")
Back to Journals » Infection and Drug Resistance » Volume 8
Profile of tedizolid phosphate and its potential in the treatment of acute bacterial skin and skin structure infections
Authors Hall RG, Michaels H
Received 3 February 2015
Accepted for publication 4 March 2015
Published 22 April 2015 Volume 2015:8 Pages 75—82
DOI https://doi.org/10.2147/IDR.S56691
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Ronald G Hall 2nd, Heidi N Michaels
Texas Tech University Health Sciences Center, Dallas, TX, USA
Abstract: Tedizolid phosphate is the first once-daily oxazolidinone approved by the United States Food and Drug Administration for the treatment of acute bacterial skin and skin structure infections (ABSSSI). It is more potent in vitro than linezolid against methicillin-resistant Staphylococcus aureus (MRSA) and other gram-positive pathogens causing ABSSSI, even retaining activity against some linezolid-resistant strains. Tedizolid is approximately 90% protein bound, leading to lower free-drug concentrations than linezolid. The impact of the effect of food, renal or hepatic insufficiency, or hemodialysis on tedizolid's pharmacokinetic have been evaluated, and no dosage adjustment is needed in these populations. In animal and clinical studies, tedizolid's effect on bacterial killing is optimized by the free-drug area under the curve to minimum inhibitory concentration ratio (fAUC/MIC). The 200 mg once-daily dose is able to achieve the target fAUC/MIC ratio in 98% of simulated patients. Two Phase III clinical trials have demonstrated the noninferiority of tedizolid 200 mg once daily for 6 days to linezolid 600 mg twice daily for 10 days. In vitro, animal, and clinical studies have failed to demonstrate that tedizolid inhibits monoamine oxidase to a clinically relevant extent. Tedizolid has several key advantages over linezolid including once daily dosing, decreased treatment duration, minimal interaction with serotonergic agents, possibly associated with less adverse events associated with the impairment of mitochondrial protein synthesis (eg, myelosuppression, lactic acidosis, and peripheral/optic neuropathies), and retains in vitro activity against linezolid-resistant gram-positive bacteria. Economic analyses with tedizolid are needed to describe the cost-effectiveness of this agent compared with other options used for ABSSSI, particularly treatment options active against MRSA.
Keywords: tedizolid, oxazolidinone, ABSSSI, MRSA, VRE, monoamine oxidase
Introduction
Hospital admissions due to acute bacterial skin and skin structure infections (ABSSSI), including cellulitis/erysipelas, wound infection, and major cutaneous abscess, continue to rise.1,2 In the year 2012, the CDC estimated 15,138 patients were infected with community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) with an incidence rate of 4.82 per 100,000 population per year (confidence interval [CI]: 3.69–6.42).3 Overall MRSA infections were estimated at 75,309 patients with an incidence rate of 23.99 per 100,000 population per year (CI: 20.64–28.10). In a population study conducted from 2009 to 2011, MRSA was found in nearly half of the S. aureus skin and soft tissue infections (46% of the 81% S. aureus isolates).4 It is widely recognized that novel treatment options for MRSA are needed as S. aureus is one of the pathogens target by the Infectious Diseases Society of America (IDSA) 10×′20 initiative.
