")
Back to Journals » OncoTargets and Therapy » Volume 8
Profile of veliparib and its potential in the treatment of solid tumors
Authors Wagner L
Received 28 April 2015
Accepted for publication 18 June 2015
Published 29 July 2015 Volume 2015:8 Pages 1931—1939
DOI https://doi.org/10.2147/OTT.S69935
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Lars M Wagner
Division of Pediatric Hematology/Oncology, University of Kentucky, Lexington, KY, USA
Abstract: Inhibition of poly(ADP-ribose) polymerase (PARP) is an attractive therapeutic strategy because of the importance of this pathway in restoring DNA damage. Small-molecule inhibitors of PARP appear most effective when used to treat tumors with underlying defects in DNA repair, or when combined with DNA-damaging agents. Veliparib is one of several recently developed oral inhibitors of PARP currently in clinical trials. This review summarizes the pharmacology, mechanisms of action, toxicity, and activity of veliparib seen in clinical trials to date. Also discussed are proposed mechanisms of resistance, potential biomarkers of activity, and issues regarding patient selection and combination therapies that may optimize use of this exciting new agent.
Keywords: veliparib, solid tumors, PARP inhibitor, BRCA
Introduction
The poly(ADP-ribose) polymerase (PARP) family of proteins consists of over 15 different enzymes, which engage in a variety of cellular functions, including cell cycle regulation, transcription, and repair of DNA damage.1 PARP-1 is the most abundant and best characterized protein in this group and is critical to the repair of single-strand DNA breaks through the base excision repair pathway. Effective inhibition of PARP-1 leads to the accumulation of single-strand breaks, which ultimately results in double-strand breaks. Usually such double-strand breaks are repaired by homologous recombination (HR), but in cells with defective HR, PARP inhibition can result in chromosomal instability, cell cycle arrest, and subsequent apoptosis.
The inability of HR to correct double-stranded breaks has been observed in tumors with mutations in the breast cancer-related genes BRCA1 and BRCA2, which code for proteins essential for normal HR function. The use of small-molecule PARP inhibitors to exploit this genetic vulnerability in DNA damage repair is an example of synthetic lethality, in which the simultaneous inhibition of two pathways leads to cell death, whereas blocking either pathway alone is not lethal. Encouraging preclinical results for PARP inhibitors in the treatment of BRCA-mutated tumor cells provided strong rationale for the clinical testing of these agents in patient populations most likely to carry these mutations, such as those with breast or ovarian cancer. This therapeutic strategy has now been validated by the recent US Food and Drug Administration (FDA)-accelerated approval for the PARP inhibitor olaparib as monotherapy to treat patients with BRCA-mutated advanced ovarian cancer who have been treated with three prior lines of chemotherapy.2
This review highlights the development of another PARP inhibitor, veliparib (ABT-888; AbbVie Pharmaceuticals, Chicago, IL, USA). Concepts general to all PARP inhibitors are discussed, with specific attention to how veliparib is being developed in clinical trials.
Biochemistry and pharmacology of veliparib
The complete chemical name of veliparib is 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide, and the chemical structure is shown in Figure 1. Veliparib is able to potently inhibit both PARP-1 and PARP-2, with Kis (inhibitory constants) of 5.2 and 2.9 nmol/L, respectively.3 As seen with many PARP inhibitors, this activity is generally selective, and veliparib does not appear to have substantial effects on other receptors or ion channels at pharmacologically relevant concentrations.
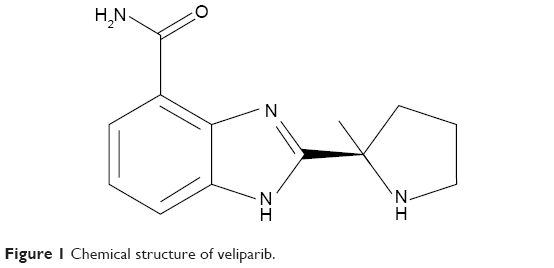 | Figure 1 Chemical structure of veliparib. |
In a 2009 Phase 0 clinical trial of veliparib in adults with advanced cancers, patients received single oral doses of 10, 25, or 50 mg veliparib.4 Veliparib showed good oral bioavailability, with peak absorption between 0.5 and 1.5 hours, and a maximum concentration of 0.45 μM after a single dose of 50 mg. Significant inhibition of PARP levels in both tumor tissue and peripheral blood mononuclear cells was observed 3–6 hours after administration, with recovery at 24 hours in both preclinical models and patients. These findings led to the recommendation of twice-daily (bid) administration, in order to ensure adequate PARP inhibition over longer periods of time.
