenshields AL, Fernando W, Hoskin DW. The anti-malarial drug artesunate causes cell cycle arrest and apoptosis of triple-negative MDA-MB-468 and HER2-enriched SK-BR-3 breast cancer cells. Exp Mol Pathol. 2019;107:10–22. doi:10.1016/j.yexmp.2019.01.006
36. Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020;5(1):108. doi:10.1038/s41392-020-00216-5
37. Kapralov AA, Yang Q, Dar HH, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. 2020;16(3):278–290. doi:10.1038/s41589-019-0462-8
38. Lorente J, Velandia C, Leal JA, et al. The interplay between autophagy and tumorigenesis: exploiting autophagy as a means of anticancer therapy. Biol Rev Camb Philos Soc. 2018;93(1):152–165. doi:10.1111/brv.12337
39. Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. J Hematol Oncol. 2017;10(1):67. doi:10.1186/s13045-017-0436-9
40. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. doi:10.1038/s41580-018-0003-4
41. Wang Z, Hu W, Zhang JL, Wu XH, Zhou HJ. Dihydroartemisinin induces autophagy and inhibits the growth of iron-loaded human myeloid leukemia K562 cells via ROS toxicity. FEBS Open Bio. 2012;2(1):103–112. doi:10.1016/j.fob.2012.05.002
42. Sun X, Yan P, Zou C, et al. Targeting autophagy enhances the anticancer effect of artemisinin and its derivatives. Med Res Rev. 2019;39(6):2172–2193. doi:10.1002/med.21580
43. Xu HD, Qin ZH. Beclin 1, Bcl-2 and autophagy. Adv Exp Med Biol. 2019;1206:109–126.
44. Hill SM, Wrobel L, Rubinsztein DC. Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death Differ. 2019;26(4):617–629. doi:10.1038/s41418-018-0254-9
45. Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. doi:10.1016/j.molcel.2008.06.001
46. Thongchot S, Vidoni C, Ferraresi A, et al. Dihydroartemisinin induces apoptosis and autophagy-dependent cell death in cholangiocarcinoma through a DAPK1-BECLIN1 pathway. Mol Carcinog. 2018;57(12):1735–1750. doi:10.1002/mc.22893
47. Xu Z, Han X, Ou D, et al. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl Microbiol Biotechnol. 2020;104(2):575–587. doi:10.1007/s00253-019-10257-8
48. Cheng C, Wang T, Song Z, et al. Induction of autophagy and autophagy-dependent apoptosis in diffuse large B-cell lymphoma by a new antimalarial artemisinin derivative, SM1044. Cancer Med. 2018;7(2):380–396. doi:10.1002/cam4.1276
49. Ma Q, Liao H, Xu L, et al. Autophagy-dependent cell cycle arrest in esophageal cancer cells exposed to dihydroartemisinin. Chin Med. 2020;15(1):37. doi:10.1186/s13020-020-00318-w
50. Li B, Bu S, Sun J, Guo Y, Lai D. Artemisinin derivatives inhibit epithelial ovarian cancer cells via autophagy-mediated cell cycle arrest. Acta Biochim Biophys Sin (Shanghai). 2018;50(12):1227–1235. doi:10.1093/abbs/gmy125
51. Jiang F, Zhou JY, Zhang D, Liu MH, Chen YG. Artesunate induces apoptosis and autophagy in HCT116 colon cancer cells, and autophagy inhibition enhances the artesunate‑induced apoptosis. Int J Mol Med. 2018;42(3):1295–1304. doi:10.3892/ijmm.2018.3712
52. Ganguli A, Choudhury D, Datta S, Bhattacharya S, Chakrabarti G. Inhibition of autophagy by chloroquine potentiates synergistically anti-cancer property of artemisinin by promoting ROS dependent apoptosis. Biochimie. 2014;107(Pt):