Structure and mechanism of action
Tedizolid phosphate (formerly torezolid phosphate, R-701, DA-7218) is the prodrug of tedizolid (formerly torezolid, TR-700, DA-7157). Tedizolid and linezolid are both oxazolidinones with similar chemical structures. Tedizolid has phosphate added to the A-ring which increases its water solubility. Tedizolid’s D-ring increases its in vitro potency through increased interaction with additional sites on the ribosome. Tedizolid’s activity against linezolid-resistant strains is due to the replacement of the acetamide group with a hydroxymethyl group. This replacement increases potency against bacterial strains containing the cfr gene.5
Oxazolidinones, including tedizolid, bind to the 50S ribosomal subunit to inhibit bacterial protein synthesis. Additionally, tedizolid interacts with the peptidyl transferase binding region of 23S rRNA which may increase its in vitro potency.6
Spectrum of activity, in vitro potency, and resistance
Tedizolid is active against the most common gram-positive pathogens in patients with ABSSSI. The MIC50, MIC90, and range of MICs observed for tedizolid and linezolid against these pathogens are shown in Table 1.6–12
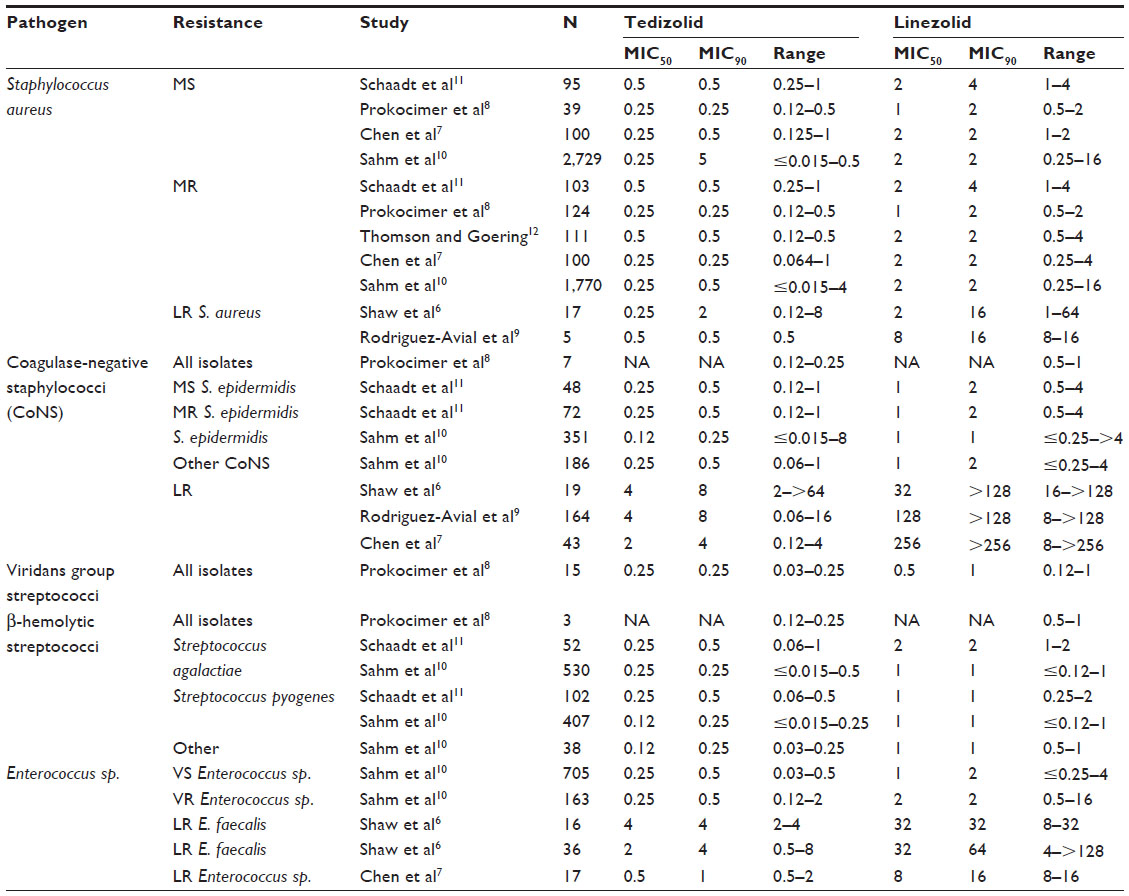 | Table 1 In vitro activity of oxazolidinones against common acute bacterial skin and skin structure infection pathogens (μg/mL) |
Staphylococci
Tedizolid is more potent than linezolid against S. aureus regardless of methicillin susceptibility (MIC50: 0.25–0.5 vs 1–2 μg/mL; MIC90: 0.25–0.5 vs 2–4 μg/mL).7,8,10–12 Small studies have suggested that the MIC50 of tedizolid (0.25–0.5 μg/mL) is unaffected by linezolid-resistance.6,9 However, the MIC90 may increase to 2 μg/mL.
Tedizolid is also more potent than linezolid in vitro against coagulase-negative staphylococci (MIC50: 0.12–0.25 vs 1 μg/mL; MIC90: 0.25–5 vs 1–2 μg/mL).8,10,11 Tedizolid susceptibility is more adversely affected by linezolid-resistance in coagulase-negative staphylococci (MIC50: 2–4 μg/mL; MIC90: 4–8 μg/mL).6,7,9
The postantibiotic effect (PAE) of tedizolid was similar to linezolid (0.05–0.7 hours vs 0.1–1.3 hours).13 A prolonged subinhibitory minimum inhibitory concentration effect (SME) and postantibiotic subinhibitory effect (PA-SME) was observed, which could inhibit bacterial growth during trough levels of drug. Methicillin resistance did not impact the PAE, SME, or PA-SME of tedizolid.
Streptococci
Studies evaluating the in vitro potency of oxazolidinones have consistently found tedizolid to be more potent against β-hemolytic streptococci than linezolid (MIC50: 0.12–0.25 vs 1–2 μg/mL; MIC90: 0.25–0.5 vs 1–2 μg/mL). Prokocimer et al8 evaluated 15 isolates of viridans group streptococci and observed similar results to those for β-hemolytic streptococci (MIC50: 0.25 vs 0.5 μg/mL; MIC90: 0.25 vs 1 μg/mL).
