")
Back to Journals » Cancer Management and Research » Volume 10
Prognostic factors for postoperative survival among patients with rhabdomyosarcoma of the limbs
Authors Ren S , Wang Z, Huang X, Sun L, Shao J, Ye Z
Received 30 May 2018
Accepted for publication 4 August 2018
Published 3 October 2018 Volume 2018:10 Pages 4181—4189
DOI https://doi.org/10.2147/CMAR.S175734
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Antonella D'Anneo
Shihong Ren,1 Zhan Wang,2 Xin Huang,2 Lingling Sun,2 Jinxiang Shao,1 Zhaoming Ye2
1Department of Orthopedics, The First People’s Hospital of Wenling, Wenling 317500, China; 2Department of Orthopedics, Centre for Orthopedic Research, Orthopedics Research Institute of Zhejiang University, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310009, China
Background: Rhabdomyosarcoma (RMS) is a rare malignant soft tissue sarcoma with a poor outcome and unclear prognostic factors. The purpose of this study was to analyze the prognostic postoperative survival factors among patients with RMS of the limbs.
Methods: Postoperative data on patients with RMS of the limbs from 1983 to 2013 were retrieved from the Surveillance, Epidemiology, and End Results (SEER) database of the US National Cancer Institute. Both overall survival (OS) and cancer-specific survival (CSS) were assessed using the Kaplan–Meier method (to obtain OS and CSS curves) and a Cox proportional hazards regression model.
Results: In total, 385 cases were obtained from the SEER database. The 5- and 10-year OS rates of the entire group were 51.5% and 42.2%, respectively. The 5- and 10-year CSS rates were 56.7% and 50.7%, respectively. Univariate analysis revealed that age, location, tumor stage, tumor size, and radiation therapy (RT) were associated with OS. Age, tumor stage, tumor size, and RT were associated with CSS. Age, tumor stage, tumor size, and RT were also independent predictors of both OS and CSS.
Conclusion: Young age, no metastasis at presentation, smaller tumor size, and RT are favorable factors for prolonging survival among patients with RMS of the limbs after surgery.
Keywords: rhabdomyosarcoma, SEER, prognosis, survival
Introduction
Rhabdomyosarcoma (RMS) is a type of tumor formed from embryonic precursors of the striated muscle lineage, due to aberrant differentiation during embryonal development.1 It is the most common soft tissue sarcoma, accounting for >50% of soft tissue sarcomas in childhood and adolescence.2
RMS can affect almost any anatomic site of the body, but it commonly manifests in the head and neck (28%–40%), genitourinary system (25%), and extremities (19%–20%).3–5 RMS has a poor prognosis and unclear prognostic factors. The primary treatment of RMS includes chemotherapy, surgery, and radiation. Identifying prognostic factors of RMS may help in optimizing treatment protocols. Many studies have reported prognostic factors for RMS of the head, neck, and urogenital system.6–13 Oberlin et al14 analyzed prognostic factors of RMS of limbs in children and adolescents from four international cooperative groups, including tumor invasiveness, tumor size, lymph-node involvement, and initial surgery completeness. But prognostic factors for RMS of the limbs after surgery have rarely been reported. Thus, a large population-based study is important for identifying the prognostic factors for postoperative survival among patients with RMS of the limbs.
Materials and methods
Data of patients diagnosed with RMS from 1983 to 2013 were extracted from the Surveillance, Epidemiology, and End Results (SEER) database. The SEER database holds only anonymized data, which are freely available. We used a SEER*Stat (version 8.3.5) Case Listing session to generate a matrix of all individuals diagnosed with RMS in the database. A selection query was designed to retrieve all RMS cases based on the ICD for Oncology, version 3 (ICD-O-3) histology codes. This involved the following: RMS, not otherwise specified (NOS) 8900/3; pleomorphic RMS, adult-type 8901/3; mixed-type RMS 8902/3; embryonal RMS 8910/3; spindle cell RMS 8912/3; and alveolar RMS 8920/3. The site codes were C49.1 (upper limb and shoulder) and C49.2 (lower limb and hip).
All included patients have been diagnosed by histological confirmation based on biopsy or surgical pathology. The other exclusion criteria were as follows: unknown SEER tumor stage, unknown tumor size, and unknown therapeutic information.
SPSS statistical software (version 21.0) and Microsoft Excel 2016 were used to analyze the data. Overall survival (OS) was defined as the time from diagnosis to death from any cause, and cancer-specific survival (CSS) was defined as the time from diagnosis to death specific to the cancer-related diagnosis. Demographic and clinical characteristics in the analysis included age at diagnosis (<20, 20–40, and >40 years), gender, tumor location (upper and lower limbs), decade of diagnosis (before the 2000s and in or after the 2000s), tumor stage (localized, regional, and distant), tumor size (<5, 5–10, and >10 cm), vital status, and radiation therapy (RT). We used “SEER Historic Stage A” to define the tumor stage. “Localized” was defined as an invasive neoplasm confined entirely to the organ of origin, but including intraluminal extension where specified. “Regional” was defined as a neoplasm that had extended 1) beyond the limits of the organ of origin directly into surrounding organs or tissues; 2) into regional lymph nodes via the lymphatic system; or 3) by a combination of extension and regional lymph nodes. “Distant” was defined as a neoplasm that had spread to parts of the body remote from the primary tumor either by direct extension or by discontinuous metastasis (eg, implantation or seeding) to distant organs, tissues, or (via the lymphatic system) to distant lymph nodes.