Population modeling from 325 adult patients receiving veliparib bid in one of four clinical trials showed that this drug’s pharmacokinetics are best described with a one-compartment model with first-order absorption and elimination.5 Veliparib is predominantly eliminated in the urine as the unchanged parent drug. This process is facilitated by drug uptake via the organic cation transporter OCT2 into the renal tubule. Although mostly eliminated by renal clearance, an estimated 13% of veliparib also undergoes hepatic metabolism by CYP2D6,6 producing the lactam metabolite M8, which is a much weaker PARP inhibitor than the parent compound.7
Dose adjustments of veliparib on the basis of body size, sex, age, ethnicity, or liver function do not appear routinely necessary. However, creatinine clearance can affect veliparib exposures, and modifications should be considered in patients with impaired renal function. Patients who have certain CYP2D6 polymorphisms, or who are receiving coadministration of OCT2 inhibitors such as cimetidine, may also be at risk for poor clearance and a clinically relevant increase in veliparib exposure.8 However, it is felt that veliparib has a generally low likelihood for meaningful drug–drug interactions.9
Mechanisms of action
A comprehensive understanding of the possible mechanisms of action of PARP inhibitors helps provide rationale for patient selection and study design. BRCA-mutated tumors are well established to have inadequate DNA repair machinery, and so be sensitive to PARP inhibition through the concept of synthetic lethality. Importantly, HR deficiencies can also be seen in other contexts as well, including tumors with defects in the DNA damage sensors ATM (ataxia-telangiectasia mutated) and ATR (ATM- and RAD3-related protein),10 PTEN mutations,11 or defects in the Fanconi repair pathway.12 This information has been used to expand the rationale for treatment to include tumors that may have limited capacity for DNA repair (also termed “BRCAness”) that could predict the activity of PARP inhibitors.
The genetic knockout of PARP-1 substantially impairs DNA repair following damage from radiation or cytotoxic chemotherapy agents;13 accordingly, investigators have combined PARP inhibitors with conventional cancer treatments known to damage DNA. As will be discussed, this approach has been or is being investigated with therapeutic irradiation as well as a wide variety of cytotoxic agents, including temozolomide, cisplatin, carboplatin, doxorubicin, paclitaxel, and topotecan.
In addition to these mechanisms of action, PARP inhibitors may also poison DNA by stabilizing PARP-1 and 2 at sites of DNA damage, generating complexes that may be even more toxic than the unrepaired single-strand breaks which result from PARP inhibition. This concept was termed “PARP trapping” by Murai et al14 and its characterization impacted PARP inhibitor development in two important ways. First, this work showed that pathways other than HR may be essential for repairing the PARP–DNA complexes, therefore providing rationale for treating tumors with defects in the FEN1, polymerase β, postreplication repair, and Fanconi anemia pathways. Secondly, these investigators demonstrated a difference between PARP inhibitors in the ability to trap PARP, despite similarities in the ability to inhibit PARP catalytic activity. In this regard, veliparib was inferior to both niraparib and olaparib in trapping PARP. This finding may be related to the period of time that PARP is “trapped” onto the DNA, and it could have implications for dosing and toxicity of the various agents.15
Preclinical activity
Donawho et al3 produced one of the earliest and most complete assessments of the preclinical activity of veliparib, and showed that veliparib potentiated the activity of temozolomide, cisplatin, carboplatin, and cyclophosphamide in a variety of tumors, including melanoma, glioma, lymphoma, colon carcinoma, and breast carcinoma. They also demonstrated that veliparib crosses the blood–brain barrier, providing further rationale for its pairing with temozolomide to treat intracranial tumors. Further, veliparib also potentiated the effect of fractionated radiation through its impairment of both single- and double-strand break repair pathways.
Additional studies have built on these earlier preclinical observations. Palma et al16 expanded the scope of tumors and showed combinatorial activity of veliparib and temozolomide in multiple types of lung cancer as well as in pancreatic and prostate cancer xenografts. Interestingly, activity was demonstrated in models that had acquired resistance to single-agent temozolomide, and conventional measures of temozolomide resistance such as expression of methylguanine methyltransferase (MGMT) or mismatch repair proteins did not correlate with the degree of sensitivity to the combination of temozolomide + veliparib. Additional work by Palma et al16 showed that potentiation of temozolomide toxicity was dose-dependent and that extended veliparib scheduling was not more beneficial than limiting administration to be simultaneous with 5-day courses of temozolomide.17
Lin et al11 further explored genetic predictors of veliparib in glioblastoma models, demonstrating that veliparib activity may be greatest in cells with PTEN deficiency, which characterizes up to one-third of gliomas. They also demonstrated the importance of using doses in laboratory experiments that are clinically relevant and can achieve serum concentrations that are feasible in humans, which is a key point also emphasized by other investigators.18
As mentioned above, PARP inhibitors appear to work in different ways, including interfering with the repair of DNA breaks as well as by stabilizing the PARP–DNA complex and inducing cytotoxicity through PARP trapping. In a recent article, Murai et al19 reported that synergy with conventional cytotoxic agents can be affected by which mechanism of action is greatest for a particular inhibitor. For example, while olaparib and veliparib have similar inhibitory effects on PARP catalytic activity, the degree of PARP trapping is greater with olaparib. This mechanism appears to be particularly important when a PARP inhibitor is combined with temozolomide, as the combination of olaparib + temozolomide has greater in vitro activity than that of veliparib + temozolomide. However, both inhibitors showed robust synergy in combination with camptothecin, suggesting that activity with that particular combination may be mediated more by downregulating direct PARP catalytic activity.