Enterococci
Vancomycin resistance has little effect on the potency of either of the oxazolidinones (MIC50: 0.25 vs 1–2 μg/mL; MIC90: 0.5 vs 2 μg/mL). Shaw et al6 published the first in vitro evaluation of tedizolid in linezolid-resistant Enterococcus sp. (MIC50: 2–4 μg/mL; MIC90: 4 μg/mL). Chen et al’s7 subsequent work suggests a lesser impact of linezolid-resistance on tedizolid MICs (MIC50: 0.5 μg/mL; MIC90: 1 μg/mL). However, the finding may be due to the lower linezolid MICs compared to Shaw et al’s6 study. The PAE of tedizolid was similar to linezolid (0.1–1.3 hours vs 0.15–1.1 hours).13
Other pathogens
Other pathogens tested against tedizolid included Moraxella catarrhalis, Haemophilus influenzae, Listeria monocytogenes, Legionella pneumophila, and Nocardia brasiliensis.11,13 Tedizolid was more active than linezolid against M. catarrhalis (MIC90 4 vs 16) and H. influenza (MIC90 16 vs 32). Listeria or Legionella-infected macrophages or human umbilical vein endothelial cells showed tedizolid to be more active than linezolid. When incubated from 0.25×MIC up to 16×MIC, tedizolid inhibited intracellular Nocardia growth.
Pharmacokinetics
General population
The pharmacokinetics of tedizolid and linezolid are summarized elsewhere.14 Tedizolid has excellent oral bioavailability (>90%) and large volume of distribution. Tedizolid’s half-life allows for once-daily dosing. The protein binding of tedizolid is 70%–90%.15,16 Administration of tedizolid with food results in a lower Cmax (4.7 vs 6.4 μg/mL) and longer Tmax (8.0 vs 2.0 hours), but does not meaningfully alter the AUC0–∞ (81.8 vs 79.9 μg · h/mL). Therefore, tedizolid can be administered without regard to food.
Skin and soft tissue concentrations
A microdialysis study of 12 healthy adults was conducted by Sahre et al15 to evaluate the unbound tissue concentrations of tedizolid. A single dose of 600 mg was administered with microdialysis samples collected every 20 minutes for the first 12 hours postdose. The mean free AUC0–12 h was 4.9 mg · h/L for plasma, 5.3 mg · h/L for adipose tissue, and 5.9 mg · h/L for muscle. The resulting fAUCtissue/fAUCplasma was 1.1±0.2 for adipose tissue and 1.2±0.2 for muscle.
Renal dysfunction and dialysis
A pharmacokinetic study was performed to evaluate the effect of severe renal impairment and dialysis on tedizolid exposure.17 Patients with severe renal insufficiency (estimated glomerular filtration rate [eGFR] <30.0 mL/min/1.73 m2) or those with end-stage renal disease requiring dialysis (eGFR <15.0 mL/min/1.73 m2) were compared with a control group (eGFR ≥80 mL/min/1.73 m2). Severe renal impairment had no meaningful effect on tedizolid exposure as estimated by AUC0–∞ (geometric mean ratio 0.925; 90% CI: 0.698–1.227). Similarly, tedizolid’s AUC0–∞ was unaffected by the use of hemodialysis (geometric mean ratio 0.913; 90% CI: 0.827–1.007). The conclusions from these data are reflected in the FDA-approved prescribing information.16
Hepatic dysfunction
Tedizolid’s pharmacokinetics have been evaluated in patients with moderate and severe hepatic impairment.17 Patients with a Child-Pugh classification B (score 7–9) were defined as having moderate hepatic impairment and those under classification C (score 10–15) were defined as having severe hepatic impairment. Patients with moderate hepatic impairment had a 22% higher AUC0–∞ than controls (geometric mean ratio 1.216; 90% CI: 0.862–1.716) and patients with severe hepatic impairment had a 34% AUC0–∞ higher than controls (geometric mean ratio 1.341; 90% CI: 0.927–1.939). Since the geometric mean ratio crosses 1, no dosage adjustment is required. This conclusion is reflected in the FDA-approved prescribing information.16
Obesity
Phase I, II, and III studies of tedizolid reported median weights of 76.10–80.30 kg.18 Obese patients represented 193 of the 647 patients receiving tedizolid in Phase III clinical studies.19 The heaviest patient enrolled in any of these studies weighed 226.4 kg. A population pharmacokinetic study was performed with the data from these trials with the authors concluding that no clinically significant covariates affected tedizolid exposure.
A recent study of obese persons receiving linezolid suggested that a dose adjustment is not needed for persons weighing ≤150 kg. Given the similarities in the two drugs, a dosing alteration for tedizolid is not currently recommended in obese patients. A study evaluating the pharmacokinetics of a single dose study of tedizolid administered intravenously in morbidly obese patients is planned, but recruitment is not open at this time (NCT02342418).20
Pharmacodynamics
Animal studies
Louie et al21 conducted a dose ranging and fractionation study with tedizolid in a neutropenic mouse thigh model. For methicillin-sensitive Staphylococcus aureus (MSSA), the dose and fAUC/MIC at 24 hours (37.7 mg/kg/day, 49.1) and 48 hours (35.3 mg/kg/day, 46) were quite similar. The doses and fAUC/MIC for MRSA were similar to those observed for MSSA at 24 hours (36.2 mg/kg/day, 47.1) and 48 hours (39.8 mg/kg/day, 51.8). In the same animal model, a linezolid dose of 120 mg/kg/day did not achieve bacterial stasis at 24 or 48 hours. The dose fractionation study determined that fAUC/MIC ratio (r2=0.984) best predicted treatment efficacy, compared with fCmax/MIC (r2=0.757) or fT>MIC (r2=0.624).