The Kaplan–Meier method was used to construct the OS and CSS curves. The log-rank test was used to compare survival curves. The effects of demographic, tumor, and treatment variables were compared using log-rank tests for categorical variables. Observations were censored if the patients were alive at the time of the last follow-up. The Cox proportional hazards regression model was used to determine the independent prognostic factors for OS and CSS. Hazard ratios (HRs) with corresponding 95% CIs were used to show the effect of factors on OS and CSS. Differences were deemed statistically significant if P<0.05.
Results
In total, 385 patients diagnosed between 1983 and 2013 were identified from the SEER database. The patient demographics are listed in Table 1. About one-third of the cases were alveolar subtype (143, 37.1%) and only three cases were mixed subtype (0.8%) (Table S1). Altogether, 164 of the patients (42.6%) were aged <20 years, 52 (13.5%) were aged 20–40 years, and 169 (43.9%) were aged >40 years. The study included 161 (41.8%) female and 224 (58.2%) male patients. Approximately three-quarters of the patients (74.3%) were born in or after the 2000s. It was found that 135 tumors (35.1%) occurred in upper limbs and 250 (64.9%) occurred in lower limbs. Based on SEER staging, 194 (50.4%) of the patients had a localized tumor, 125 (32.5%) were at the regional stage, and 66 (17.1%) were at the distant stage. Overall, 103 tumors (26.8%) were <5 cm, 186 (48.3%) were 5–10 cm, and 96 (24.9%) were >10 cm. More than half (62.3%) of the patients received RT. A total of 196 patients (50.9%) died, 140 of them due to RMS-related reasons. The 5- and 10-year OS rates for the entire cohort were 51.5% and 42.2%, respectively. The 5- and 10-year CSS rates were 56.7% and 50.7%, respectively.
 | Table 1 Clinical characteristics of 385 patients with RMS of the limbs after surgery identified in the SEER database from 1983 to 2013 Note: a2000–2013 year. Abbreviations: SEER, Surveillance, Epidemiology, and End Results; OS, overall survival; CSS, cancer-specific survival, RMS, Rhabdomyosarcoma. |
We performed univariate analysis using the log-rank test to analyze prognostic factors (Table 2, Figures 1 and 2). The test revealed that older age was significantly associated with a worse OS and CSS (Table 2, Figures 1A and 2A). Neither decade of diagnosis nor gender was significantly associated with OS, which was also the case for CSS. Tumor location was associated with significant differences in OS (P=0.008), but not in CSS (P=0.072). There were significant differences in both OS and CSS based on the extent of disease at presentation (OS: distant vs localized, P<0.001; distant vs regional, P<0.001; CSS: distant vs localized, P<0.001; distant vs regional, P<0.001). However, there was no significant difference between regional and localized (OS: P=0.292; CSS: P=0.205) (Table 2, Figures 1B and 2B). Smaller tumor size was a favorable prognostic factor for both OS and CSS (OS: >10 vs 5–10 cm, P<0.001; >10 vs <5 cm, P<0.001; CSS: >10 vs 5–10 cm, P<0.001; >10 vs <5 cm, P<0.001; 5–10 vs <5 cm, P=0.032) (Table 2, Figures 1C and 2C). However, for OS, the difference between the 5–10 cm group and the <5 cm group was not significant. In terms of treatment, patients who did not receive RT had worse OS and CSS than patients who underwent RT (P<0.001 and P=0.006, respectively) (Table 2, Figures 1D and 2D).