Several studies have also reported the radiosensitizing effect of veliparib in a variety of solid tumors,20–23 including under the hypoxic conditions often found in larger tumors.24 In cultured glioblastoma cells, veliparib enhanced the lethality of radiation, especially in combination with temozolomide. Interestingly, this effect again was seen irrespective of the MGMT status of the tumor cells.25 Similar combinatorial efficacy has also been seen with veliparib and radiation combined with oxaliplatin, 5-fluorouracil, or irinotecan in cultured colorectal carcinoma cells.26
There have been a limited number of direct preclinical comparisons between PARP inhibitors. In addition to the studies reported above, Shen et al27 reported that the newer-generation PARP inhibitor BMN 673 exhibited selective cytotoxicity and elicited DNA repair biomarkers at much lower concentrations than olaparib or veliparib. A further study suggests that PARP inhibitors may vary in their “off-target” effects, and this may significantly impact their efficacy against certain tumor types. For example, Jelinic and Levine28 showed that olaparib reduced DNA damage repair activity via G2 cell cycle arrest in a p53-dependent manner, an effect not seen with veliparib.
In summary, the preclinical studies provide rationale for various clinical applications, including the targeting of specific tumor types and possible therapeutic combinations. These studies also provide some insight into possible mechanisms of action and the relative efficacy of different agents, but are ultimately limited somewhat by the artificial nature of the preclinical models used. Only through rigorous clinical trials will the ultimate utility, or futility, of a particular agent be decided.
Clinical trials
Veliparib has already been studied in a variety of Phase I and II trials, and currently there are five Phase III trials ongoing. Results from several reported studies are summarized in Table 1. To date, there have been no head-to-head clinical studies of different PARP inhibitors, which therefore limits the assessment of how veliparib compares to the six other agents currently in clinical trials.
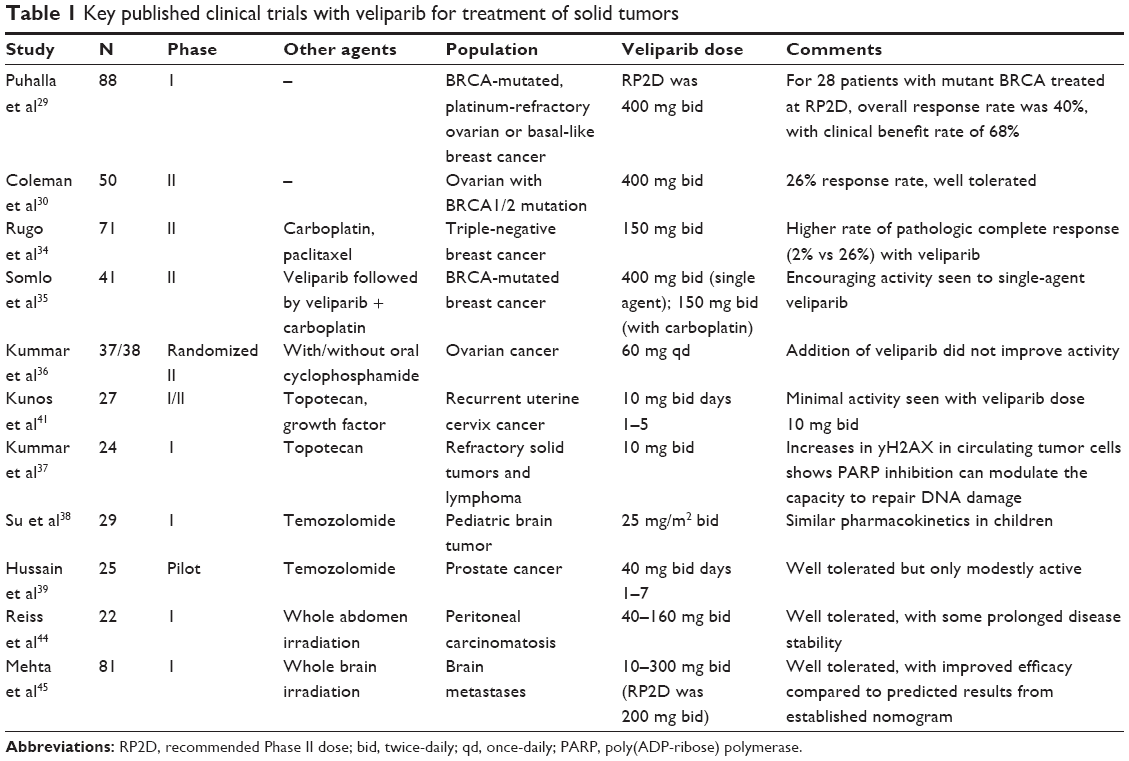 | Table 1 Key published clinical trials with veliparib for treatment of solid tumors |
Dose-finding and toxicity of single-agent trials of veliparib
Puhalla et al29 have reported in abstract form the Phase I trial of veliparib in adults with relapsed cancers, with doses ranging from 50 to 500 mg bid being studied. They defined the recommended Phase II dose (RP2D) of single-agent veliparib as 400 mg bid. Given that veliparib has been studied using tablet strengths of 10 and 40 mg, patients receiving the RP2D dose will take up to 20 tablets bid, which compares to 16 tablets bid of olaparib at its RP2D. The toxicity of this dose is best estimated from a Phase II trial in 50 patients with ovarian cancer conducted by Coleman et al.30 In that study, the most common side effects were gastrointestinal, with half of the patients having nausea of at least grade 2 (46%) or grade 3 (4%) severity. An additional 18% of these patients had grade 2 vomiting. In general, gastrointestinal toxicity was seen primarily in the earlier courses, and was manageable with aggressive antiemetics, delays, and dose reductions. Fatigue was seen in one-third of patients, but was generally grade 2. Myelosuppression was modest, with only 2% having either grade 3–4 neutropenia or thrombocytopenia. The median dose intensity was 78%. The overall toxicity profile seen with veliparib is somewhat similar to that reported in Phase II trials of other PARP inhibitors such as olaparib. For example, in a recent trial of 46 women with ovarian cancer treated with single-agent olaparib, grade 3 fatigue was seen in 11% of patients.31 While three-fourths of patients experienced nausea, only 26% of patients had grade 2 nausea, and grade 3 nausea was not reported. Serious late effects such as secondary leukemia and myelodysplastic syndrome have occasionally been observed following treatment with PARP inhibitors,32 although the extensive pretreatment of patients included in these studies makes attribution of this complication very difficult. However, given that double-strand breaks may build up in normal tissues following treatment with PARP inhibitors over time,33 continued surveillance for second malignancies is reasonable.