Drusano et al22 evaluated the impact of granulocytes on staphylococcal cell killing. They found that a “human-equivalent” dose of less than 200 mg/day was needed for stasis when granulocytes were present in the mouse thigh infection model. However, the “human-equivalent” dose increased to approximately 2,000 mg/day when the mice were rendered granulocytopenic. The investigators also noted that the extent of enhancement increased over time (16-fold at 24 hours, 25-fold at 48 hours, and 35-fold at 72 hours). Therefore, additional studies are needed before tedizolid is routinely used in patients with granulocytopenia.
Keel et al23 compared the bacterial killing of linezolid and tedizolid against four strains of MRSA and one strain of MSSA in an immunocompetent murine thigh infection model. There were no significant differences in the two treatments that persisted throughout the 72-hour treatment period. Both agents achieved bacteriostatic effects by 24 hours and were bactericidal by 72 hours.
Clinical studies
A Phase II study evaluated torezolid phosphate doses of 200, 300, and 400 mg daily for the treatment of complicated skin and skin structure infections (cSSSI).24 Clinical cure rates in the modified intention to treat (MITT) population were similar for the 200 mg (89%), 300 mg (89%), and 400 mg (86%) daily regimens. The subset analysis of patients with a severe cSSSI was also similar with 92% (200 mg), 100% (300 mg), and 93% (400 mg) clinical success rates for the MITT population. The similar clinical success rates for all three dosing regimens led to the selection of the 200 mg once-daily dose for the Phase III clinical trials with tedizolid.
A target attainment analysis revealed that 98% of simulated patients who receive tedizolid 200 mg once daily should achieve the fAUC0–24/MIC ratio of ≥3.18 The target attainment rate drops to 71% when simulating a MIC of 1 μg/mL and is 1% for MICs of ≥2 μg/mL. If the fAUC0–24/MIC ratio is adjusted to 2 or 4, the target attainment rate changes to 99.5% or 95.5%. The investigators also found that >99% of patients with tedizolid pharmacokinetic and MIC data in Phase III studies met the pharmacodynamic target.
Efficacy in clinical trials
ESTABLISH-1
The Efficacy and Safety of 6-day Oral Tedizolid in ABSSSI versus 10-day Oral Linezolid Therapy (ESTABLISH-1) trial was held in 81 different study centers from August 2010 through September 2011.25 This Phase III, randomized, double-blind, double-dummy, noninferiority trial evaluated the efficacy and safety of tedizolid 200 mg PO daily for 6 days (n=332) versus linezolid 600 mg PO twice daily for 10 days (n=335) in patients 18 years or older with ABSSSI. The primary outcome was early clinical response at 48–72 hours, which was defined as no increase in lesion surface area from baseline and oral temperature of ≤37.6°C (with recheck to confirm in 3–24 hours). The predefined 10% noninferiority margin was met with an absolute treatment difference of 0.1% (95% CI: −6.1 to 6.2). The results of the secondary outcomes evaluated in ESTABLISH-1 are shown in Table 2. These included clinical response at the end of treatment (day 11 for both groups) and posttherapy evaluation (7–14 days after the end of treatment). The secondary outcomes failed to show statistical significance between the two groups, further supporting the results of the primary outcome. These findings lead to the determination of noninferiority of 200 mg of tedizolid daily for 6 days compared to 600 mg of linezolid twice daily for 10 days.