53. Zhang L, Guo M, Li J, et al. Systems biology-based discovery of a potential Atg4B agonist (Flubendazole) that induces autophagy in breast cancer. Mol Biosyst. 2015;11(11):2860–2866. doi:10.1039/C5MB00466G
54. Chauhan S, Ahmed Z, Bradfute SB, et al. Pharmaceutical screen identifies novel target processes for activation of autophagy with a broad translational potential. Nat Commun. 2015;6(1):8620. doi:10.1038/ncomms9620
55. Lin S, Yang L, Yao Y, et al. Flubendazole demonstrates valid antitumor effects by inhibiting STAT3 and activating autophagy. J Exp Clin Cancer Res. 2019;38(1):293. doi:10.1186/s13046-019-1303-z
56. Zhen Y, Zhao R, Wang M, et al. Flubendazole elicits anti-cancer effects via targeting EVA1A-modulated autophagy and apoptosis in triple-negative breast cancer. Theranostics. 2020;10(18):8080–8097. doi:10.7150/thno.43473
57. Hu J, Li G, Qu L, et al. TMEM166/EVA1A interacts with ATG16L1 and induces autophagosome formation and cell death. Cell Death Dis. 2016;7(8):e2323. doi:10.1038/cddis.2016.230
58. Sung SJ, Kim HK, Hong YK, Joe YA. Autophagy is a potential target for enhancing the anti-angiogenic effect of mebendazole in endothelial cells. Biomol Ther (Seoul). 2019;27(1):117–125. doi:10.4062/biomolther.2018.222
59. Draganov D, Gopalakrishna-Pillai S, Chen YR, et al. Modulation of P2X4/P2X7/Pannexin-1 sensitivity to extracellular ATP via ivermectin induces a non-apoptotic and inflammatory form of cancer cell death. Sci Rep. 2015;5(1):16222. doi:10.1038/srep16222
60. Dou Q, Chen HN, Wang K, et al. Ivermectin induces cytostatic autophagy by blocking the PAK1/Akt axis in breast cancer. Cancer Res. 2016;76(15):4457–4469. doi:10.1158/0008-5472.CAN-15-2887
61. Liu J, Liang H, Chen C, et al. Ivermectin induces autophagy-mediated cell death through the AKT/mTOR signaling pathway in glioma cells. Biosci Rep. 2019;39(12). doi:10.1042/BSR20192489
62. Um JH, Yun J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017;50(6):299–307. doi:10.5483/BMBRep.2017.50.6.056
63. Zachari M, Gudmundsson SR, Li Z, et al. Selective autophagy of mitochondria on a ubiquitin-endoplasmic-reticulum platform. Dev Cell. 2019;50(5):627–643.e625. doi:10.1016/j.devcel.2019.06.016
64. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–542. doi:10.1038/nrc.2017.53
65. Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–1455. doi:10.1080/15548627.2018.1474314
66. Rebecca VW, Nicastri MC, Fennelly C, et al. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov. 2019;9(2):220–229. doi:10.1158/2159-8290.CD-18-0706
67. Rebecca VW, Nicastri MC, McLaughlin N, et al. A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov. 2017;7(11):1266–1283. doi:10.1158/2159-8290.CD-17-0741
68. Liu T, Zhang J, Li K, Deng L, Wang H. Combination of an autophagy inducer and an autophagy inhibitor: a smarter strategy emerging in cancer therapy. Front Pharmacol. 2020;11:408. doi:10.3389/fphar.2020.00408
69. Xie Y, Zhang J, Lu B, et al. Mefloquine inhibits esophageal squamous cell carcinoma tumor growth by inducing mitochondrial autophagy. Front Oncol. 2020;10:1217. doi:10.3389/fonc.2020.01217
70. Takeda M, Koseki J, Takahashi H, et al. Disruption of endolysosomal RAB5/7 efficiently eliminates colorectal cancer stem cells. Cancer Res. 2019;79(7):1426–1437. doi:10.1158/0008-5472.CAN-18-2192