Combination trials using veliparib and cytotoxic chemotherapy have used either the full RP2D as above, or lower dosing, depending in part on the expected toxicity related to the conventional agent. This has led to a wide range of doses being studied, as noted in Table 1. In general, no unusual toxicities have been encountered in these trials to date, although myelosuppression may be enhanced when veliparib is combined with a drug known to cause this effect.
Activity of veliparib in clinical trials
The single-agent Phase I trial conducted by Puhalla et al29 involved 88 patients, and was designed to enrich the population with patients more likely to respond to PARP inhibitors. Eligibility criteria included patients with BRCA-mutated tumors as well as those with BRCA-like tumors, such as serous ovarian cancer and basal-like breast cancer. At the RP2D, 28 BRCA-mutated patients were evaluable, and the response rate and clinical benefit rate (complete + partial responses + stable disease) were 40% and 68%, respectively. This compares to 4% and 38%, respectively, of patients with tumors wild-type for BRCA.
Coleman et al30 then performed a Phase II study using the same dose of 400 mg veliparib bid in patients with BRCA-mutated ovarian cancer. Of the 50 evaluable patients, 30 (60%) were platinum resistant. The study was designed to identify with 90% power a response rate of 25%. For all patients, the response rate was 26%, thus meeting the predefined definition of activity in this multicenter prospective study. For platinum-resistant and platinum-sensitive patients, the response rate was 20% and 35%, respectively, (P=0.33).
Another encouraging Phase II result came from the I-SPY 2 trial for patients with triple-negative breast cancer.34 Patients in the experimental group received veliparib plus carboplatin and paclitaxel, while the control group was assigned to standard paclitaxel followed by anthracycline chemotherapy. Women in the veliparib group were twice as likely to have a pathologic complete response compared to those receiving standard therapy (52% vs 26%). Researchers then used this data to calculate a 92% Bayesian predictive probability that the veliparib regimen would be statistically superior to standard therapy alone for women with triple-negative disease in a Phase III trial enrolling 300 patients.
Additional studies combining veliparib with conventional chemotherapy agents have also been reported. A multicenter Phase II study recently reported in abstract form by Somlo et al35 involved patients with metastatic BRCA-mutated breast cancer. Patients received veliparib 400 mg bid daily until progression, at which time carboplatin was added and the veliparib dose reduced to 150 mg bid. A partial response rate of 20% was seen in patients receiving four cycles of single-agent veliparib, and larger trials of veliparib are planned both alone and in combination with chemotherapy.
Unfortunately, compelling clinical activity has not been demonstrated in all trials to date. In a randomized Phase II trial conducted by Kummar et al36 ovarian cancer patients received the combination of veliparib 60 mg once daily together with daily oral cyclophosphamide, which had previously been established as the RP2D based on an earlier Phase I trial of this combination.37 Control patients received cyclophosphamide alone. While the combination was well tolerated, there was no improvement seen from the addition of this dose of veliparib to metronomic administration of cyclophosphamide.
The combination of veliparib and temozolomide has been described, although many reports are preliminary. Myelotoxicity can be considerable with this combination, and so the dose of veliparib is often as low as one-tenth of the usual single-agent dose. Veliparib doses of 40 mg bid (and an equivalent dose of 25 mg/m2 in a pediatric trial) together with temozolomide 150 mg/m2/day for 5 days appear tolerable and are associated with some extent of disease stabilization in glioma38 and prostate cancer,39 although minimal activity was observed for those with refractory hepatocellular carcinoma.40 Other combinations with topotecan have been reported41,42 and continue to be under investigation (Table 2).