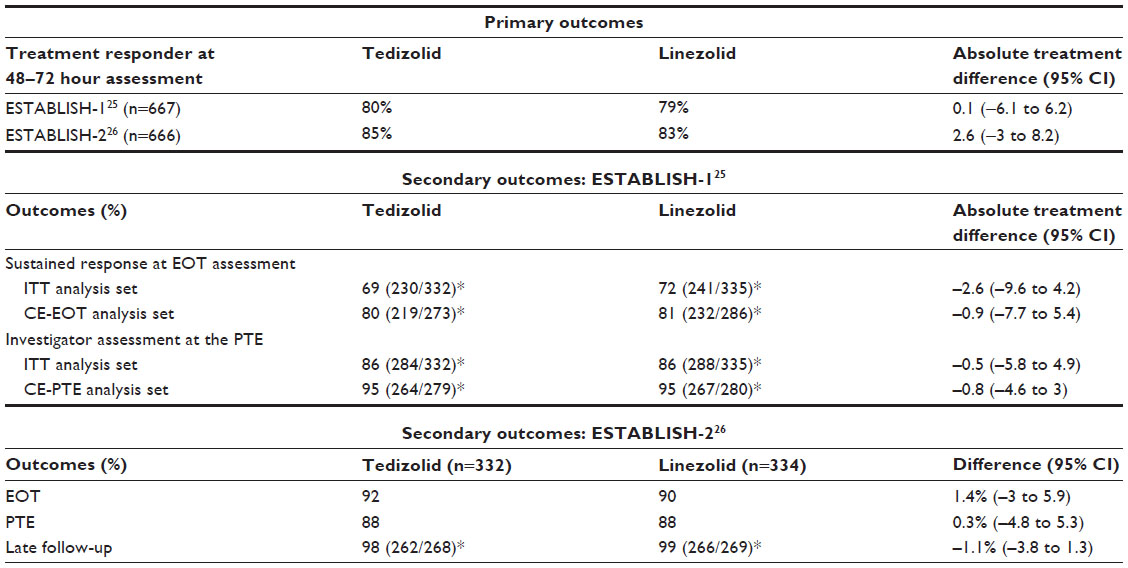 | Table 2 Phase III trials evaluating the safety and efficacy of tedizolid for acute bacterial skin and skin structure infections |
ESTABLISH-2
The Tedizolid for 6 days versus Linezolid for 10 days for ABSSSI (ESTABLISH-2) trial was conducted in 58 institutions from September 2011 to January 2013.26 This randomized, double-blind, Phase III, noninferiority trial of patients ≥12 years of age compared the efficacy and safety of tedizolid 200 mg for 6 days (n=332) compared to linezolid 600 mg twice daily for 10 days (n=334) in patients switching from intravenous (IV) to oral formulation. The primary outcome of early clinical response was defined as at least a 20% reduction in lesion area at 48–72 hours after treatment initiation as compared to baseline. The predefined noninferiority margin of 10% for early clinical response rates was met (treatment difference 2, 95% CI: −3.0 to 8.2). Secondary outcomes (Table 2) included clinical success rates at the end of treatment (day 11), posttherapy assessment (7–14 days after end of treatment), and late follow-up (18–25 days after end of treatment). The secondary outcomes were not statistically significant, further supporting the primary outcome. These findings lead to the determination of noninferiority between IV to oral tedizolid 200 mg daily for 6 days against linezolid 600 mg twice a day for 10 days.
Safety data with tedizolid
ESTABLISH-1
The majority of adverse events reported were gastrointestinal, infections and infestations, and nervous system disorders.25 No statistical analysis was performed on the adverse effects. Gastrointestinal disorder (nausea, vomiting, diarrhea, and dyspepsia) occurred less frequently in patients receiving tedizolid (16% vs 25%). The rates of all other adverse effects were similar for both groups. Low platelet count (<75% the lower limit of normal and <75% of a patient’s abnormally low baseline count) occurred infrequently (tedizolid =2.3%, linezolid =4.9%) and resolved without intervention. Alanine aminotransferase elevations (defined at ≥2× upper limit of normal and ≥2× baseline value) were found in 24 total patients (34% had hepatitis C virus), however none of the patients discontinued the study drug, and the investigators determined that liver dysfunction or toxicity did not exist. The death in the tedizolid group was considered to be unrelated to study treatment (occurred 49 days after last tedizolid dose).
ESTABLISH-2
The lower rates of gastrointestinal disorders with tedizolid observed in the ESTABLISH-1 trial were confirmed in ESTABLISH-2 (16% vs 20%).26 Most adverse events were mild to moderate and similar between the two groups. The incidence of low platelet counts were similar between the tedizolid and linezolid (9% vs 13%, P=0.071). A mild infusion-site reaction occurred in 5 patients in the tedizolid group as compared to 7 in the linezolid group (difference of −0.6, 95% CI: −3 to 1.6). A single patient receiving tedizolid had a myocardial infarction and died. The patient’s death was determined to be not related to tedizolid. None of the adverse events were statistically significant between tedizolid and linezolid.
Serotonin syndrome
In vitro data shows that tedizolid, like linezolid, is a weak and reversible monoamine oxidase inhibitor.27 Two human, randomized, double-blind, placebo-controlled, crossover studies analyzed the combination of tedizolid with oral tyramine or pseudoephedrine and its effect on patient blood pressure. Patients (n=30) were given either placebo or tedizolid 200 mg once daily for the first 2 days of the trial, then on day 3 were started on tyramine 25 mg (increasing by 50 mg/day until an increase in systolic blood pressure of ≥30 mmHg or 575 mg tyramine of day 14). Seven patients had an increase of at least 30 mmHg in systolic blood pressure during both placebo and tedizolid treatment phases. The geometric mean ratio (placebo:tedizolid) was 1.33 (95% CI: 1.05–1.69), indicating no significant interaction since the ratio is below 2.
In the pseudoephedrine trial (n=18), patients that had systolic blood pressure increase of ≥15 mmHg were similar between the tedizolid (22%) and placebo (28%) groups. The maximum changes in systolic blood pressure (tedizolid 11.6 vs placebo 12.1, P=0.73), diastolic blood pressure (tedizolid 6.7 vs placebo 6.8, P=0.90), and heart rate (tedizolid 13.6 vs placebo 15.2, P=0.17) were not statistically significant.