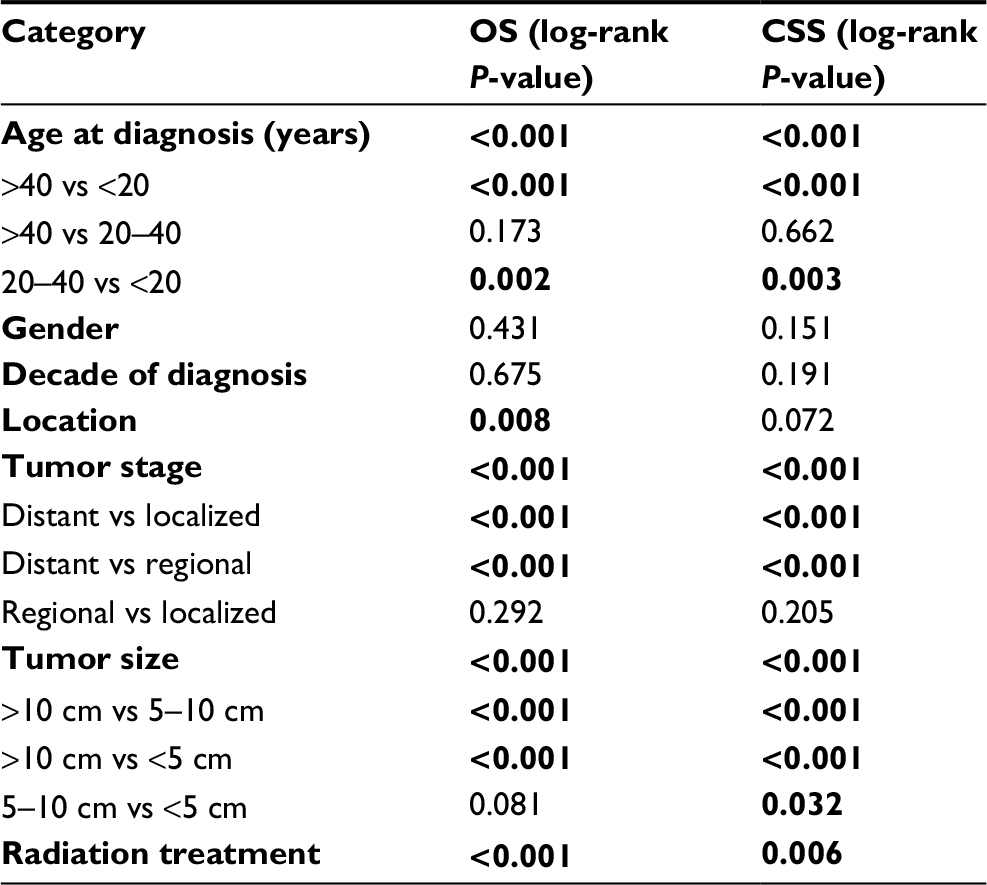 | Table 2 Univariate analysis of variables in patients with RMS of the limbs after surgery using the Kaplan–Meier method Note: Statistically significant values (P<0.05) are indicated in bold. Abbreviations: OS, overall survival; CSS, cancer-specific survival; RMS, Rhabdomyosarcoma. |
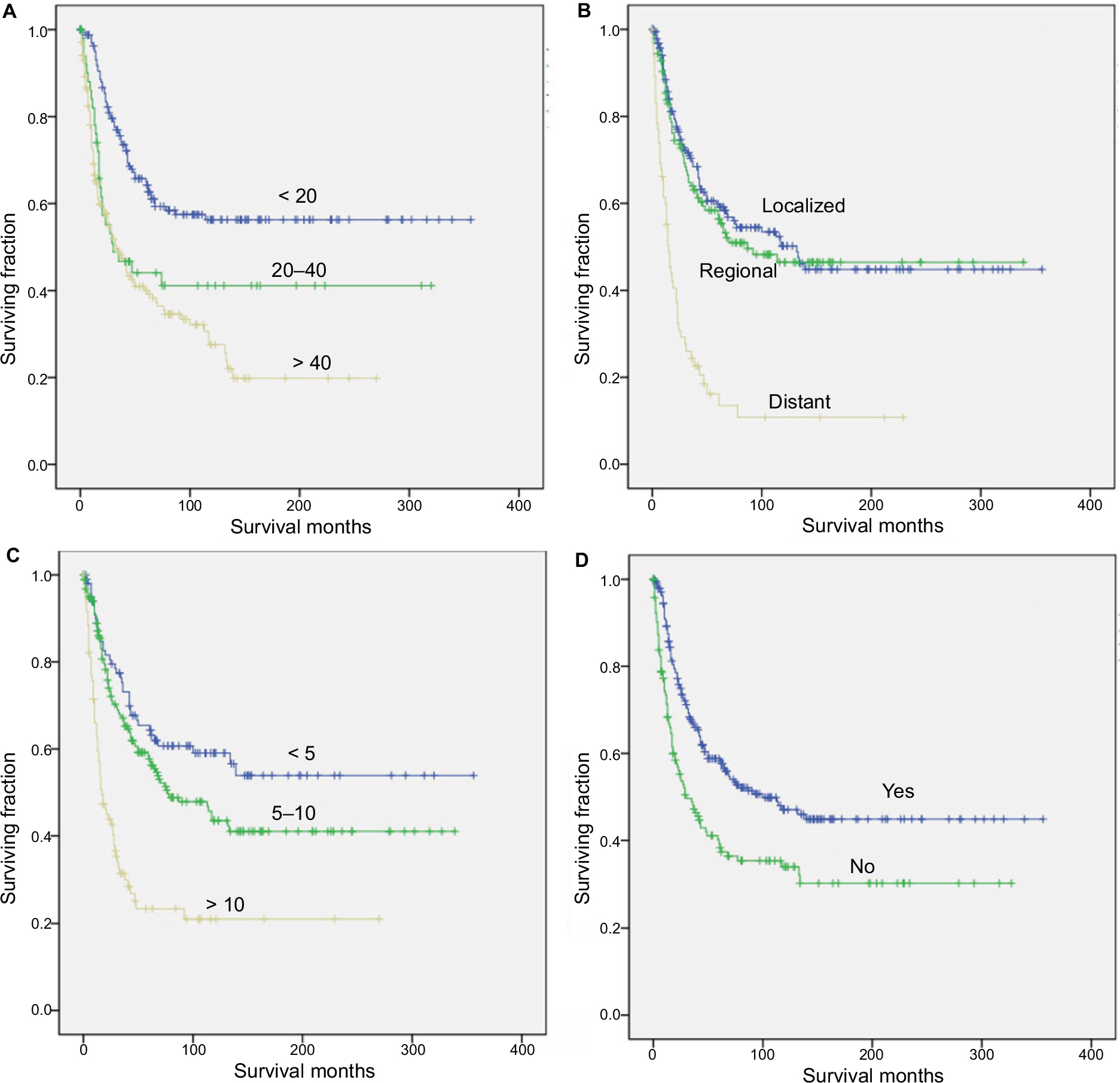 | Figure 1 Kaplan–Meier method estimated OS in patients with RMS of the limbs after surgery, stratified by (A) age at diagnosis (years), (B) tumor stage, (C) tumor size (cm), and (D) radiation treatment. Abbreviations: OS, overall survival; RMS, rhabdomyosarcoma. |