71. Wang X, Shen C, Liu Z, et al. Nitazoxanide, an antiprotozoal drug, inhibits late-stage autophagy and promotes ING1-induced cell cycle arrest in glioblastoma. Cell Death Dis. 2018;9(10):1032. doi:10.1038/s41419-018-1058-z
72. Lin YC, Lin JF, Wen SI, et al. Chloroquine and hydroxychloroquine inhibit bladder cancer cell growth by targeting basal autophagy and enhancing apoptosis. Kaohsiung J Med Sci. 2017;33(5):215–223. doi:10.1016/j.kjms.2017.01.004
73. Nordstrøm LU, Sironi J, Aranda E, et al. Discovery of autophagy inhibitors with antiproliferative activity in lung and pancreatic cancer cells. ACS Med Chem Lett. 2015;6(2):134–139. doi:10.1021/ml500348p
74. New J, Arnold L, Ananth M, et al. Secretory autophagy in cancer-associated fibroblasts promotes head and neck cancer progression and offers a novel therapeutic target. Cancer Res. 2017;77(23):6679–6691. doi:10.1158/0008-5472.CAN-17-1077
75. Zhao Z, Bai S, Wang R, et al. Cancer-associated fibroblasts endow stem-like qualities to liver cancer cells by modulating autophagy. Cancer Manag Res. 2019;11:5737–5744. doi:10.2147/CMAR.S197634
76. Folkerts H, Hilgendorf S, Vellenga E, Bremer E, Wiersma VR. The multifaceted role of autophagy in cancer and the microenvironment. Med Res Rev. 2019;39(2):517–560. doi:10.1002/med.21531
77. Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019;9(9):1167–1181. doi:10.1158/2159-8290.CD-19-0292
78. Jiang GM, Tan Y, Wang H, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer. 2019;18(1):17. doi:10.1186/s12943-019-0944-z
79. Poillet-Perez L, Xie X, Zhan L, et al. Autophagy maintains tumour growth through circulating arginine. Nature. 2018;563(7732):569–573. doi:10.1038/s41586-018-0697-7
80. Li H, Jiao S, Li X, Banu H, Hamal S, Wang X. Therapeutic effects of antibiotic drug mefloquine against cervical cancer through impairing mitochondrial function and inhibiting mTOR pathway. Can J Physiol Pharmacol. 2017;95(1):43–50. doi:10.1139/cjpp-2016-0124
81. Liang DH, Choi DS, Ensor JE, Kaipparettu BA, Bass BL, Chang JC. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016;376(2):249–258. doi:10.1016/j.canlet.2016.04.002
82. Vessoni AT, Quinet A, de Andrade-lima LC, et al. Chloroquine-induced glioma cells death is associated with mitochondrial membrane potential loss, but not oxidative stress. Free Radical Biol Med. 2016;90:91–100. doi:10.1016/j.freeradbiomed.2015.11.008
83. Frayha GJ, Smyth JD, Gobert JG, Savel J. The mechanisms of action of antiprotozoal and anthelmintic drugs in man. Gen Pharmacol. 1997;28(2):273–299. doi:10.1016/S0306-3623(96)00149-8
84. Shangguan F, Liu Y, Ma L, et al. Niclosamide inhibits ovarian carcinoma growth by interrupting cellular bioenergetics. J Cancer. 2020;11(12):3454–3466. doi:10.7150/jca.41418
85. Alasadi A, Chen M, Swapna GVT, et al. Effect of mitochondrial uncouplers niclosamide ethanolamine (NEN) and oxyclozanide on hepatic metastasis of colon cancer. Cell Death Dis. 2018;9(2):215. doi:10.1038/s41419-017-0092-6
86. Kumar R, Coronel L, Somalanka B, et al. Mitochondrial uncoupling reveals a novel therapeutic opportunity for p53-defective cancers. Nat Commun. 2018;9(1):3931. doi:10.1038/s41467-018-05805-1
