")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 3
Profile of tivantinib and its potential in the treatment of hepatocellular carcinoma: the evidence to date
Authors Pievsky D, Pyrsopoulos N
Received 8 September 2016
Accepted for publication 14 October 2016
Published 15 November 2016 Volume 2016:3 Pages 69—76
DOI https://doi.org/10.2147/JHC.S106072
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ahmed Kaseb
Daniel Pievsky,1 Nikolaos Pyrsopoulos2
1Department of Internal Medicine, 2Division of Gastroenterology and Hepatology, Rutgers New Jersey Medical School, University Hospital, Newark, NJ, USA
Abstract: Hepatocellular carcinoma (HCC) is the fastest rising cause of cancer-related death in the United States and carries a very poor prognosis, with a median survival time of <50% at 1 year for advanced disease. To date, sorafenib is the only therapy approved by the Food and Drug Administration for the treatment of advanced HCC. Tivantinib (ARQ-197), a non-ATP competitive inhibitor of cellular mesenchymal–epithelial transcription factor (c-MET), has shown a survival benefit in patients with advanced HCC who have failed or are intolerant to sorafenib in Phase I and II trials. Those patients who have tumors with high concentrations of MET (MET-high) appear to derive the greatest benefit from tivantinib therapy. Currently, two large randomized double-blind placebo-controlled Phase III trials (METIV-HCC [NCT01755767] and JET-HCC [NCT02029157]) are evaluating tivantinib in patients with MET-high advanced HCC, with the primary end points of overall survival and progression-free survival, respectively. This study reviews the evidence for the use of tivantinib in advanced HCC. Specific topics addressed include the pharmacology, dosing, toxicity, and biomarkers associated with tivantinib use.
Keywords: tivantinib, ARQ-197, hepatocellular carcinoma, met inhibitor
Introduction
Liver cancer is the second most common cause of cancer-related death worldwide, the fifth most common cancer among men and the ninth most common cancer among women.1 Hepatocellular carcinoma (HCC) is responsible for ~90% of primary liver cancers.2 In the United States, HCC is the fastest rising cause of cancer-related death, and its incidence has nearly tripled since the 1980s.3,4 Despite increased focus and research, the overall prognosis for HCC remains poor. Curative modalities, such as liver transplant, resection and radiofrequency ablation, do exist when HCC is diagnosed early; however, only ~15% of patients are eligible for such treatments (Table 1).3,5,6 The majority of patients are diagnosed with advanced-stage HCC, for which the median survival time is <50% at 1 year and only 12% at 5 years.7,8 Chemotherapy is rarely used for the treatment of HCC due to its minimal success rate of 10–20% and high levels of toxicity.2
 | Table 1 Survival rate with different treatment modalities for HCC Notes: aVery early-stage HCC with nodule <2 cm without vascular or distant metastases. bVery early-stage HCC meeting Milan Criteria for transplantation. cEarly-stage HCC but not candidates for resection or transplant. Abbreviation: HCC, hepatocellular carcinoma. |
The only therapy currently approved by the Food and Drug Administration (FDA) for the treatment of advanced HCC is sorafenib, a tyrosine kinase inhibitor that has been shown to decrease cell proliferation and tumor angiogenesis.9,10 While sorafenib represents a major advancement in the treatment of HCC, its effects are modest, with the SHARP trial demonstrating an increase in overall survival from 7.9 months to 10.7 months and an increase from 2.8 months to 5.5 months in the median time to radiological progression.10 It demonstrated even less robust benefits in the Asia-Pacific trial, with overall survival in the treatment group of 6.5 months and median time to radiological progression of 2.8 months.9 Furthermore, after experiencing an initial response to treatment, most HCC patients develop a decrease in efficacy with sorafenib.11 Many novel therapies are currently under investigation in clinical trials. In a recently published Phase III trial, regorafenib, an oral multikinase inhibitor, has demonstrated an increase in median overall survival (10.6 months vs. 7.8 months) and an increase in median progression-free survival (PFS; 3.1 months vs. 1.5 months) compared to placebo in patients with advanced HCC who have progressed on sorafenib.12 To date, however, there are no FDA-approved second-line or salvage therapies for those patients who progress with or are intolerant to sorafenib.13
One of the most promising new therapies under investigation is tivantinib (ARQ 197), which has demonstrated positive results in early-phase clinical trials as a second-line agent for multiple solid tumors. This review focuses on tivantinib and its therapeutic profile in the management of HCC.