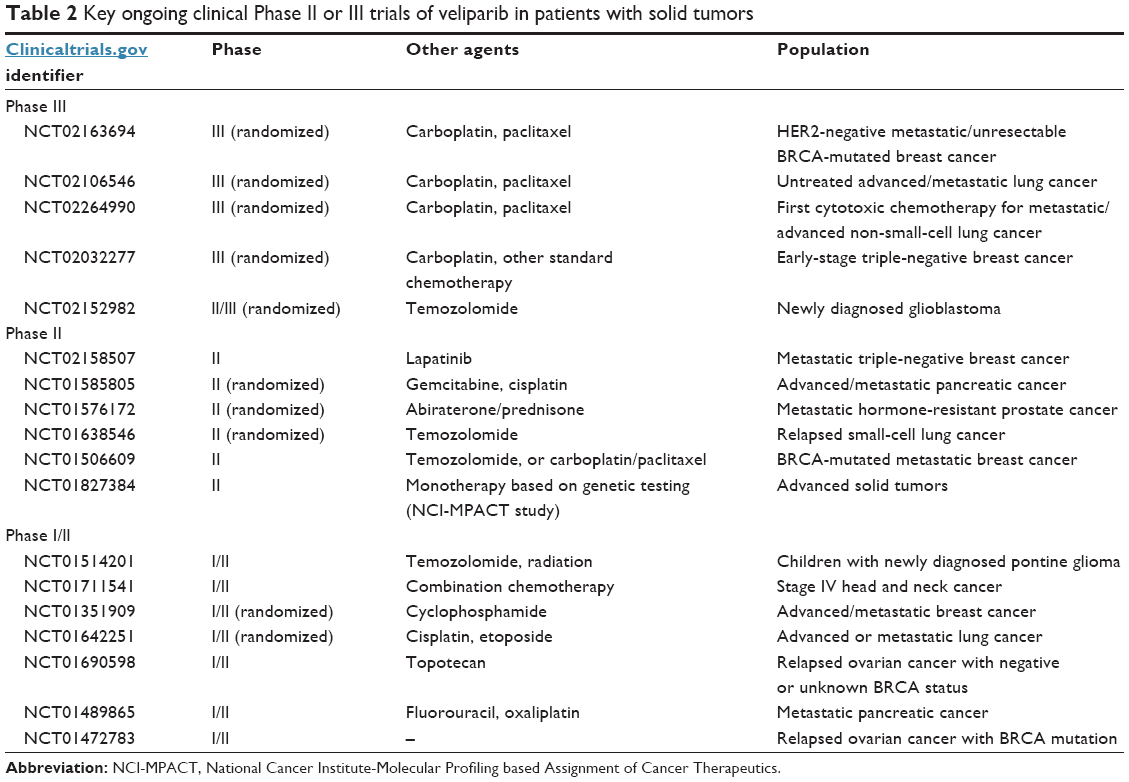 | Table 2 Key ongoing clinical Phase II or III trials of veliparib in patients with solid tumors |
In regard to combination studies, a recent preclinical assessment of PARP inhibitors combined with temozolomide and irinotecan to treat an orthotopic mouse model of Ewing sarcoma may possibly inform further decisions about dosing and combination with chemotherapy agents.15 PARP inhibitors are an attractive option for the treatment of Ewing sarcoma because the characteristic EWS–FLI1 fusion protein that drives tumor growth interacts with PARP-1 through a proposed positive feedback loop,43 which may make Ewing sarcoma cells particularly sensitive to PARP inhibitors in vitro. Although limited single-agent activity in the orthotopic model was seen from any of the three agents tested (olaparib, BMN-673, or veliparib), there was synergy with the combined use of PARP inhibitors + temozolomide, and especially with the further addition of irinotecan.15 Interestingly, veliparib was the least active of the three PARP inhibitors at the dosages used, although later testing of veliparib at higher doses demonstrated both tolerability as well as similar efficacy to both olaparib and BMN-673. Although the previous clinical studies mentioned above had tried to maximize the temozolomide dose while escalating veliparib dose,38–40 the Ewing sarcoma data would suggest that the opposite should be done, at least for that tumor type. It is not clear whether this strategy should be employed when treating other cancers, although these interesting results do raise questions when combinatorial therapies fail to produce the desired level of activity.
Finally, some early trials have investigated the combination of veliparib and therapeutic irradiation. In the Phase I setting, veliparib was studied with whole abdominal radiation in patients with peritoneal carcinomatosis.44 The highest studied dose of veliparib (160 mg bid) was well tolerated, with some suggestion of disease stability seen. When given in combination with whole-brain radiotherapy for patients with metastatic cancer, Mehta et al45 reported the dose of 200 mg bid as the RP2D.
Ongoing clinical trials
As seen in Table 2, clinicaltrials.gov lists at least 19 Phase II or III trials using veliparib, currently recruiting patients. These studies include such cancers as breast, prostate, head and neck, lung, pancreatic, ovarian, colorectal, and glioma. While many of these studies couple veliparib with conventional cytotoxic chemotherapy, some involve combination with radiation or other targeted agents such as lapatinib. The five open Phase III trials are focused on lung cancer, breast cancer, and glioblastoma. In addition to these, there are multiple Phase I trials studying an even broader array of combinations and tumor types.