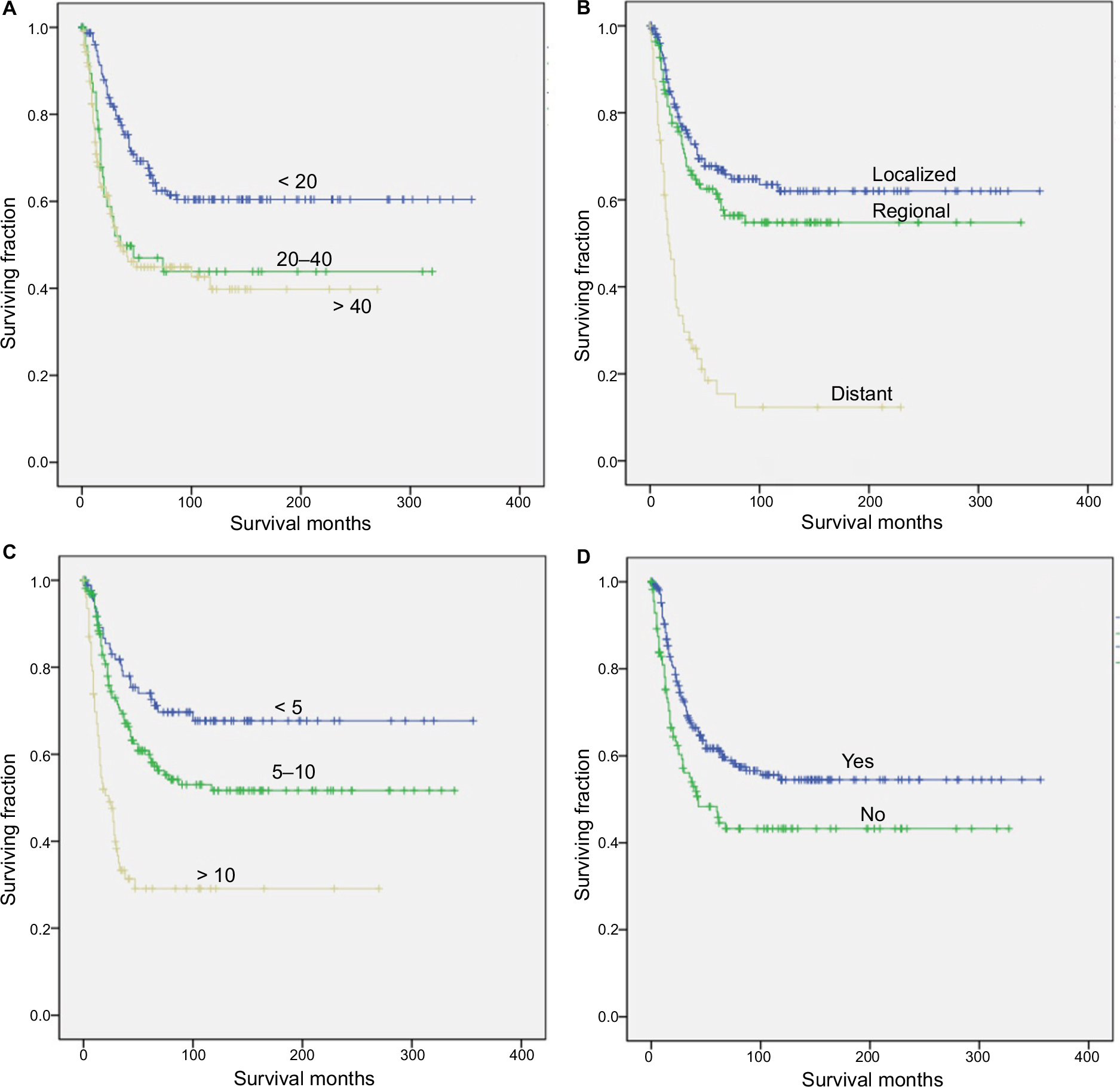 | Figure 2 Kaplan–Meier method estimated CSS in patients with RMS of the limbs after surgery, stratified by (A) age at diagnosis (years), (B) tumor stage, (C) tumor size (cm), and (D) radiation treatment. Abbreviations: CSS, cancer-specific survival; RMS, rhabdomyosarcoma. |
The results of the multivariate analyses for all patients are shown in Table 3. The independent predictors of OS and CSS were age at diagnosis, tumor stage, tumor size, and RT. However, multivariate analysis showed no significant difference in OS by primary tumor location (OS: P=0.327).