Development and pharmacology
Tivantinib is an oral, highly selective, non-ATP competitive inhibitor of c-MET receptor tyrosine kinase.14 The MET proto-oncogene was initially discovered in human osteosarcoma, and it encodes for cellular mesenchymal–epithelial transition factor (c-MET).15 c-MET and hepatocyte growth factor (HGF), its only known ligand, are necessary for normal embryonic development and tissue repair; however, their expression is normally very low and only found in cells of epithelial or mesenchymal origin.16–18 When HGF binds to c-MET, there is activation of multiple metabolic pathways, including the mitogen-activated protein kinase, protein kinase B, mammalian target of rapamycin, phophatidylinositol 3-kinase and focal adhesion kinase, which can lead to tumor growth, proliferation, cell invasion and distant metastasis (Figure 1).19–21 Upregulation of the c-MET pathway is found in multiple cancers, including HCC, where it is present 20–40% of the time and is associated with increased risk for metastases and poor prognosis.22,23 HGF is also over-expressed in HCC, which results in increased hepatic regeneration and suppression of hepatocyte apoptosis.24,25 Similarly, livers with HCC have been shown to have high levels of mitogen-activated protein kinase, which can go on to activate the Raf/MEK/ERK signaling pathway, leading to unopposed cellular proliferation, migration and survival.25,26 The inhibition of c-MET by tivantinib disrupts the deregulated signaling cascade, promotes apoptosis and inhibits cellular growth in those cells that express c-MET.
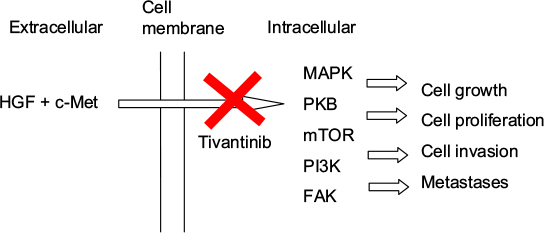 | Figure 1 Mechanism of action of tivantinib. Abbreviations: HGF, hepatocyte growth factor; c-MET, cellular mesenchymal–epithelial transition factor; MAPK, mitogen-activated protein kinase; PKB, protein kinase B; mTOR, mammalian target of rapamycin; PI3K, phophatidylinositol 3-kinase; FAK, focal adhesion kinase. |
Munshi et al14 demonstrated the selectivity of tivantinib by showing that tivantinib inhibited only four of 229 human kinases with a Ki for c-MET of 355 nmol/L. It was noted that while a Ki of 355 nmol/L is less than that of other available c-MET inhibitors, cellular assays demonstrated similar biochemical potency. Tivantinib targets the inactive, unphosphorylated form of c-MET, locking it in the inactive configuration and preventing downstream signaling.27 Controversy does exist regarding the mechanism of action of tivantinib and whether c-MET inhibition is the major anti-tumor effect of the drug. Several studies have shown that tivantinib possesses cytotoxic activity, even in cells that lack c-MET.28–32 Other proposed mechanisms include microtubule, glycogen synthase kinase 3, cyclin B1 and proteasome inhibition. Microtubule inhibition is the most widely theorized alternative mechanism of action, with three independent in vitro studies corroborating this hypothesis.29–31 Significant challenges to this alternative hypothesis have been raised. Notably, in vivo studies have not documented evidence of neurotoxicity, which is known to be a major adverse effect of microtubule inhibitors.23 Similarly, tivantinib has demonstrated only minimal, if any, response in patients who have tumors with low levels of MET expression, while those tumors with high levels of MET have had the most profound responses to the drug.33,34
From a pharmacokinetic standpoint, tivantinib has an oral bioavailability of >20%, with a half life of 29 minutes. It is metabolized by the CYP2C19 and CYP3A4 pathways, with 19% elimination via the kidneys and 68% via the stool.35 Possible drug interactions could occur with known CYP2C19 inhibitors, such as clopidogrel, esomeprazole, fluconazole, fluoxetine and omeprazole, or with known CYP2C19 inducers, such as carbamazepine, phenytoin and rifampin. Maximum inhibition of c-MET is obtained after just 24 hours of treatment, and this effect is sustained for 12 hours after the drug is withdrawn.36
Clinical studies
Three Phase I trials have evaluated tivantinib monotherapy in samples without HCC and found tivantinib to be well tolerated with mild clinical benefit.37–39 The first of these was conducted as an open-label, sequential 3 + 3 dose escalation design with an initial dose of 100 mg BID at a single center in the United Kingdom.37 Fifty-one patients with advanced solid organ tumors including prostate, melanoma, gastric, colorectal, sarcoma and breast were included in the trial. No patients with HCC were enrolled. The primary objective was to evaluate safety and tolerability and determine the maximum tolerated dose (MTD), establish a recommended Phase II dose (RP2D) and define dose-limited toxicities. Tivantinib was deemed safe and well tolerated, with an MTD/RP2D of 360 mg BID. Stable disease was observed in 14 patients, as defined by Response Evaluation Criteria in Solid Tumors.40
Similarly, a multicenter open-label single-arm dose escalation trial with an initial dose of 10 mg BID was conducted in the United States.38 Seventy-nine patients with advanced solid organ tumors, including colorectal, renal, ovarian, pancreatic, sarcoma and thyroid among others, were enrolled. No HCC patients were included in the sample. Tivantinib was well tolerated, with an MTD/RP2D of 360 mg BID. The PFS for all 79 patients was 77% at 6 weeks, 52% at 12 weeks and 34% at 21 weeks.