Investigators also performed a murine serotonergic model where head twitches (serving as a measure of serotonin receptor 2A activation) were counted in mice treated with moclobemide 10 mg/kg, fluoxetine 20 mg/kg, linezolid 50 mg/kg, or tedizolid at doses of 10, 30, 100, or 300 mg/kg. The moclobemide (a potent monoamine oxidase inhibitor), fluoxetine (a potent serotonin reuptake inhibitor), and linezolid (a weak monoamine oxidase inhibitor) served as positive controls for head twitch. Unlike these agents, tedizolid did not increase head twitch response at any dose administered. As a result of these three studies tedizolid was concluded to have a low probability of monoamine oxidase related problems, including serotonin syndrome.
Mitochondrial toxicity
Linezolid’s effects on myelosuppression, lactic acidosis, and peripheral/optic neuropathies are thought to be due to its ability to impair mitochondrial protein synthesis (MPS).28 An in vitro study of tedizolid and linezolid with heart mitochondria from rabbits suggested that tedizolid inhibited MPS more potently (50% inhibitory concentration [IC50] 0.31 vs 6.4 μM). Therefore, a 9-month study of rats to compare placebo and high-dose tedizolid was conducted. None of the rats experienced any evidence of neuropathy at tedizolid doses that were approximately eightfold higher than those achieved in humans with therapeutic dosing. Further studies in mice found that tedizolid did not form a stable associated with eukaryotic mitochondria in macrophages. The IC50 was not reached in 84% of simulated patients using data from a population pharmacokinetic study of tedizolid, while only 36% of simulated patients fell below the IC50 threshold for linezolid. While these results suggest tedizolid may cause fewer MPS-related adverse events, clinical data through Phase IV studies and adverse event reporting mechanisms are needed to confirm these findings.
Thrombocytopenia
Thrombocytopenia is typically a concern with oxazolidinones after therapy over a long period of time. Lodise et al5 combined platelet data from both the ESTABLISH-1 and ESTABLISH-2 trials. The investigators analyzed platelet counts at the day 7–9 visit (tedizolid n=555, linezolid n=552), day 11–13 visit (tedizolid n=552, linezolid n=538), and posttherapy evaluation (n=545 in both groups). At the day 7–9 visit, thrombocytopenia was similar between the two groups, and fewer patients had thrombocytopenia in the tedizolid group (3.2% vs 5.6%, relative risk 0.58, 95% CI: 0.33–1.02). At the end of treatment (days 11–13), thrombocytopenia was less common in the tedizolid group (4.9% vs 10.8%, relative risk 0.45, 95% CI: 0.29–0.71). The posttherapy evaluation showed similar results as the previous evaluations, with tedizolid causing less thrombocytopenia (4.2% vs 7.7%, relative risk 0.55, 95% CI: 0.33–0.90). Concern of thrombocytopenia is falsely elevated in oxazolidinones. In a cohort study comparing platelet outcomes in patients receiving either linezolid or vancomycin, platelet counts of ≤50,000 cells/mm3 (linezolid 3.6%, vancomycin 1.2% at 28 days) and ≤20,000 cells/mm3 (linezolid 0.8%, vancomycin 1.2% at 28 days) had low incidence and were not statistically significant between the two groups.29 The incidence of a ≥50% decline in platelet count from baseline was higher in the vancomycin group (31% vs 17%; relative risk 0.55, 95% CI: 0.40–0.77). The investigators noted a positive correlation between vancomycin trough concentrations and the incidence of patients with ≥50% decline in platelet count from baseline.
Pregnancy
Tedizolid is currently a pregnancy category C drug.16 Animal studies have shown an increase in fetal developmental effects when supratherapeutic doses are administered. In mice, doses that are fourfold the estimated human exposure based on AUC resulted in reduced fetal weights and an increase of costal cartilage anomalies. In rats, the incidence of skeletal variations and reduced fetal body weight when exposed to a dose that is sixfold the estimated human exposure, based on AUC. The no observed adverse effect levels for fetal toxicity when tedizolid doses administered to mice or rats were approximately equivalent to human exposures. In addition, tedizolid is excreted in breast milk of rats.
Economic analyses
There are no pharmacoeconomic analyses of tedizolid published in the peer-reviewed literature to date. However, there are several pharmacoeconomic evaluations of linezolid to vancomycin and/or daptomycin.30–37 These studies consistently conclude that linezolid, especially use of the oral formulation, is associated with significant cost savings. While some of these studies suffer from significant design flaws and/or potential conflicts of interest, the cost savings associated with a decreased length of stay and the lack of outpatient parenteral therapy (and the health care personnel required to deliver it) make sense intuitively. However, none of the pharmacoeconomic studies performed to date have compared the oxazolidinones to generic oral treatment (eg, clindamycin, minocycline, trimethoprim/sulfamethoxazole) for the treatment of MRSA ABSSSI.