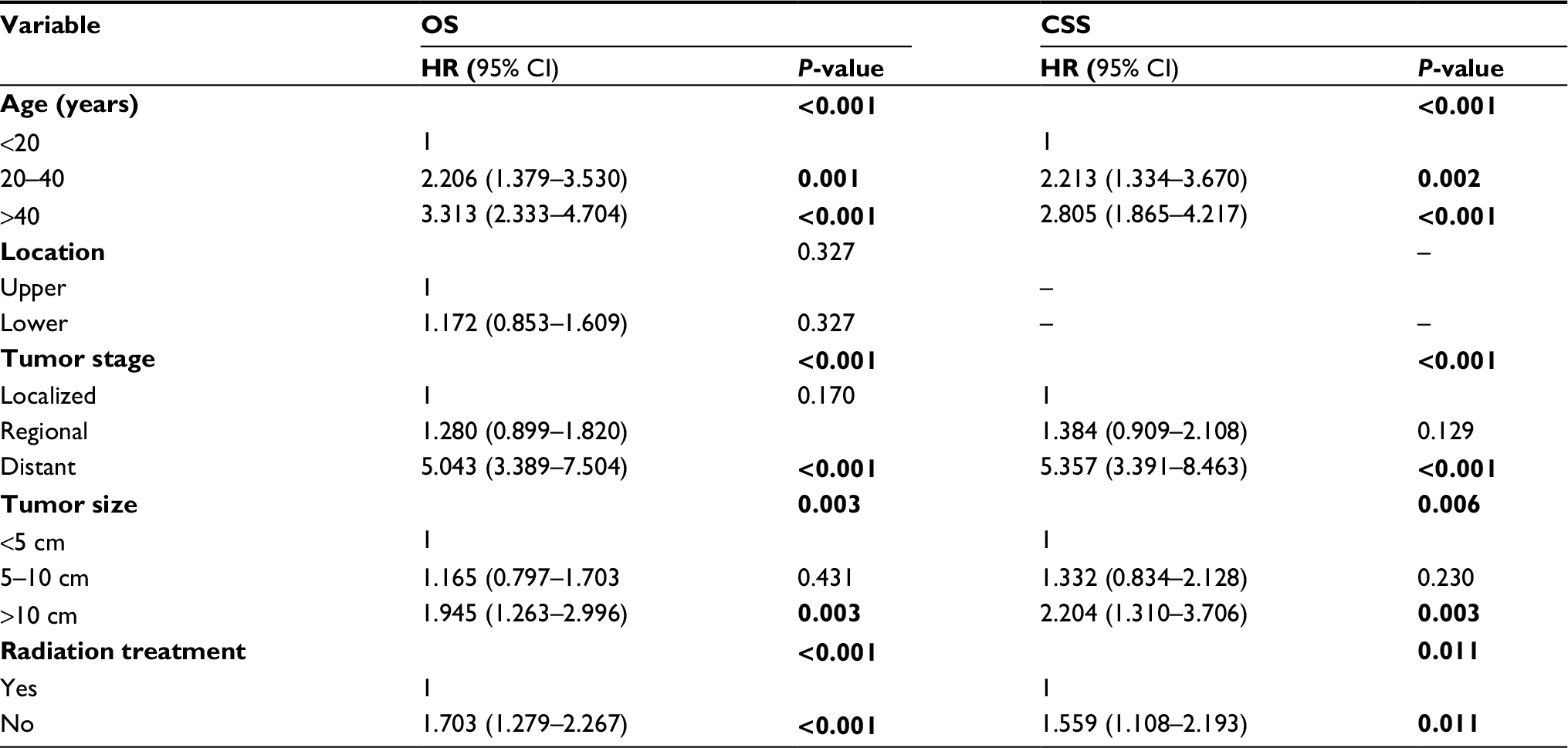 | Table 3 Multivariate analysis for OS and CSS for patients with RMS of the limbs after surgery Note: Statistically significant values (P<0.05) are indicated in bold. Abbreviations: OS, overall survival; CSS, cancer-specific survival; HR, hazard ratio; RMS, rhabdomyosarcoma. |
Discussion
RMS is a rare malignant tumor. It can infiltrate adjacent tissues and metastasize distally. Prognostic factors for RMS of the head, neck, and other sites have been highlighted in many reports. However, there are rarely corresponding studies on RMS of the limbs after surgery. To the best of our knowledge, this study has the largest sample among studies on postoperative survival prognosis among patients with RMS of the limbs. The data were retrieved from the US National Cancer Institute (NCI) SEER database, which is the largest registry of cancer incidence and survival. SEER data are collected in a unified standard manner and are of high quality, which ensures that the SEER cancer registry has a low rate of errors. Furthermore, unlike most analyses in clinical studies, our population-based analysis included all cases of RMS of the limbs after surgery, regardless of whether formal treatment protocols were used, comorbidity, or other prognostic or personal factors. Moreover, the study used multivariate regression analysis to identify possible independent prognostic factors for survival.