The same concept was evaluated in Japan in a multicenter dose-escalating open-label trial with a 3 + 3 design at an initial dose of 70 mg BID.39 Forty-seven Japanese patients with solid organ tumors were enrolled, although no HCC patients were included in the cohort. It is of interest that these patients were enrolled based on their CYP2C19 polymorphism status. The CYP2C19 enzyme plays a key role in the metabolism of tivantinib. It has been shown that 30–80% of Asians have a genetic polymorphism of CYP2C19, making them very poor metabolizers of tivantinib. This same polymorphism, in contrast, is found in only 12–19% of Whites or Blacks.41 Tivantinib was well tolerated by both normal and poor metabolizers of CYP2C19, although a dose adjustment down to 240 mg BID was recommended for poor metabolizers.
The first study of tivantinib monotherapy in patients with HCC was a Phase IB study conducted by Santoro et al.42 Three hundred and sixty milligrams of tivantinib was administered twice per day to 21 adult patients with Child-Pugh A and B cirrhosis and HCC in 28-day treatment cycles. All 21 patients eventually discontinued the treatment, 17 due to disease progression and four due to adverse events. Only 16 patients were able to be evaluated for tumor response, and while none of them demonstrated any objective response to therapy, nine did show stable disease (Table 2). The overall median time to disease progression was 3.3 months (range 1.47–5.3 months) among those who were able to be evaluated and 1.8 months (range 1.6–5.3 months) in the intention-to-treat analysis. Pharmacokinetic analysis found significant tivantinib accumulation in the plasma of HCC patients compared to the accumulation in other solid tumors, but no correlation was found between tivantinib exposure and adverse events. Several years later, a Phase I trial using tivantinib monotherapy was performed in a Japanese sample with advanced HCC.43 Only 28 patients were included in the trial, and though the authors found no complete or partial responses, they did see stable disease in 20 of the 28 patients.
 | Table 2 Tumor response to tivantinib in Phase 1B triala Notes: aOnly 16 of 21 patients were able to be evaluated for tumor response. Data from Santoro et al.42 |
Multiple Phase I studies have been performed using tivantinib as part of a combination therapy with sorafenib, sunitinib, erlotinib and gemcitabine.44–47 A preclinical study showed that tivantinib and sorafenib had an additive effect in three HCC cell lines (JHH-4, PLC/PRF/5 and SK-Hep-1) but did not find any degree of synergy between tivantinib and sunitinib in any of the 87 cancer cell lines tested.48 One of the clinical trials, a Phase I trial, assessed the combination of tivantinib and sorafenib in 87 adults with advanced solid organ tumors.44 Twenty patients had HCC with Child-Pugh A (n=14) and B (n=6) cirrhosis. An overall response rate of 11.5% and a 10% response in the HCC population were observed. Among the HCC patients, there was one complete response, one partial response and 12 cases of stable disease. Interestingly, both the complete response and the partial response were noted in patients who had a high concentration of tumor MET, and overall three of four patients with high concentrations of tumor MET achieved disease control. The median PFS was 3.5 months (95% confidence interval [95% CI]: 3–11.1), and the disease control rate was 70%. Patients with HCC who had received prior treatment with vascular endothelial growth factor (VEGF) therapy, sorafenib and/or sunitinib had a longer PFS than those who did not receive prior VEGF therapy (15.9 months [95% CI: 1.7–15.9] vs. 3.5 months [95% CI: 3–7.4]).
A pooled analysis was conducted by Chai et al46 on all Phase I studies with tivantinib in HCC and biliary tract cancer patients. Overall, 53 patients were treated with tivantinib: 42 were diagnosed with HCC, ten with cholangiocarcinoma and one with gallbladder adenoma. Twenty-three patients received tivantinib monotherapy, and 30 received a combination of tivantinib and either sorafenib (n=20), gemcitabine (n=8) or erlotinib (n=2). The overall response rate was 6%, but the disease control rate was 62%, with one complete response, two partial responses and 30 cases of stable disease.