Mechanisms of resistance
Although exciting activity has been seen with the use of PARP inhibitors in treating BRCA-deficient tumors, some patients still do not respond initially or develop acquired resistance with continued treatment. There are likely several potential mechanisms that may explain resistance in these patients. First, there may be secondary genetic and/or epigenetic events that restore functional HR in tumors that were once HR deficient.46 Secondary mutations that restore BRCA protein function and lead to cisplatin resistance have been reported in patients with recurrent ovarian cancer,47 and such secondary mutations have been seen in patients who initially respond but then develop resistance to olaparib.48
Other potential mechanisms of resistance include somatic mutations of the p53 binding protein TP53BP1,49 which can result in partial restoration in HR. Drug efflux through transporters such as the multidrug resistance protein 1 (P-glycoprotein) has also been implicated in resistance, with some suggestion that cotreatment with medications to block P-glycoprotein can help reverse resistance to PARP inhibitors.50 Finally, loss or even reduction of PARP1 expression may also be associated with acquired resistance.51 Further prospective studies of all of these potential mechanisms may ultimately help identify which patients are most likely to benefit from PARP inhibition. It is unclear at this point whether there are specific mechanisms of resistance that differ between individual PARP inhibitors, and no mechanism to date appears necessarily unique to veliparib.
Potential biomarkers
As discussed above, the hallmark of sensitivity to PARP inhibition is deficient DNA repair. The most compelling clinical benefit to date in single-agent studies has been in trials selecting for patients whose tumors have either confirmed or suspected HR deficiencies, such as BRCA mutations, patients with BRCA-like tumors such as basal- or triple-negative breast cancer, or patients who are platinum sensitive. However, it is clear that a subset of ovarian and breast cancer patients who lack BRCA mutations can respond to PARP inhibitors, and so there is no absolute correlation between these predictors and clinical response.29,52 Other specific genes involved in the DNA damage response, such as HPIβ, have also been reported as putative biomarkers of sensitivity to veliparib, either given alone or in combination with chemotherapy.53 Given the complexity of the DNA repair process and the complicating factor of tumor heterogeneity, the search for genetic biomarkers to predict sensitivity to PARP inhibitors remains quite complicated, such that single biomarker models may not ultimately prove beneficial.54
PARP expression level and/or PARP activity in tumor tissue may also play a role in determining the sensitivity to PARP inhibitors,51 and they are being prospectively studied in clinical trials. Elevated levels of PAR-related proteins as assessed by Western blotting or immunohistochemistry have also been shown to predict sensitivity of human cancer cells to PARP inhibitors55 and could be another avenue of investigation in clinical trials.
In acute myeloid leukemia cells, BRCA1 protein levels inversely correlate with PARP inhibitory activity, with the majority of cell lines having low BRCA1 levels, presumably due to posttranscriptional regulatory mechanisms.56 This raises the possibility that immunostaining of tumors for BRCA1 could potentially be a useful biomarker, although this approach has not yet been reported in a clinical trial. One biomarker combination predicted to be exquisitely sensitive to PARP inhibitors is high 53BP1 expression coupled with methylation of BRCA1, although these findings were only noted in a small subset of patients with triple-negative breast cancers.57 It is hoped that further molecular characterization of tumors using genetic profiling techniques will identify biomarkers that can be validated in clinical trials, and this remains a focus of continued research.
Conclusions and challenges ahead
PARP inhibitors represent an exciting demonstration of the potential for targeted therapy and genetic selection of patients. The recent FDA approval of olaparib, and the encouraging data from clinical trials of related compounds such as veliparib, suggests the worthiness of pursuing this therapeutic strategy. Many questions remain regarding the use of these agents, including their proper sequence in treatment. For example, in ovarian cancer, there is debate about whether these targeted agents should be used before or after platinum chemotherapy, in combination with conventional cytotoxic agents, or as maintenance therapy for high-risk, genetically susceptible patients following standard treatment.58 These issues will be better clarified in the multiple Phase II and III trials, which are already underway.
Although the PARP inhibitors now in clinical trials have shown some preclinical differences, especially in the degree of PARP trapping, the true clinical significance of these differences is not yet clear. There have been no head-to-head clinical trials of different agents in the same class, and comparisons between trials are complicated. As noted above, some preclinical studies suggest that veliparib is not the most robust PARP inhibitor in its class, and in fact there is now a clinical trial of BMN-673 open for patients with BRCA1/2-mutated ovarian cancer who have failed other PARP inhibitors (clinicaltrials.gov identifier NCT02326844). Nevertheless, the activity and tolerability of veliparib seen in early phase studies is exciting, and the likelihood of FDA approval will hinge on the results of the ongoing Phase III trials. Whether more than one targeted agent in a class will receive licensure, as occurred with the EGFR inhibitors erlotinib and gefitinib, remains to be seen.
Substantial challenges lie ahead for the further development of veliparib. For the translational scientist, the identification of reliable biomarkers will be critical for the success of this targeted agent. For the clinical scientist, opportunities exist for expanding veliparib treatment for tumors beyond those already studied, including for Ewing sarcoma,15 lymphoma,42 and even leukemia.59 Finally, thoughtful trial design regarding the dosing and sequence of veliparib and its combination with radiation or other chemotherapy agents will be necessary to realize the full potential of this drug.
Disclosure
The author reports no conflicts of interest in this work.