In the current study, we retrieved data on 385 cases of RMS of the limbs after surgery from the SEER database between 1983 and 2013. The 5- and 10-year OS rates (51.5% and 42.2%, respectively) of RMS of the limbs in this study were consistent with previous study results (50% and 44%, respectively).4 We found that age at diagnosis, stage of disease, tumor size, and RT were independently associated with patient survival time in terms of both OS and CSS. Older age at diagnosis, metastasis, larger tumor size (>10 cm), and no RT led to a poor prognosis.
Survival by age
Different survival rates by age of diagnosis have been observed in many studies.5–7,11 Sultan et al4 studied 2,600 patients with a diagnosis of RMS and revealed that the outcome for adults is consistently worse than that for children regardless of clinical characteristics. Our study found similar results in that the older the patient, the higher the risk of poor OS. In the multivariate analysis of OS, for patients aged 20–40 years, the HR was 2.206 (P=0.001), and for those aged >40 years, the HR was 3.313 (P<0.001). The reason for the different OS rates remains unknown. Komdeur et al15 observed an increased expression in adult RMS patients of multidrug-resistant proteins, such as lung resistance-related protein, which may explain their worse survival. Another possible explanation is that pediatric RMS is more sensitive to chemotherapy than adult RMS. Stevens et al found that chemotherapy led to complete remission in >93% of children.16 A single-center retrospective analysis found that adults with RMS had a poor outcome, and the outcome was similar when adults adhered to the current guidelines for the treatment of pediatric RMS, but they also found that the rate of response to chemotherapy in adults was similar to the rate typically observed among children.17 Nonetheless, age remains an important postoperative prognostic factor in RMS of the limbs. Our results indicated that patients aged <20 years have better survival.
Survival by gender, tumor location, and decade of diagnosis
Our results were consistent with previous results showing no differences in survival by gender or year of diagnosis. For example, a previous study of RMS indicated that there were no differences in survival by gender or year of diagnosis (1979–2005).18 Punyko et al5 also reported that gender and year of diagnosis (1979–2000) were not found to be significantly associated with survival time among children with RMS.
In addition, a study showed that survival rates for RMS of the female genital tract were not significantly different from RMS in different locations (cervix, uterus, and vagina).9 In contrast, a population-based study showed that mortality rates were higher for children with RMS of the non-orbital head and neck regions, extremities, and all other sites combined compared to those with genitourinary RMS.5 Other studies also reported worse survival rates for RMS at unfavorable sites.4,18 Tumor locations were classified as favorable or unfavorable sites based on criteria used for staging pediatric tumors. The favorable sites included the head and neck (non-parameningeal), genitourinary regions (non-bladder/prostate), and bile duct regions, and all other sites were classified as unfavorable. Some of the above results conflict with each other. Moreover, our univariate analysis results showed a significant difference in OS by tumor location (P=0.008), but the multivariate analysis results for both OS and CSS did not show any significant differences. Thus, whether different RMS locations have an influence on survival time needs further verification.
Survival by tumor stage
Many authors concluded that local and regional control is the most important factor in improving long-term survival.19–21 Turner and Richmon11 reported that the prognosis for RMS of the head and neck is largely dependent on the extent of disease at diagnosis. Yang et al22 showed that the 5-year OS rates of patients with localized tumors (84.0%) and regional disease (72.4%) were better than those for patients with distant metastasis (35.7%). In our study, localized and regional RMS of the limbs also led to a higher postoperative survival than distant RMS, which is consistent with previous findings which showed that distant RMS has an unfavorable prognosis.
Survival by tumor size
For smaller tumors with no evidence of metastasis, surgical extirpation alone may be the definitive treatment.23–25 Unsal et al and Panda et al have affirmed that RMS tumor size >5 cm is a prognostic factor for poor survival.7,26 Another study limited to RMS of the head and neck also supports the above finding.27 Ferrari et al also suggested that initial tumor size was a significant prognostic factor in RMS.28 Our study showed that tumor size >10 cm was a prognostic factor for both poor OS and CSS. The differences between studies may be related to the different site of the primary tumor. Orbital or sinus RMS may have more obvious clinical symptoms when the tumor is smaller. Turner and Richmon found that most orbital tumors (60.6%) presented with localized disease.11 In contrast, when the tumor occurs in the extremities, especially in muscular sites, it can be easily overlooked by patients (especially patients with low economic status) because the clinical symptoms are often negligible (when the main vasculature and nerves in the extremities are not affected).