Based on positive results from Phase I trials, a Phase II international multicenter double-blind randomized placebo-controlled trial was conducted.34 One hundred and seven adult patients with advanced-stage HCC who had previously progressed or had demonstrated intolerance to one prior systemic therapy (sorafenib or sunitinib) were randomized in a 2:1 ratio to receive tivantinib or placebo. The initial dose of tivantinib was 360 mg BID; however, this was later decreased to 240 mg BID due to a higher than expected incidence of grade 3 or higher neutropenia. In total, 38 patients received tivantinib 360 mg BID, 33 patients received tivantinib 240 mg BID and 36 patients received placebo. The median time to progression (TTP) was significantly longer in the tivantinib group (1.6 months [95% CI: 1.4–2.8]) compared to that in the placebo group (1.4 months [95% CI: 1.4–1.5]), with a hazard ratio (HR) of 0.64, 90% CI 0.43–0.94, p=0.04. The median PFS in the tivantinib group (1.5 months [95% CI: 1.4–2.7]) was longer than that in the placebo group (1.4 months [95% CI: 1.4–1.5]), with this trend approaching significance, HR 0.67, 95% CI: 0.44–1.04, p=0.06. There was no significant difference in overall survival between the two groups. According to the statistical analysis plan, the study participants were also subdivided based on their tumor MET status into high-expression (MET-high) and low-expression (MET-low) subgroups. There were 37 patients with MET-high tumors, 22 of whom received tivantinib (ten patients at 360 mg BID and 12 patients at 240 mg BID), and 40 patients with MET-low tumors, 27 of whom received tivantinib (13 patients at 360 mg BID and 14 patients at 240 mg BID). Patients who had MET-high tumors and received tivantinib had a longer median TTP (2.7 months vs. 1.4 months [HR 0.43, 95% CI: 0.19–0.97, p=0.03]), a longer median PFS (2.2 months vs. 1.4 months [HR 0.45, 95% CI: 0.21–0.95, p=0.02]) and a longer median overall survival (7.2 months vs. 3.8 months [HR 0.38, 95% CI: 0.18–0.81, p=0.01]) compared to placebo. There was no difference in those with MET-low tumors who received tivantinib and those who received placebo.
Based on the significant benefits seen in the MET-high group in the above-mentioned Phase II study, two Phase III trials have been initiated to further evaluate the benefit of tivantinib in patients with MET-high HCC tumors. The METIV-HCC trial (NCT01755767) is a large international randomized double-blind, placebo-controlled trial comparing tivantinib 120 mg BID to placebo in patients with MET-high HCC tumors who have failed or progressed with sorafenib.49 The primary end point is overall survival, and secondary end points are PFS and safety. This study has enrolled over 300 participants. A planned interim analysis was recently completed, and the study will continue to the final analysis as recommended by the data monitoring committee.50 The JET-HCC trial (NCT02029157) is a randomized, double-blind, placebo-controlled trial being conducted in Japan to evaluate the effect of tivantinib at 120 mg BID on MET-high HCC in patients previously treated with one prior course of sorafenib therapy. The primary end point of this study is PFS, and the secondary end point is overall survival. The study is currently recruiting patients, with a goal of 160 patients. The estimated study completion date is December 2016.
Dosing and toxicity
Tivantinib dosing for HCC has continued to evolve as more studies have been performed in this patient population. The two initial Phase I dosing trials did not include any patients with HCC, and both recommended a tivantinib dose of 360 mg BID.37,38 The three most common adverse events in both trials were fatigue, nausea and vomiting. Dose-limiting toxicities observed by Yap et al included one case of grade 3 fatigue at 200 mg BID, two cases of febrile neutropenia at 400 mg BID and one incidence of a combination of mucositis, palmar–plantar erythrodysesthesia and hypokalemia at 400 mg BID. Two cases of pancytopenia, vomiting and dehydration at 360 mg BID were seen in the Rosen et al trial. The hematologic toxicity associated with tivantinib is expected, as HGF promotes hematopoiesis by stimulating production of cytokines by the stromal cells in the bone marrow.51,52
The Santoro et al42 trial was the first to evaluate tivantinib dosing exclusively in HCC patients. Based on the results of prior studies, a starting dose of 360 mg BID was selected. Overall, 20 of the 21 patients developed a drug-related event, with the most frequent events being neutropenia (52%), anemia (48%), asthenia (48%) and leukopenia (38%). Eleven patients (52%) developed adverse reactions that were grade 3 or grade 4 in severity, the most common of which were neutropenia (73%), anemia (45%) and leukopenia (36%). In general, neutropenia was the main cause of dose reduction, treatment interruption and treatment discontinuation. Four patients developed a serious adverse reaction that required treatment discontinuation. These reactions included one case of grade 3 anemia, one case of grade 3 anemia and grade 4 neutropenia, one case of grade 4 leukopenia and grade 4 neutropenia, and one case of grade 4 neutropenia with grade 5 septic shock leading to death. Due to interpatient variability, the authors did not find any correlations among adverse events with tivantinib dose, exposure or Child-Pugh status; however, they did recommend that tivantinib dosing in HCC patients be reduced to 240 mg BID due to the high amount of hematologic toxicity.