References
Basu B, Sandhu SK, de Bono JS. PARP inhibitors: mechanism of action and their potential role in the prevention and treatment of cancer. Drugs. 2012;72(12):1579–1590. | ||
Olaparib approved for advanced ovarian cancer. Cancer Discov. 2015;5(3):218. | ||
Donawho CK, Luo Y, Luo Y, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13(9):2728–2737. | ||
Kummar S, Kinders R, Gutierrez ME, et al. Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol. 2009;27(16):2705–2711. | ||
Salem AH, Giranda VL, Mostafa NM. Population pharmacokinetic modeling of veliparib (ABT-888) in patients with non-hematologic malignancies. Clin Pharmacokinet. 2014;53(5):479–488. | ||
Li X, Delzer J, Voorman R, de Morais SM, Lao Y. Disposition and drug-drug interaction potential of veliparib (ABT-888), a novel and potent inhibitor of poly(ADP-ribose) polymerase. Drug Metab Dispos. 2011;39(7):1161–1169. | ||
Penning TD, Zhu GD, Gandhi VB, et al. Discovery of the Poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the treatment of cancer. J Med Chem. 2009;52(2):514–523. | ||
Li J, Kim S, Sha X, Wiegand R, Wu J, LoRusso P. Complex disease-, gene-, and drug-drug interactions: impacts of renal function, CYP2D6 phenotype, and OCT2 activity on veliparib pharmacokinetics. Clin Cancer Res. 2014;20(15):3931–3944. | ||
Kikuchi R, Lao Y, Bow DA, et al. Prediction of clinical drug-drug interactions of veliparib (ABT-888) with human renal transporters (OAT1, OAT3, OCT2, MATE1, and MATE2K). J Pharm Sci. 2013;102(12):4426–4432. | ||
McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–8115. | ||
Lin F, de Gooijer MC, Roig EM, et al. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin Cancer Res. 2014;20(10):2703–2713. | ||
Duan W, Gao L, Aguila B, Kalvala A, Otterson GA, Villalona-Calero MA. Fanconi anemia repair pathway dysfunction, a potential therapeutic target in lung cancer. Front Oncol. 2014;4:368. | ||
Shall S, de Murcia G. Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat Res. 2000;460(1):1–15. | ||
Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599. | ||
Stewart E, Goshorn R, Bradley C, et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep. 2014;9(3):829–841. | ||
Palma JP, Wang YC, Rodriguez LE, et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. 2009;15(23):7277–7290. | ||
Palma JP, Rodriguez LE, Bontcheva-Diaz VD, et al. The PARP inhibitor, ABT-888 potentiates temozolomide: correlation with drug levels and reduction in PARP activity in vivo. Anticancer Res. 2008;28(5A):2625–2635. | ||
Gupta SK, Mladek AC, Carlson BL, et al. Discordant in vitro and in vivo chemopotentiating effects of the PARP inhibitor veliparib in temozolomide-sensitive versus -resistant glioblastoma multiforme xenografts. Clin Cancer Res. 2014;20(14):3730–3741. | ||
Murai J, Zhang Y, Morris J, et al. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther. 2014;349(3):408–416. | ||
Schaefer NG, James E, Wahl RL. Poly(ADP-ribose) polymerase inhibitors combined with external beam and radioimmunotherapy to treat aggressive lymphoma. Nucl Med Commun. 2011;32(11):1046–1051. | ||
Nowsheen S, Bonner JA, Yang ES. The poly(ADP-ribose) polymerase inhibitor ABT-888 reduces radiation-induced nuclear EGFR and augments head and neck tumor response to radiotherapy. Radiother Oncol. 2011;99(3):331–338. | ||
Efimova EV, Mauceri HJ, Golden DW, et al. Poly(ADP-ribose) polymerase inhibitor induces accelerated senescence in irradiated breast cancer cells and tumors. Cancer Res. 2010;70(15):6277–6282. | ||
Albert JM, Cao C, Kim KW, et al. Inhibition of poly(ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin Cancer Res. 2007;13(10):3033–3042. | ||
Liu SK, Coackley C, Krause M, Jalali F, Chan N, Bristow RG. A novel poly(ADP-ribose) polymerase inhibitor, ABT-888, radiosensitizes malignant human cell lines under hypoxia. Radiother Oncol. 2008;88(2):258–268. | ||
Barazzuol L, Jena R, Burnet NG, et al. Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiat Oncol. 2013;8:65. | ||
Shelton JW, Waxweiler TV, Landry J, et al. In vitro and in vivo enhancement of chemoradiation using the oral PARP inhibitor ABT-888 in colorectal cancer cells. Int J Radiat Oncol Biol Phys. 2013;86(3):469–476. | ||
Shen Y, Rehman FL, Feng Y, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19(18):5003–5015. | ||
Jelinic P, Levine DA. New insights into PARP inhibitors’ effect on cell cycle and homology-directed DNA damage repair. Mol Cancer Ther. 2014;13(6):1645–1654. | ||
Puhalla S, Beumer JH, Pahuja S, et al. Final results of a phase 1 syudy of single-agent veliparib in patient swith either BRCA1/2-mutated cancer, platinum-refractory ovarian, or basal-like breast cancer [Abstract 2570]. J Clin Oncol. 2014;32:5s. | ||
Coleman RL, Sill MW, Bell-McGuinn K, et al. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation – An NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. Epub 2015 Mar 24. | ||