Survival by RT
A single-center retrospective analysis29 reported that the use of intensity-modulated radiation therapy (IMRT) was significantly associated with reduced local recurrence compared with conventional external-beam RT for primary soft tissue sarcoma of the extremities, despite a preponderance of higher-risk features (especially close/positive margin) in the IMRT group. Stevens reported that systematically using RT as a primary treatment for RMS may increase the rate of local control, but it can result in important long-term problems, particularly in very young children.30 According to the International Rhabdomyosarcoma Study Group, adjuvant RT is recommended for patients with microscopically positive margins, gross residual disease after surgery, distant metastases on initial diagnosis, or alveolar histology.31,32 A population-based study involving 1,578 patients with RMS suggested that the 5-year CSS rate would improve from 48% with surgery alone to 60% with adjuvant RT.33 In a study by Yang et al, RT showed a weak but significant association with prognosis: the 5-year OS was 65.6% in patients with RT compared with 62.7% in those without RT (P=0.045).22 Other studies also confirmed that RT was associated with improved survival.6,34
However, these results conflict with the results of other studies. Lee et al6 documented that RT was not associated with a survival advantage for patients with localized and regional RMS, but only for patients with distant RMS. Perez et al18 came to the same conclusion as Lee et al. A population-based analysis6 of patients with sinonasal RMS also showed that there were no significant differences in outcomes among patients who underwent surgery, RT, or combined treatment.
The impact of therapy is influenced by the location of the primary tumor and the amount of local disease remaining after surgical resection at the time RT is initiated.35 Our results showed that RT led to a favorable prognosis regarding both OS and CSS. There was a significant difference not only in the univariate analyses (P<0.001 and P=0.006, respectively) but also in the multivariate analyses (P<0.001 and P=0.011, respectively).
Strengths and limitations
In our study, we excluded all cases for which the tumor size, stage, and RT status were unknown, to improve the validity of our results. In addition, data from the SEER database offer high statistical power owing to the collection of data from multiple centers, which allows for the investigation of rare tumors such as RMS of the limbs. However, the study has several limitations. These include the fact that there were no data on chemotherapy, surgical margin status, surgical type, nodes status, and RT dose.
Conclusion
Significant prognostic factors that improve postoperative survival among patients with RMS of the limbs include young age, no metastasis at diagnosis, smaller tumor size (<10 cm), and RT. No significant differences in survival were observed for gender, tumor location, or decade of diagnosis. This study may provide patients and clinicians with a reference for treatment options.
Disclosure
The authors report no conflicts of interest in this work.
References
Ananthaneni A, Kuberappa PH, Srinivas GV, Kiresur MA. Alveolar rhabdomyosarcoma of maxilla. J Oral Maxillofac Pathol. 2016;20(1):164. | ||
Rodeberg D, Paidas C. Childhood rhabdomyosarcoma. Semin Pediatr Surg. 2006;15(1):57–62. | ||
Ruiz-Mesa C, Goldberg JM, Coronado Munoz AJ, Dumont SN, Trent JC. Rhabdomyosarcoma in adults: new perspectives on therapy. Curr Treat Options Oncol. 2015;16(6):27. | ||
Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. 2009;27(20):3391–3397. | ||
Punyko JA, Mertens AC, Baker KS, et al. Long-term survival probabilities for childhood rhabdomyosarcoma. Cancer. 2005;103(7):1475–1483. | ||
Lee RJ, Lee KK, Lin T, et al. Rhabdomyosarcoma of the head and neck: impact of demographic and clinicopathologic factors on survival. Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;124(3):271–279. | ||
Unsal AA, Chung SY, Unsal AB, Baredes S, Eloy JA. A Population-Based Analysis of Survival for Sinonasal Rhabdomyosarcoma. Otolaryngol Head Neck Surg. 2017;157(1):142–149. | ||
Nasioudis D, Alevizakos M, Chapman-Davis E, Witkin SS, Holcomb K. Rhabdomyosarcoma of the lower female genital tract: an analysis of 144 cases. Arch Gynecol Obstet. 2017;296(2):327–334. | ||
Kirsch CH, Goodman M, Esiashvili N. Outcome of female pediatric patients diagnosed with genital tract rhabdomyosarcoma based on analysis of cases registered in SEER database between 1973 and 2006. Am J Clin Oncol. 2014;37(1):47–50. | ||
Wu AW, Suh JD, Metson R, Wang MB. Prognostic factors in sinonasal sarcomas: analysis of the surveillance, epidemiology and end result database. Laryngoscope. 2012;122(10):2137–2142. | ||