A pooled analysis from all Phase I/IB and Phase II studies assessed the exposure response of tivantinib in 289 patients, 73 of whom had HCC.53 The pharmacokinetic analysis demonstrated that the clearance of tivantinib in HCC patients was reduced by 67%, which resulted in a threefold increase in exposure compared to other types of cancer. A significant relationship was also found between tivantinib exposure and incidence of grade ≥2/3 neutropenia. The authors suggested that a starting dose of 240 mg BID of tivantinib rather than 360 mg BID would result in a decrease in the incidence of grade ≥3 neutropenia from 28% to 16% in HCC patients.
Similar results were seen in a later Phase II trial by Santoro et al,34 in which the treatment dose was reduced from 360 mg BID to 240 mg BID due to a high incidence of grade ≥3 neutropenia. Overall, hematologic toxicities (neutropenia, anemia and leukopenia) occurred more frequently in the tivantinib group than in the placebo group, and the incidence of grade ≥3 neutropenia was higher in the tivantinib 360 mg BID group than either the tivantinib 240 mg BID or the placebo group. There was even a trend toward longer overall survival in the tivantinib 240 mg BID group (7.5 months, 95% CI 4.4–not available) compared to the 360 mg BID group (6.4 months, 95% CI 4–9). Both the currently active Phase III trials, METIV-HCC and JET-HCC, are evaluating tivantinib at 120 mg BID. The METIV-HCC trial initially started patients on 240 mg BID; however, the dose was decreased to 120 mg BID in response to a higher than expected incidence of neutropenia.54 However, it should be noted that the METIV-HCC trial used a different formulation of tivantinib than the Santoro et al Phase II trial.23 Once the dose was decreased to 120 mg BID, a safety analysis showed that a dose of 120 mg BID of the new formulation resulted in an incidence of neutropenia similar to that seen at a dose of 240 mg in the Phase II trial.55 In addition, a pharmacokinetic analysis showed that the plasma exposure of the new formulation at 120 mg BID was comparable to that of the capsule formulation used in the Santoro et al trial.55 The JET-HCC trial set its dosing at 120 mg BID based on dosing studies in Japanese samples.43
Patients with a CYP2C19 polymorphism may require even lower doses of tivantinib. Yamamoto et al39 were the first to examine the effect of the CYP2C19 polymorphism in patients taking tivantinib. Forty-seven patients were included in the study, 14 of whom were considered poor metabolizers. The study was unable to demonstrate a relationship between dose level and plasma exposure; however, it was found that the mean exposure of tivantinib in poor metabolizers at 240 mg BID was equivalent to that of normal metabolizers at a dose of 360 mg BID. Based on this finding, the authors recommended that those with the CYP2C19 polymorphism receive doses of 240 mg BID, while those with normal CYP2C19 enzymes receive 360 mg BID. This study did not include any patients with HCC. A second trial was conducted by Okusaka et al43 to specifically look at tivantinib dosing in those with the CYP2C19 polymorphism and HCC. Twenty-eight patients were included in the study, seven of whom had the CYP2C19 polymorphism. All patients tolerated tivantinib at a dose of 120 mg BID, but once the dose was increased to 240 mg BID, five of nine normal metabolizers developed dose-limiting toxicities, either neutropenia or febrile neutropenia, which resolved with drug interruption and treatment with granulocyte colony-stimulating factor medications. Because normal metabolizers were unable to tolerate a dose of 240 mg BID, those with the CYP2C19 polymorphism were never escalated to a dose higher than 120 mg BID. The authors concluded that Japanese patients with HCC were unable to tolerate a dose of tivantinib >120 mg BID, irrespective of CYP2C19 polymorphism status. This study served as the basis for the dosing regimen in the JET-HCC trial.
Future directions
The future of tivantinib, like that of many other drugs, rests on appropriate patient selection. The identification of biomarkers for tivantinib sensitivity is an active area of research with promising early results. The first biomarker for tivantinib in HCC patients was tumor MET status. It was published that patients with MET-high tumors showed a significant survival benefit, with longer median TTP (2.7 months vs. 1.4 months), longer median PFS (2.2 months vs. 1.4 months) and longer median overall survival (7.2 months vs. 3.8 months) on tivantinib compared to placebo.34 No difference in efficacy was found between tivantinib and placebo in patients with MET-low tumors. In a combination study of tivantinib with sorafenib, the combination therapy demonstrated a trend toward significance regarding increased disease control rate, defined as the combination of complete responses, partial responses and stable disease for at least 8 weeks.44 While only four cases of MET-high HCC were observed, three of the four patients achieved disease control. Based on these promising results, both of the active Phase III trials are evaluating tivantinib in HCC patients with MET-high tumor status.