Conclusion
Tedizolid phosphate is an oxazolidinone with gram-positive activity approved for ABSSSI. Tedizolid is more potent in vitro than linezolid against gram-positive pathogens that cause ABSSSIs, but this increased in vitro potency is not as important clinically due to its decreased fAUC compared with linezolid. Tedizolid is has several key advantages over linezolid including once-daily dosing, decreased treatment duration, minimal interaction with serotonergic agents, possibly associated with less adverse events associated with the impairment of MPS (eg, myelosuppression, lactic acidosis, and peripheral/optic neuropathies), and retains in vitro activity against linezolid-resistant gram-positive bacteria. Economic analyses with tedizolid are needed to describe the cost-effectiveness of this agent compared with other options used for ABSSSI, particularly treatment options active against MRSA.
Disclosure
RGH and HNM have no potential conflicts of interest associated with this work. No funding was provided for this work.
References
Edelsberg J, Taneja C, Zervos M, et al. Trends in US hospital admissions for skin and soft tissue infections. Emerg Infect Dis. 2009;15(9):1516–1518. | |
Jarvis WR, Jarvis AA, Chinn RY. National prevalence of methicillin-resistant Staphylococcus aureus in inpatients at United States health care facilities, 2010. Am J Infect Control. 2012;40(3):194–200. | |
Centers for Disease Control and Prevention. Active Bacterial Core Surveillance Report, Emerging Infections Program Network, Methicillin-Resistant Staphylococcus aureus. 2012. | |
Ray GT, Suaya JA, Baxter R. Incidence, microbiology, and patient characteristics of skin and soft-tissue infections in a US population: a retrospective population-based study. BMC Infect Dis. 2013;13:252. | |
Lodise TP, Fang E, Minassian SL, Prokocimer P. Platelet profile in patients with acute bacterial skin and skin structure infections receiving tedizolid or linezolid: findings from the phase 3 ESTABLISH Clinical Trials. Antimicrob Agents Chemother. 2014;58(12):7198–7204. | |
Shaw KJ, Poppe S, Schaadt R, et al. In vitro activity of TR-700, the antibacterial moiety of the prodrug TR-701, against linezolid-resistant strains. Antimicrob Agents Chemother. 2008;52(12):4442–4447. | |
Chen H, Yang Q, Zhang R, et al. In vitro antimicrobial activity of the novel oxazolidinone tedizolid and comparator agents against Staphylococcus aureus and linezolid-resistant Gram-positive pathogens: a multicentre study in China. Int J Antimicrob Agents. 2014;44(3):276–277. | |
Prokocimer P, Bien P, Deanda C, Pillar CM, Bartizal K. In vitro activity and microbiological efficacy of tedizolid (TR-700) against Gram-positive clinical isolates from a phase 2 study of oral tedizolid phosphate (TR-701) in patients with complicated skin and skin structure infections. Antimicrob Agents Chemother. 2012;56(9):4608–4613. | |
Rodriguez-Avial I, Culebras E, Betriu C, Morales G, Pena I, Picazo JJ. In vitro activity of tedizolid (TR-700) against linezolid-resistant staphylococci. J Antimicrob Chemother. 2012;67(1):167–169. | |
Sahm DF, Deane J, Bien PA, et al. Results of the surveillance of tedizolid activity and resistance program: in vitro susceptibility of gram-positive pathogens collected in 2011 and 2012 from the United States and Europe. Diagn Microbiol Infect Dis. 2015;81(2):112–118. | |
Schaadt R, Sweeney D, Shinabarger D, Zurenko G. In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent. Antimicrob Agents Chemother. 2009;53(8):3236–3239. | |
Thomson KS, Goering RV. Activity of tedizolid (TR-700) against well-characterized methicillin-resistant Staphylococcus aureus strains of diverse epidemiological origins. Antimicrob Agents Chemother. 2013;57(6):2892–2895. | |
Locke JB, Zurenko GE, Shaw KJ, Bartizal K. Tedizolid for the management of human infections: in vitro characteristics. Clin Infect Dis. 2014;58(Suppl 1):S35–S42. | |
Flanagan SD, Bien PA, Munoz KA, Minassian SL, Prokocimer PG. Pharmacokinetics of tedizolid following oral administration: single and multiple dose, effect of food, and comparison of two solid forms of the prodrug. Pharmacotherapy. 2014;34(3):240–250. | |
Sahre M, Sabarinath S, Grant M, et al. Skin and soft tissue concentrations of tedizolid (formerly torezolid), a novel oxazolidinone, following a single oral dose in healthy volunteers. Int J Antimicrob Agents. 2012;40(1):51–54. | |
Sivextro™ lyophilized powder for intravenous injection, oral tablets, tedizolid phosphate lyophilized powder for intravenous injection, oral tablets [Product information]. Cubist Pharmaceuticals, Lexington, MA. 2014. | |
Flanagan S, Minassian SL, Morris D, et al. Pharmacokinetics of tedizolid in subjects with renal or hepatic impairment. Antimicrob Agents Chemother. 2014;58(11):6471–6476. | |