Turner JH, Richmon JD. Head and neck rhabdomyosarcoma: a critical analysis of population-based incidence and survival data. Otolaryngol Head Neck Surg. 2011;145(6):967–973. | ||
Raney RB, Lawrence W, Maurer HM, et al. Rhabdomyosarcoma of the ear in childhood. A report from the Intergroup Rhabdomyosarcoma Study-I. Cancer. 1983;51(12):2356–2361. | ||
Sanghvi S, Misra P, Patel NR, et al. Incidence trends and long-term survival analysis of sinonasal rhabdomyosarcoma. Am J Otolaryngol. 2013;34(6):682–689. | ||
Oberlin O, Rey A, Brown KL, et al. Prognostic Factors for Outcome in Localized Extremity Rhabdomyosarcoma. Pooled Analysis from Four International Cooperative Groups. Pediatr Blood Cancer. 2015;62(12):2125–2131. | ||
Komdeur R, Klunder J, van der Graaf WT, et al. Multidrug resistance proteins in rhabdomyosarcomas: comparison between children and adults. Cancer. 2003;97(8):1999–2005. | ||
Stevens MC, Rey A, Bouvet N, et al. Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology–SIOP Malignant Mesenchymal Tumor 89. J Clin Oncol. 2005;23(12):2618–2628. | ||
Ferrari A, Dileo P, Casanova M, et al. Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer-Am Cancer Soc. 2003;98(3):571–580. | ||
Perez EA, Kassira N, Cheung MC, et al. Rhabdomyosarcoma in children: a SEER population based study. J Surg Res. 2011;170(2): e243–e251. | ||
Maurer HM, Moon T, Donaldson M, et al. The intergroup rhabdomyosarcoma study: a preliminary report. Cancer. 1977;40(5):2015–2026. | ||
Reilly BK, Kim A, Peña MT, et al. Rhabdomyosarcoma of the head and neck in children: review and update. Int J Pediatr Otorhinolaryngol. 2015;79(9):1477–1483. | ||
Radzikowska J, Kukwa W, Kukwa A, Czarnecka A, Krzeski A. Rhabdomyosarcoma of the head and neck in children. Contemp Oncol. 2015;19(2):98–107. | ||
Yang L, Takimoto T, Fujimoto J. Prognostic model for predicting overall survival in children and adolescents with rhabdomyosarcoma. BMC Cancer. 2014;14:654. | ||
Thompson CF, Kim BJ, Lai C, et al. Sinonasal rhabdomyosarcoma: prognostic factors and treatment outcomes. Int Forum Allergy Rhinol. 2013;3(8):678–683. | ||
Simon JH, Paulino AC, Smith RB, Buatti JM. Prognostic factors in head and neck rhabdomyosarcoma. Head Neck. 2002;24(5):468–473. | ||
Gillespie MB, Marshall DT, Day TA, et al. Pediatric rhabdomyosarcoma of the head and neck. Curr Treat Options Oncol. 2006;7(1):13–22. | ||
Panda SP, Chinnaswamy G, Vora T, et al. Diagnosis and Management of Rhabdomyosarcoma in Children and Adolescents: ICMR Consensus Document. Indian J Pediatr. 2017;84(5):393–402. | ||
Wu Y, Li C, Zhong Y, Guo W, Ren G. Head and neck rhabdomyosarcoma in adults. J Craniofac Surg. 2014;25(3):922–925. | ||
Ferrari A, Miceli R, Meazza C, et al. Comparison of the prognostic value of assessing tumor diameter versus tumor volume at diagnosis or in response to initial chemotherapy in rhabdomyosarcoma. J Clin Oncol. 2010;28(8):1322–1328. | ||
Folkert MR, Singer S, Brennan MF, et al. Local Control Comparison of Conventional and Intensity Modulated Radiation Therapy (IMRT) for Primary Soft-Tissue Sarcomas of the Extremity. Int J Radiat Oncol Biol Phys. 2013;87(2):S63. | ||
Stevens MC. Treatment for childhood rhabdomyosarcoma: the cost of cure. Lancet Oncol. 2005;6(2):77–84. | ||
Wolden SL, Anderson JR, Crist WM, et al. Indications for radiotherapy and chemotherapy after complete resection in rhabdomyosarcoma: A report from the Intergroup Rhabdomyosarcoma Studies I to III. J Clin Oncol. 1999;17(11):3468–3475. | ||
Regine WF, Fontanesi J, Kumar P, et al. Local tumor control in rhabdomyosarcoma following low-dose irradiation: comparison of group II and select group III patients. Int J Radiat Oncol Biol Phys. 1995;31(3):485–491. | ||
Shen W, Sakamoto N, Yang L. Model to predict the survival benefit of radiation for patients with rhabdomyosarcoma after surgery: a population-based study. Int J Oncol. 2014;45(2):549–557. | ||
Mizumoto M, Murayama S, Akimoto T, et al. Preliminary results of proton radiotherapy for pediatric rhabdomyosarcoma: a multi-institutional study in Japan. Cancer Med. 2018;7(5):1870–1874. | ||
Wharam MD, Hanfelt JJ, Tefft MC, et al. Radiation therapy for rhabdomyosarcoma: local failure risk for Clinical Group III patients on Intergroup Rhabdomyosarcoma Study II. Int J Radiat Oncol Biol Phys. 1997;38(4):797–804. |
Supplementary material
 | Table S1 Subtypes of 385 patients with RMS of the limbs after surgery identified in the SEER database from 1983 to 2013 Abbreviations: SEER, Surveillance, Epidemiology, and End Results; RMS, rhabdomyosarcoma; NOS, not otherwise specified. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.