")
Back to Journals » Patient Preference and Adherence » Volume 11
Profile of sodium phenylbutyrate granules for the treatment of urea-cycle disorders: patient perspectives
Authors Peña-Quintana L , Llarena M, Reyes-Suárez D, Aldámiz-Echevarria L
Received 11 March 2017
Accepted for publication 1 August 2017
Published 6 September 2017 Volume 2017:11 Pages 1489—1496
DOI https://doi.org/10.2147/PPA.S136754
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Johnny Chen
Luis Peña-Quintana,1–3 Marta Llarena,2 Desiderio Reyes-Suárez,2 Luis Aldámiz-Echevarria4
1Pediatric Gastroenterology, Hepatology, and Nutrition Unit, Universitario Materno-Infantil Hospital de Canarias, University of Las Palmas de Gran Canaria, 2Research Institute of Biomedical and Health Sciences, University of Las Palmas de Gran Canaria, Las Palmas, 3CIBEROBN, Madrid, 4Unit of Metabolism, Cruces University Hospital, BioCruces Health Research Institute, GCV-CIBER de Enfremedades Raras (CIBERER), Barakaldo, Spain
Abstract: Urea-cycle disorders are a group of rare hereditary metabolic diseases characterized by deficiencies of one of the enzymes and transporters involved in the urea cycle, which is necessary for the removal of nitrogen produced from protein breakdown. These hereditary metabolic diseases are characterized by hyperammonemia and life-threatening hyperammonemic crises. Pharmacological treatment of urea-cycle disorders involves alternative nitrogen-scavenging pathways. Sodium benzoate combines with glycine and phenylacetate/phenylbutyrate with glutamine, forming, respectively, hippuric acid and phenylacetylglutamine, which are eliminated in the urine. Among the ammonia-scavenging drugs, sodium phenylbutyrate is a well-known long-term treatment of urea-cycle disorders. It has been used since 1987 as an investigational new drug, and was approved for marketing in the US in 1996 and the EU in 1999. However, sodium phenylbutyrate has an aversive odor and taste, which may compromise patients’ compliance, and many patients have reported difficulty in taking this drug. Sodium phenylbutyrate granules are a new tasteless and odor-free formulation of sodium phenylbutyrate, which is indicated in the treatment of urea-cycle disorders. This recently developed taste-masked formulation of sodium phenylbutyrate granules was designed to overcome the considerable issues that taste has on adherence to therapy. Several studies have reported the clinical experience of patients with urea-cycle disorders treated with this new tasteless formulation of sodium phenylbutyrate. Analysis of the data indicated that this taste-masked formulation of sodium phenylbutyrate granules improved quality of life for urea-cycle disorder patients. Furthermore, a postmarketing report on the use of the product has confirmed the previous observations of improved compliance, efficacy, and safety with this taste-masked formulation of sodium phenylbutyrate.
Keywords: sodium phenylbutyrate granules, urea-cycle disorders, treatment adherence, quality of life
Urea-cycle disorders
Urea-cycle disorders (UCDs) are a group of rare hereditary metabolic diseases characterized by deficiencies of one of the enzymes and transporters involved in the UC. Six disorders involving different defects in the biosynthesis of these enzymes have been described: CPS1 deficiency1 (MIM 237300), NAGS deficiency (MIM 237310), OTC deficiency2 (MIM 311250), ASS1 deficiency,3 citrullinemia type I (MIM 215700), ASL deficiency4 (MIM 207900), and ARG1 deficiency5 (MIM 207800). Except for OTC deficiency, which is X-linked, these disorders display an autosomal-recessive inheritance.6 Additionally, there are three transporter defects: mitochondrial ornithine carrier (hyperornithinemia–hyperammonemia–homocitrullinuria syndrome; MIM 238970), mitochondrial aspartate–glutamate carrier (citrullinemia type II; MIM 605814, 603471), and dibasic amino-acid carrier (hyperdibasic aminoaciduria or lysinuric protein intolerance; MIM 222700).6 The incidence of UCDs is estimated to 1:8,000–1:44,000 births.7,8 However, these numbers may underestimate prevalence, due to unreliable newborn-screening programs and underdiagnosis of these disorders in fatal cases.