Liu JF, Barry WT, Birrer M, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014;15(11):1207–1214. | ||
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–250. | ||
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. | ||
Rugo HS, Olopade O, DeMichele A, et al. Veliparib/carboplatin plus standard neoadjuvant therapy for high-risk breast cancer: first efficacy results from the I-SPY 2 trial. Cancer Res. 2013;73(24 Suppl):Abstract S5-02. | ||
Somlo G, Frankel PH, Luu TH, et al. Phase II trial of single-agent PARP inhibitor ABT-888 (veliparib) followed by post-progression therapy of veliparib with carboplatin in patients with BRCA-associated metastatic breast cancer [Abstract 1021]. J Clin Oncol. 2014;32:5s. | ||
Kummar S, Oza AM, Fleming GF, et al. Randomized trial of oral cyclophosphamide and veliparib in high-grade serous ovarian, primary peritoneal, or fallopian tube cancers, or BRCA-mutant ovarian cancer. Clin Cancer Res. 2015;21(7):1574–1582. | ||
Kummar S, Ji J, Morgan R, et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res. 2012;18(6):1726–1734. | ||
Su JM, Thompson P, Adesina A, et al. A phase I trial of veliparib (ABT-888) and temozolomide in children with recurrent CNS tumors: a pediatric brain tumor consortium report. Neuro Oncol. 2014;16(12):1661–1668. | ||
Hussain M, Carducci MA, Slovin S, et al. Targeting DNA repair with combination veliparib (ABT-888) and temozolomide in patients with metastatic castration-resistant prostate cancer. Invest New Drugs. 2014;32(5):904–912. | ||
He AR, Tesfaye A, Smith D, et al. Phase II trial of temozolomide and veliparib combination therapy for sorafenib-refractory advanced hepatocellular carcinoma [Abstract 240]. J Clin Oncol. 2014;32(3 Suppl). | ||
Kunos C, Deng W, Dawson D, et al. A phase I-II evaluation of veliparib (NSC #737664), topotecan, and filgrastim or pegfilgrastim in the treatment of persistent or recurrent carcinoma of the uterine cervix: an NRG Oncology/Gynecologic Oncology Group study. Int J Gynecol Cancer. 2015;25(3):484–492. | ||
Kummar S, Chen A, Ji J, et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011;71(17):5626–5634. | ||
Brenner JC, Feng FY, Han S, et al. PARP-1 inhibition as a targeted strategy to treat Ewing’s sarcoma. Cancer Res. 2012;72(7):1608–1613. | ||
Reiss KA, Herman JM, Zahurak M, et al. A phase I study of veliparib (ABT-888) in combination with low-dose fractionated whole abdominal radiation therapy in patients with advanced solid malignancies and peritoneal carcinomatosis. Clin Cancer Res. 2015;21(1):68–76. | ||
Mehta MP, Wang D, Wang F, et al. Veliparib in combination with whole brain radiation therapy in patients with brain metastases: results of a phase 1 study. J Neurooncol. 2015;122(2):409–417. | ||
Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–1115. | ||
Norquist B, Wurz KA, Pennil CC, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29(22):3008–3015. | ||
Barber LJ, Sandhu S, Chen L, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229(3):422–429. | ||
Jaspers JE, Kersbergen A, Boon U, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68–81. | ||
Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deificent mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079–17084. | ||
Pettitt SJ, Rehman FL, Bajrami I, et al. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLoS One. 2013;8(4):e61520. | ||
Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–861. | ||
Lee YH, Liu X, Qiu F, O’Connor TR, Yen Y, Ann DK. HP1β is a biomarker for breast cancer prognosis and PARP inhibitor therapy. PLoS One. 2015;10(3):e0121207. | ||
Wang X, Weaver DT. The ups and downs of DNA repair biomarkers for PARP inhibitor therapies. Am J Cancer Res. 2011;1(3):301–327. | ||
Michels J, Vitale I, Galluzzi L, et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res. 2013;73(7):2271–2280. | ||
Faraoni I, Compagnone M, Lavorgna S, et al. BRCA1, PARP1 and γH2AX in acute myeloid leukemia: role as biomarkers of response to the PARP inhibitor olaparib. Biochim Biophys Acta. 2015;1852(3):462–472. | ||
Jacot W, Thezenas S, Senal R, et al. BRCA1 promoter hypermethylation, 53BP1 protein expression and PARP-1 activity as biomarkers of DNA repair deficit in breast cancer. BMC Cancer. 2013;13:523. | ||
Liu JF, Konstantinopoulos PA, Matulonis UA. PARP inhibitors in ovarian cancer: current status and future promise. Gynecol Oncol. 2014;133(2):362–369. | ||
Horton TM, Jenkins G, Pati D, et al. Poly(ADP-ribose) polymerase inhibitor ABT-888 potentiates the cytotoxic activity of temozolomide in leukemia cells: influence of mismatch repair status and O6-methylguanine-DNA methyltransferase activity. Mol Cancer Ther. 2009;8(8):2232–2242. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.