87. Yu K, Wang T, Li Y, et al. Niclosamide induces apoptosis through mitochondrial intrinsic pathway and inhibits migration and invasion in human thyroid cancer in vitro. Biomed Pharmacother. 2017;92:403–411. doi:10.1016/j.biopha.2017.05.097
88. Liu Y, Fang S, Sun Q, Liu B. Anthelmintic drug ivermectin inhibits angiogenesis, growth and survival of glioblastoma through inducing mitochondrial dysfunction and oxidative stress. Biochem Biophys Res Commun. 2016;480(3):415–421. doi:10.1016/j.bbrc.2016.10.064
89. Wang J, Xu Y, Wan H, Hu J. Antibiotic ivermectin selectively induces apoptosis in chronic myeloid leukemia through inducing mitochondrial dysfunction and oxidative stress. Biochem Biophys Res Commun. 2018;497(1):241–247. doi:10.1016/j.bbrc.2018.02.063
90. Zhu M, Li Y, Zhou Z. Antibiotic ivermectin preferentially targets renal cancer through inducing mitochondrial dysfunction and oxidative damage. Biochem Biophys Res Commun. 2017;492(3):373–378. doi:10.1016/j.bbrc.2017.08.097
91. Zhang P, Zhang Y, Liu K, et al. Ivermectin induces cell cycle arrest and apoptosis of HeLa cells via mitochondrial pathway. Cell Prolif. 2019;52(2):e12543. doi:10.1111/cpr.12543
92. Tantawy MA, El-Sherbeeny NA, Helmi N, Alazragi R, Salem N, Elaidy SM. Synthetic antiprotozoal thiazolide drug induced apoptosis in colorectal cancer cells: implications of IL-6/JAK2/STAT3 and p53/caspases-dependent signaling pathways based on molecular docking and in vitro study. Mol Cell Biochem. 2020;469(1–2):143–157. doi:10.1007/s11010-020-03736-4
93. Ripani P, Delp J, Bode K, et al. Thiazolides promote G1 cell cycle arrest in colorectal cancer cells by targeting the mitochondrial respiratory chain. Oncogene. 2020;39(11):2345–2357. doi:10.1038/s41388-019-1142-6
94. Cao Y, Feng YH, Gao LW, et al. Artemisinin enhances the anti-tumor immune response in 4T1 breast cancer cells in vitro and in vivo. Int Immunopharmacol. 2019;70:110–116. doi:10.1016/j.intimp.2019.01.041
95. Qian P, Zhang YW, Zhou ZH, et al. Artesunate enhances γδ T-cell-mediated antitumor activity through augmenting γδ T-cell function and reversing immune escape of HepG2 cells. Immunopharmacol Immunotoxicol. 2018;40(2):107–116. doi:10.1080/08923973.2017.1386212
96. Sebestyen Z, Prinz I, Déchanet-Merville J, Silva-Santos B, Kuball J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat Rev Drug Discov. 2020;19(3):169–184. doi:10.1038/s41573-019-0038-z
97. Houh YK, Kim KE, Park S, et al. The effects of artemisinin on the cytolytic activity of natural killer (NK) cells. Int J Mol Sci. 2017;18(7):1600. doi:10.3390/ijms18071600
98. Lu Z, Bi J, Wan X. Artemisinin sensitizes tumor cells to NK cell-mediated cytolysis. Biochem Biophys Res Commun. 2020;524(2):418–423. doi:10.1016/j.bbrc.2020.01.094
99. Vesely MD, Schreiber RD. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Ann N Y Acad Sci. 2013;1284(1):1–5. doi:10.1111/nyas.12105
100. Zamame Ramirez JA, Romagnoli GG, Falasco BF, et al. Blocking drug-induced autophagy with chloroquine in HCT-116 colon cancer cells enhances DC maturation and T cell responses induced by tumor cell lysate. Int Immunopharmacol. 2020;84:106495. doi:10.1016/j.intimp.2020.106495