A preclinical study evaluated whether HGF tumor concentration could be used as a marker for HCC, but no significant differences were found between HGF-high and HGF-low groups in clinicopathological factors or patient outcomes.56 A more recent study that specifically evaluated MET, HGF and AFP as circulating biomarkers (by 75 percentile) found that they were prognostic markers for overall survival in patients with HCC.57 Circulating MET was also a pharmacodynamic biomarker for tivantinib: patients on treatment with a ≥10% decrease in circulating MET levels demonstrated increased overall survival compared to those with a <10% decrease (13.3 months vs. 6.3 months; HR: 0.46 [95% CI: 0.24–0.86], p=0.01). Additionally, tumor MET levels were correlated with response to tivantinib treatment, the only biomarker thus far to predict response to treatment. These results support the use of tivantinib exclusively in MET-high tumor patients, although more definitive data will be forthcoming with the results of the METIV-HCC trial, which will analyze over 900 tumor samples and validate the use of select biomarkers in HCC.
Additional studies are needed to assess the use of tivantinib in those with more severe cirrhosis. The vast majority of trials have evaluated tivantinib in patients who are Child-Pugh Class A, with only a few trials including patients who are Child-Pugh Class B. Although theoretically tivantinib could also be used for those who are Child-Pugh Class C, this has not yet been studied. Moreover, it may not be safe to attempt such a trial given the anticipated side effects. Nevertheless, future development of more specific targeted biologic markers may enable treating physicians to administer agents such as tivantinib in a safer and more effective way.
Conclusion
Tivantinib, a potent inhibitor of c-MET, appears to be a promising therapy for those with advanced-stage HCC that has progressed or is intolerant to sorafenib. Tivantinib has demonstrated increased overall survival, PFS and TTP compared to placebo in patients with MET-high tumors. Serious hematologic side effects (neutropenia, anemia and leukopenia) are anticipated to be less prominent at lower dosing regimens (120 mg BID), at which dose good treatment efficacy is still maintained. Two large Phase III trials, METIV-HCC and JET-HCC, are currently ongoing. To date, there is no FDA-approved salvage or second-line therapy for patients with HCC who have failed or progressed with sorafenib. Whether tivantinib will be able to fill that need still remains to be determined.
Disclosure
The authors report no conflicts of interest in this work.
References
Fact Sheets by Cancer [webpage on the Internet]. All Cancers (Excluding Non-Melanoma Skin Cancer) Estimated Incidence, Mortality and Prevalence Worldwide in 2012. Available from: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx. Accessed June 12, 2016. | ||
Deng G-L, Zeng S, Shen H. Chemotherapy and target therapy for hepatocellular carcinoma: new advances and challenges. World J Hepatol. 2015;7(5):787–798. | ||
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–1127. | ||
National Cancer Institute [webpage on the Internet]. A Snapshot of Liver Cancers. Available from: http://www.cancer.gov/research/progress/snapshots/liver. Accessed August 23, 2016. | ||
Finn RS. Development of molecularly targeted therapies in hepatocellular carcinoma: where do we go now? Clin Cancer Res. 2010;16(2):390–397. | ||
Cho YK, Kim JK, Kim MY, Rhim H, Han JK. Systematic review of randomized trials for hepatocellular carcinoma treated with percutaneous ablation therapies. Hepatology. 2009;49(2):453–459. | ||
Nordenstedt H, White DL, El-Serag HB. The changing pattern of epidemiology in hepatocellular carcinoma. Dig Liver Dis. 2010;42(suppl 3):S206–S214. | ||
Artinyan A, Mailey B, Sanchez-Luege N, et al. Race, ethnicity, and socioeconomic status influence the survival of patients with hepatocellular carcinoma in the United States. Cancer. 2010;116(5):1367–1377. | ||
Cheng A-L, Kang Y-K, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34. | ||
Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | ||