Clinical symptoms
UCDs may present during the neonatal period, after a variable symptom-free interval. In these cases, they are characterized by overwhelming illness, which rapidly progresses from poor feeding, vomiting, lethargy, and/or irritability to coma and/or death.9–14 However, most of these disorders usually display a late-onset form (childhood and/or adulthood), which is caused by partial enzymatic activity deficiencies. Their symptoms, which are less severe and more variable, include poor developmental progress, behavioral problems, hepatomegaly, and gastrointestinal disorders. In patients with UCDs, acute episodes of hyperammonemia are recurrent and can be triggered by metabolic stress in response to infection, trauma, surgery, or pregnancy.9–14 In addition, patients with UCDs often present chronic neurological illness. In this regard, hyperammonemia-related neurologic injury ranges from lethal cerebral edema to mild or subclinical cognitive impairment among individuals with milder phenotypes. Furthermore, abnormalities in executive function, manifested by difficulty in goal setting, planning, monitoring progress, and purposeful problem solving significantly impair day-to-day function among children with UCDs, even those with milder disease who present beyond the neonatal period.15
The pathogenesis of hyperammonemia in UCDs is produced in part by altered ammonia detoxification to urea and in part by enhanced catabolism of glutamine, mostly in skeletal muscle, but also to a lesser degree in the brain and likely in the lungs. The majority of glutamine released into circulation is catabolized back into ammonia in enterocytes and the kidneys.16
Management and treatment
Timely diagnosis and prompt treatment are crucial for the management of UCDs. The absence of effective early treatment results in ammonia accumulation, which leads to irreversible brain damage. The main goal in the management of patients with UCD is to achieve good metabolic control of the disease, while ensuring that nutritional requirements are met. Therefore, the long-term treatment of patients with UCDs is based on dietary protein restriction and dietary supplements that consist of all essential amino acids, vitamins, and minerals.17 This therapy is complemented with the use of ammonia-scavenging drugs, such as benzoate and phenylacetate (PA)/phenylbutyrate (PB). These drugs prevent the accumulation of ammonia