Flanagan S, Passarell J, Lu Q, Fiedler-Kelly J, Ludwig E, Prokocimer P. Tedizolid population pharmacokinetics, exposure response, and target attainment. Antimicrob Agents Chemother. 2014;58(11):6462–6470. | |
Flanagan S, Minassian SL, Passarell J, Fiedler-Kelly J, Prokocimer P. Tedizolid plasma pharmacokinetics are comparable in obese and nonobese patients and healthy subjects. Paper presented at: 24th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID); May 10–13, 2014; Barcelona, Spain. | |
Single Dose PK of IV Tedizolid Phosphate in Morbidly Obese and Non-Obese Adults. Available from: https://www.clinicaltrials.gov/ct2/show/NCT02342418?term=tedizolid&rank=3. Accessed January 30, 2015. | |
Louie A, Liu W, Kulawy R, Drusano GL. In vivo pharmacodynamics of torezolid phosphate (TR-701), a new oxazolidinone antibiotic, against methicillin-susceptible and methicillin-resistant Staphylococcus aureus strains in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(7):3453–3460. | |
Drusano GL, Liu W, Kulawy R, Louie A. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(11):5300–5305. | |
Keel RA, Tessier PR, Crandon JL, Nicolau DP. Comparative efficacies of human simulated exposures of tedizolid and linezolid against Staphylococcus aureus in the murine thigh infection model. Antimicrob Agents Chemother. 2012;56(8):4403–4407. | |
Prokocimer P, Bien P, Surber J, et al. Phase 2, randomized, double-blind, dose-ranging study evaluating the safety, tolerability, population pharmacokinetics, and efficacy of oral torezolid phosphate in patients with complicated skin and skin structure infections. Antimicrob Agents Chemother. 2011;55(2):583–592. | |
Prokocimer P, De Anda C, Fang E, Mehra P, Das A. Tedizolid phosphate vs linezolid for treatment of acute bacterial skin and skin structure infections: the ESTABLISH-1 randomized trial. JAMA. 2013;309(6):559–569. | |
Moran GJ, Fang E, Corey GR, Das AF, De Anda C, Prokocimer P. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): a randomised, double-blind, phase 3, non-inferiority trial. Lancet Infect Dis. 2014;14(8):696–705. | |
Flanagan S, Bartizal K, Minassian SL, Fang E, Prokocimer P. In vitro, in vivo, and clinical studies of tedizolid to assess the potential for peripheral or central monoamine oxidase interactions. Antimicrob Agents Chemother. 2013;57(7):3060–3066. | |
Flanagan S, McKee EE, Das D, et al. Nonclinical and pharmacokinetic assessments to evaluate the potential of tedizolid and linezolid to affect mitochondrial function. Antimicrob Agents Chemother. 2015;59(1):178–185. | |
Patel N, VanDeWall H, Tristani L, et al. A comparative evaluation of adverse platelet outcomes among Veterans’ Affairs patients receiving linezolid or vancomycin. J Antimicrob Chemother. 2012;67(3):727–735. | |
Athanasakis K, Petrakis I, Ollandezos M, et al. Antibacterial treatment of meticillin-resistant staphylococcus aureus complicated skin and soft tissue infections: a cost and budget impact analysis in Greek hospitals. Infect Dis Ther. 2014;3(2):257–268. | |
Barron J, Turner R, Jaeger M, Adamson W, Singer J. Comparing the use of intravenous antibiotics under the medical benefit with the use of oral antibiotics under the pharmacy benefit in treating skin and soft tissue infections. Manag Care. 2012;21(9):44–52. | |
Menzin J, Marton JP, Meyers JL, Carson RT, Rothermel CD, Friedman M. Inpatient treatment patterns, outcomes, and costs of skin and skin structure infections because of Staphylococcus aureus. Am J Infect Control. 2010;38(1):44–49. | |
Schurmann D, Sorensen SV, De Cock E, Duttagupta S, Resch A. Cost-effectiveness of linezolid versus vancomycin for hospitalised patients with complicated skin and soft-tissue infections in Germany. Eur J Health Econ. 2009;10(1):65–79. | |
De Cock E, Sorensen S, Levrat F, et al. Cost-effectiveness of linezolid versus vancomycin for hospitalized patients with complicated skin and soft-tissue infections in France. Med Mal Infect. 2009;39(5):330–340. | |
McCollum M, Sorensen SV, Liu LZ. A comparison of costs and hospital length of stay associated with intravenous/oral linezolid or intravenous vancomycin treatment of complicated skin and soft-tissue infections caused by suspected or confirmed methicillin-resistant Staphylococcus aureus in elderly US patients. Clin Ther. 2007;29(3):469–477. | |
Sharpe JN, Shively EH, Polk HC Jr. Clinical and economic outcomes of oral linezolid versus intravenous vancomycin in the treatment of MRSA-complicated, lower-extremity skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus. Am J Surg. 2005;189(4):425–428. | |
Rosner AJ, Becker DL, Wong AH, Miller E, Conly JM. The costs and consequences of methicillin-resistant Staphylococcus aureus infection treatments in Canada. Can J Infect Dis Med Microbiol. 2004;15(4):213–220. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.