101. Han B, Zhao Y, Lin Y, et al. Hydroxychloroquine sensitizes chronic myeloid leukemia cells to Vγ9Vδ2 T cell-mediated lysis independent of autophagy. Int J Oncol. 2017;50(5):1810–1820. doi:10.3892/ijo.2017.3934
102. Zhang Y, Cao Y, Sun X, et al. Chloroquine (CQ) exerts anti-breast cancer through modulating microenvironment and inducing apoptosis. Int Immunopharmacol. 2017;42:100–107. doi:10.1016/j.intimp.2016.11.027
103. Masuelli L, Granato M, Benvenuto M, et al. Chloroquine supplementation increases the cytotoxic effect of curcumin against Her2/neu overexpressing breast cancer cells in vitro and in vivo in nude mice while counteracts it in immune competent mice. Oncoimmunology. 2017;6(11):e1356151. doi:10.1080/2162402X.2017.1356151
104. Takakura A, Nelson EA, Haque N, et al. Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum Mol Genet. 2011;20(21):4143–4154. doi:10.1093/hmg/ddr338
105. Khan MW, Saadalla A, Ewida AH, et al. The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer. Cancer Immunol Immunother. 2018;67(1):13–23. doi:10.1007/s00262-017-2057-0
106. Mi YJ, Geng GJ, Zou ZZ, et al. Dihydroartemisinin inhibits glucose uptake and cooperates with glycolysis inhibitor to induce apoptosis in non-small cell lung carcinoma cells. PLoS One. 2015;10(3):e0120426. doi:10.1371/journal.pone.0120426
107. Li Y, Lu J, Chen Q, et al. Artemisinin suppresses hepatocellular carcinoma cell growth, migration and invasion by targeting cellular bioenergetics and Hippo-YAP signaling. Arch Toxicol. 2019;93(11):3367–3383. doi:10.1007/s00204-019-02579-3
108. Zhu W, Li Y, Zhao D, et al. Dihydroartemisinin suppresses glycolysis of LNCaP cells by inhibiting PI3K/AKT pathway and downregulating HIF-1alpha expression. Life Sci. 2019;233:116730. doi:10.1016/j.lfs.2019.116730
109. Vatsveen TK, Myhre MR, Steen CB, et al. Artesunate shows potent anti-tumor activity in B-cell lymphoma. J Hematol Oncol. 2018;11(1):23. doi:10.1186/s13045-018-0561-0
110. Zhou F, Du J, Wang J. Albendazole inhibits HIF-1α-dependent glycolysis and VEGF expression in non-small cell lung cancer cells. Mol Cell Biochem. 2017;428(1–2):171–178. doi:10.1007/s11010-016-2927-3
111. Dogra N, Kumar A, Mukhopadhyay T. Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways. Sci Rep. 2018;8(1):11926. doi:10.1038/s41598-018-30158-6
112. Efferth T. Cancer combination therapies with artemisinin-type drugs. Biochem Pharmacol. 2017;139:56–70. doi:10.1016/j.bcp.2017.03.019
113. Zhang L, Bochkur Dratver M, Yazal T, et al. Mebendazole potentiates radiation therapy in triple-negative breast cancer. Int J Radiat Oncol Biol Phys. 2019;103(1):195–207. doi:10.1016/j.ijrobp.2018.08.046
114. Caliskan B, Sinoplu E, Ibis K, Akhan Guzelcan E, Cetin Atalay R, Banoglu E. Synthesis and cellular bioactivities of novel isoxazole derivatives incorporating an arylpiperazine moiety as anticancer agents. J Enzyme Inhib Med Chem. 2018;33(1):1352–1361. doi:10.1080/14756366.2018.1504041
115. Liu H, Qin Y, Zhai D, et al. Antimalarial drug pyrimethamine plays a dual role in antitumor proliferation and metastasis through targeting DHFR and TP. Mol Cancer Ther. 2019;18(3):541–555. doi:10.1158/1535-7163.MCT-18-0936
116. Baritchii A, Jurj A, Soritau O, et al. Sensitizer drugs for the treatment of temozolomide-resistant glioblastoma. J buon. 2016;21(1):199–207.