Zhao X, Tian C, Puszyk WM, et al. OPA1 downregulation is involved in sorafenib-induced apoptosis in hepatocellular carcinoma. Lab Invest. 2013;93(1):8–19. | ||
Bruix J, Merle P, Granito A, et al. LBA-03Efficacy and safety of regorafenib versus placebo in patients with hepatocellular carcinoma (HCC) progressing on sorafenib: results of the international, randomized phase 3 RESORCE trial. Ann Oncol. 2016;27(suppl 2):ii140–ii141. | ||
Porta C, Paglino C. Medical treatment of unresectable hepatocellular carcinoma: going beyond sorafenib. World J Hepatol. 2010;2(3):103–113. | ||
Munshi N, Jeay S, Li Y, et al. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther. 2010;9(6):1544–1553. | ||
Cooper CS, Park M, Blair DG, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311(5981):29–33. | ||
Bottaro DP, Rubin JS, Faletto DL, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251(4995):802–804. | ||
Birchmeier C, Gherardi E. Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol. 1998;8(10):404–410. | ||
Sonnenberg E, Meyer D, Weidner KM, Birchmeier C. Scatter factor/hepatocyte growth factor and its receptor, the c-met tyrosine kinase, can mediate a signal exchange between mesenchyme and epithelia during mouse development. J Cell Biol. 1993;123(1):223–235. | ||
Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4(12):915–925. | ||
Blumenschein GR, Mills GB, Gonzalez-Angulo AM. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J Clin Oncol. 2012;30(26):3287–3296. | ||
Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003;22(4):309–325. | ||
Gao JJ, Inagaki Y, Xue X, Qu XJ, Tang W. c-Met: a potential therapeutic target for hepatocellular carcinoma. Drug Discov Ther. 2011;5(1):2–11. | ||
Porta C, Giglione P, Ferrari A, et al. Tivantinib (ARQ197) in hepatocellular carcinoma. Expert Rev Anticancer Ther. 2015;15(6):615–622. | ||
Venepalli NK, Goff L. Targeting the HGF-cMET axis in hepatocellular carcinoma. Int J Hepatol. 2013;2013:341636. | ||
Au J, Frenette C. Development of tivantinib as treatment for hepatocellular carcinoma. J Clin Transl Hepatol. 2013;1(1):75–78. | ||
Peyssonnaux C, Eychène A. The Raf/MEK/ERK pathway: new concepts of activation. Biol Cell. 2001;93(1–2):53–62. | ||
Eathiraj S, Palma R, Volckova E, et al. Discovery of a novel mode of protein kinase inhibition characterized by the mechanism of inhibition of human mesenchymal-epithelial transition factor (c-Met) protein autophosphorylation by ARQ 197. J Biol Chem. 2011;286(23):20666–20676. | ||
Aoyama A, Katayama R, Oh-hara T, Sato S, Okuno Y, Fujita N. Tivantinib (ARQ 197) exhibits antitumor activity by directly interacting with tubulin and overcomes abc transporter–mediated drug resistance. Am Assoc Cancer Res. 2014;13(12):2978–2990. | ||
Basilico C, Pennacchietti S, Vigna E, et al. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Am Assoc Cancer Res. 2013;19(9):2381–2392. | ||
Katayama R, Aoyama A, Yamori T, et al. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 2013;73(10):3087–3096. | ||
Xiang Q, Zhen Z, Deng DY, et al. Tivantinib induces G2/M arrest and apoptosis by disrupting tubulin polymerization in hepatocellular carcinoma. J Exp Clin Cancer Res. 2015;34:118. | ||
Remsing Rix LL, Kuenzi BM, Luo Y, et al. GSK3 alpha and beta are new functionally relevant targets of tivantinib in lung cancer cells. ACS Chem Biol. 2014;9(2):353–358. | ||
Tolaney SM, Tan S, Guo H, et al. Phase II study of tivantinib (ARQ 197) in patients with metastatic triple-negative breast cancer. Invest New Drugs. 2015;33(5):1108–1114. | ||
Santoro A, Rimassa L, Borbath I, et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013;14(1):55–63. | ||
Lu S, Rizzani A, Kolligs FT, et al. Anti-proliferative mechanisms downstream of c-MET of the kinase inhibitor tivantinib (ARQ 197). Hepatology. 2013;58(S1):180A. | ||
Gu X, Wang C, Yu Y, et al. Abstract #1748: inhibition of HGF/c-Met pathway by ARQ 197: characterization of pharmacodynamic markers in vitro and in vivo. Cancer Res. 2009;69(9 suppl):1748. | ||
Yap TA, Olmos D, Brunetto AT, et al. Phase I trial of a selective c-MET inhibitor ARQ 197 incorporating proof of mechanism pharmacodynamic studies. J Clin Oncol. 2011;29(10):1271–1279. | ||