117. Deeken JF, Wang H, Hartley M, et al. A phase I study of intravenous artesunate in patients with advanced solid tumor malignancies. Cancer Chemother Pharmacol. 2018;81(3):587–596. doi:10.1007/s00280-018-3533-8
118. von Hagens C, Walter-Sack I, Goeckenjan M, et al. Prospective open uncontrolled phase I study to define a well-tolerated dose of oral artesunate as add-on therapy in patients with metastatic breast cancer (ARTIC M33/2). Breast Cancer Res Treat. 2017;164(2):359–369. doi:10.1007/s10549-017-4261-1
119. König M, von Hagens C, Hoth S, et al. Investigation of ototoxicity of artesunate as add-on therapy in patients with metastatic or locally advanced breast cancer: new audiological results from a Prospective, Open, Uncontrolled, Monocentric Phase I Study. Cancer Chemother Pharmacol. 2016;77(2):413–427. doi:10.1007/s00280-016-2960-7
120. Yao Y, Guo Q, Cao Y, et al. Artemisinin derivatives inactivate cancer-associated fibroblasts through suppressing TGF-β signaling in breast cancer. J Exp Clin Cancer Res. 2018;37(1):282. doi:10.1186/s13046-018-0960-7
121. Pourgholami MH, Szwajcer M, Chin M, et al. Phase I clinical trial to determine maximum tolerated dose of oral albendazole in patients with advanced cancer. Cancer Chemother Pharmacol. 2010;65(3):597–605. doi:10.1007/s00280-009-1157-8
122. Chen H, Weng Z, Xu C. Albendazole suppresses cell proliferation and migration and induces apoptosis in human pancreatic cancer cells. Anticancer Drugs. 2020;31(5):431–439. doi:10.1097/CAD.0000000000000914
123. Pourgholami MH, Akhter J, Wang L, Lu Y, Morris DL. Antitumor activity of albendazole against the human colorectal cancer cell line HT-29: in vitro and in a xenograft model of peritoneal carcinomatosis. Cancer Chemother Pharmacol. 2005;55(5):425–432. doi:10.1007/s00280-004-0927-6
124. Guerini AE, Triggiani L, Maddalo M, et al. Mebendazole as a candidate for drug repurposing in oncology: an extensive review of current literature. Cancers (Basel). 2019;11(9):1284. doi:10.3390/cancers11091284
125. Juarez M, Schcolnik-Cabrera A, Dueñas-Gonzalez A. The multitargeted drug ivermectin: from an antiparasitic agent to a repositioned cancer drug. Am J Cancer Res. 2018;8(2):317–331.
126. Liu J, Zhang K, Cheng L, Zhu H, Xu T. Progress in understanding the molecular mechanisms underlying the antitumour effects of ivermectin. Drug Des Devel Ther. 2020;14:285–296. doi:10.2147/DDDT.S237393
127. Muñoz J, Ballester MR, Antonijoan RM, et al. Safety and pharmacokinetic profile of fixed-dose ivermectin with an innovative 18mg tablet in healthy adult volunteers. PLoS Negl Trop Dis. 2018;12(1):e0006020. doi:10.1371/journal.pntd.0006020
128. Buonfrate D, Salas-Coronas J, Muñoz J, et al. Multiple-dose versus single-dose ivermectin for strongyloides stercoralis infection (strong treat 1 to 4): a multicentre, open-label, Phase 3, randomised controlled superiority trial. Lancet Infect Dis. 2019;19(11):1181–1190. doi:10.1016/S1473-3099(19)30289-0
129. Sotelo J, Briceño E, López-González MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2006;144(5):337–343. doi:10.7326/0003-4819-144-5-200603070-00008
130. Compter I, Eekers DBP, Hoeben A. et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: a phase IB trial. Autophagy. 2020;1–9. doi:10.1080/15548627.2020.1816343
131. Gao JL, Zhao J, Zhu HB, et al. Characterizations of mitochondrial uncoupling induced by chemical mitochondrial uncouplers in cardiomyocytes. Free Radical Biol Med. 2018;124:288–298. doi:10.1016/j.freeradbiomed.2018.06.020
132. Schweizer MT, Haugk K, McKiernan JS, et al. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PLoS One. 2018;13(6):e0198389. doi:10.1371/journal.pone.0198389
133. Barbosa EJ, Löbenberg R, de Araujo GLB, Bou-Chacra NA. Niclosamide repositioning for treating cancer: challenges and nano-based drug delivery opportunities. Eur J Pharm Biopharm. 2019;141:58–69. doi:10.1016/j.ejpb.2019.05.004
134. Brown JR, Walker SR, Heppler LN, et al. Targeting constitutively active STAT3 in chronic lymphocytic leukemia: a clinical trial of the STAT3 inhibitor pyrimethamine with pharmacodynamic analyses. Am J Hematol. 2021;96(4):E95–e98. doi:10.1002/ajh.26084
135. Walsh KF, McAulay K, Lee MH, et al. Early bactericidal activity trial of nitazoxanide for pulmonary tuberculosis. Antimicrob Agents Chemother. 2020;64(5). doi:10.1128/AAC.01956-19
136. Stockis A, Allemon AM, De Bruyn S, Gengler C. Nitazoxanide pharmacokinetics and tolerability in man using single ascending oral doses. Int J Clin Pharmacol Ther. 2002;40(5):213–220. doi:10.5414/CPP40213
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.