Rosen LS, Senzer N, Mekhail T, et al. A phase I dose-escalation study of tivantinib (ARQ 197) in adult patients with metastatic solid tumors. Clin Cancer Res. 2011;17(24):7754–7764. | ||
Yamamoto N, Murakami H, Nishina T, et al. The effect of CYP2C19 polymorphism on the safety, tolerability, and pharmacokinetics of tivantinib (ARQ 197): results from a phase I trial in advanced solid tumors. Ann Oncol. 2013;24(6):1653–1659. | ||
Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–216. | ||
Kubota T, Chiba K, Iga T. Frequency distribution of CYP2C19, CYP2D6, and CYP2C9 mutant-alleles in several different populations. Xenobio Metab Dispos. 2001;16:69–74. | ||
Santoro A, Simonelli M, Rodriguez-Lope C, et al. A Phase-1b study of tivantinib (ARQ 197) in adult patients with hepatocellular carcinoma and cirrhosis. Br J Cancer. 2013;108(1):21–24. | ||
Okusaka T, Aramaki T, Inaba Y, et al. Phase I study of tivantinib in Japanese patients with advanced hepatocellular carcinoma: distinctive pharmacokinetic profiles from other solid tumors. Cancer Sci. 2015;106(5):611–617. | ||
Puzanov I, Sosman J, Santoro A, et al. Phase 1 trial of tivantinib in combination with sorafenib in adult patients with advanced solid tumors. Invest New Drugs. 2015;33(1):159–168. | ||
Adjei AA, Sosman JA, Martell RE, et al. Efficacy in selected tumor types in a phase I study of the c-MET inhibitor ARQ 197 in combination with sorafenib. ASCO Meet Abstr. 2011;29(15_suppl):3034. | ||
Chai F, Abbadessa G, Savage R, et al. Phase 1 experience of tivantinib in patients with hepatocellular carcinoma (HCC) or biliary tract cancer (BTC). Ann Oncol. 2012;23(9 suppl):ix245. | ||
Goldman JW, Laux I, Chai F, et al. Phase 1 dose-escalation trial evaluating the combination of the selective MET (mesenchymal-epithelial transition factor) inhibitor tivantinib (ARQ 197) plus erlotinib. Cancer. 2012;118(23):5903–5911. | ||
Chen C-R, Szwaya J, Rojnuckarin A, et al. Abstract #820: combination studies of tyrosine kinase inhibitors (TKIs): assessment of potential cytotoxic synergy of ARQ 197 with sorafenib or sunitinib. Cancer Res. 2009;69(9 suppl):820. | ||
Santoro A, Porta C, Rimassa L, et al. Metiv-HCC: a phase III clinical trial evaluating tivantinib (ARQ 197), a MET inhibitor, versus placebo as second-line in patients (pts) with MET-high inoperable hepatocellular carcinoma (HCC). ASCO Meet Abstr. 2013;31(15_suppl):TS4159. | ||
METIV-HCC_Interim_Analysis_Press_Release__cs.pdf. Available from: http://files.shareholder.com/downloads/ARQL/2313700819x0x882287/84DDD033-9DCA-46B6-9556-D4E90448754B/METIV-HCC_Interim_Analysis_Press_Release__cs.pdf. Accessed October 2, 2016. | ||
Takai K, Hara J, Matsumoto K, et al. Hepatocyte growth factor is constitutively produced by human bone marrow stromal cells and indirectly promotes hematopoiesis. Blood. 1997;89(5):1560–1565. | ||
Matsuda-Hashii Y, Takai K, Ohta H, et al. Hepatocyte growth factor plays roles in the induction and autocrine maintenance of bone marrow stromal cell IL-11, SDF-1 alpha, and stem cell factor. Exp Hematol. 2004;32(10):955–961. | ||
Zahir H, Kastrissios H, Carothers T, et al. Exposure-response relationship to assess the risk of neutropenia in patients with hepatocellular carcinoma (HCC) treated with tivantinib. Ann Oncol. 2012;23(9 suppl):ix244. | ||
ArQule [webpage on the Internet]. ArQule Provides Updates on Clinical Trials in Hepatocellular Carcinoma and Non-Small Cell Lung Cancer with Tivantinib (NASDAQ:ARQL). Available from: http://investors.arqule.com/releasedetail.cfm?ReleaseID=819847. Accessed July 4, 2016. | ||
ARQL_News_2014_1_16_General_Releases.pdf. Available from: http://files.shareholder.com/downloads/ARQL/2313700819x0x719482/5FB4DEC7-86CB-411D-8072-F5E523DBFC73/ARQL_News_2014_1_16_General_Releases.pdf. Accessed October 6, 2016. | ||
Ueki T, Fujimoto J, Suzuki T, Yamamoto H, Okamoto E. Expression of hepatocyte growth factor and its receptor c-met proto-oncogene in hepatocellular carcinoma. Hepatology. 1997;25(4):862–866. | ||
Rimassa L, Abbadessa G, Personeni N, et al. Tumor and circulating biomarkers in patients with second-line hepatocellular carcinoma from the randomized phase II study with tivantinib. Oncotarget. Epub 2016 Aug 25. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.