providing an alternative pathway for nitrogen disposal through the combination with glycine in the case of benzoate and glutamine for PA/PB, forming hippuric acid and phenylacetylglutamine, respectively. For each mole of benzoate and PA/PB, 1 and 2 mol of ammonia are eliminated easily in the urine. On the other hand, the UC could be relieved by diverting its substrates. By this way of avoiding the UC, urea synthesis is enhanced, in order to support nitrogen homeostasis further.7,18,19 Figure 1 shows the mechanism by which PA/PB decreases ammonia. In addition, patients of all ages with NAGS deficiency are treated with carglumic acid, which is a synthetic form of NAG. This chemical compound acts as a replacement for the cofactor NAG, which is the key to starting the UC and thus the process of removing excess ammonia.20
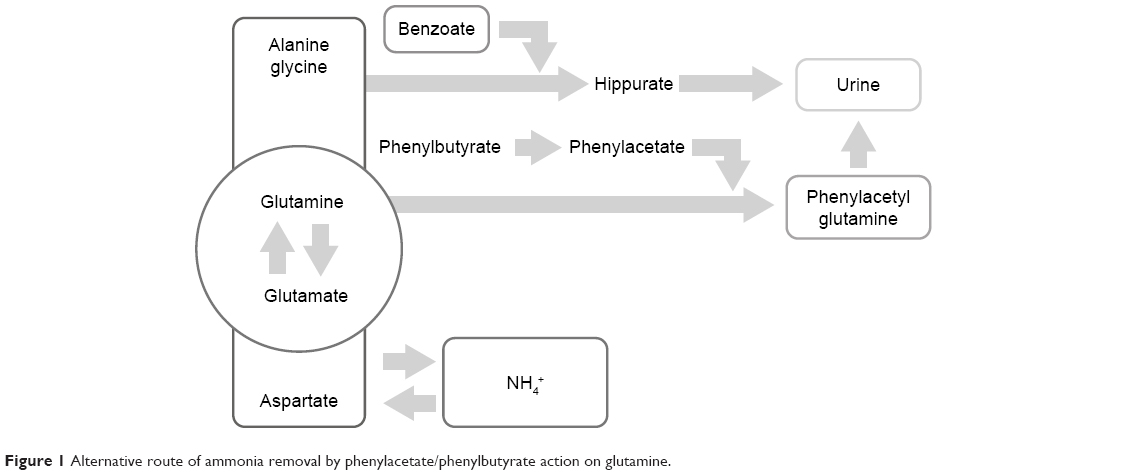 | Figure 1 Alternative route of ammonia removal by phenylacetate/phenylbutyrate action on glutamine. |
Ammonia-scavenging drugs
As mentioned, disruptions of nitrogen homeostasis lead to an excessive and noxious accumulation of ammonia. It is well known that elevated ammonia levels (>100 μmol/L) are extremely toxic to the central nervous system. Indeed, patient prognosis is considered to be very poor when a hyperammonemic coma has lasted more than 3 days, intracranial pressure is markedly elevated, or when ammonia levels are >1,000 μmol/L.21,22 For this reason, the use of drugs that are able to lower ammonia levels and act as ammonia scavengers are included in the therapeutic regimens for acute and chronic treatment of UCDs (Table 1).
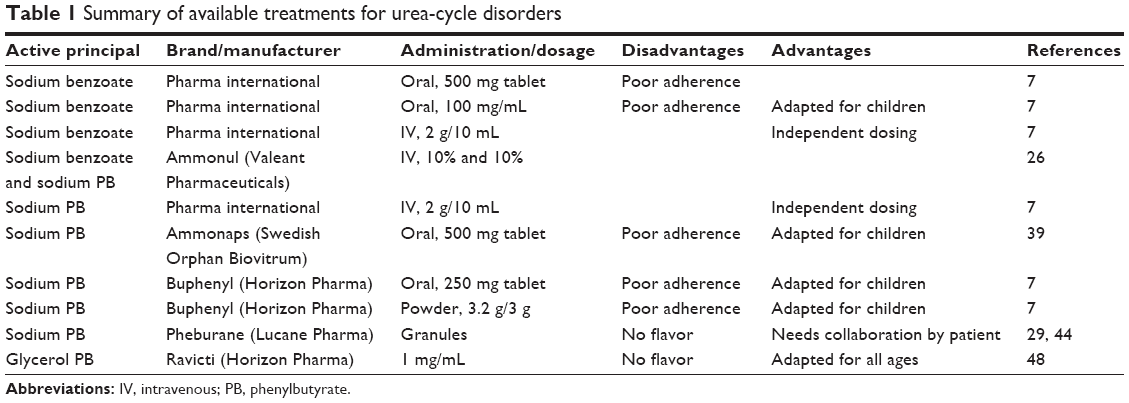 | Table 1 Summary of available treatments for urea-cycle disorders |
Benzoate was the first-described ammonia-scavenging drug. Its use was proposed in 1914.23 Based on previously published studies, sodium benzoate was administered as a supplement to 26 children with defective ureagenic pathways. The results indicated that benzoate could be used to control defects in nitrogen excretion.24 Moreover, the same authors also showed that sodium PA supplementation caused ammonia elimination by its conjugation to glutamine and phenylacetylglutamine excretion in patients with UCDs.25 These studies led to the approval in the early 1980s by the US Food and Drug Administration (FDA) of combined therapy with sodium benzoate and sodium PA to treat hyperammonemia in UCDs. In 2005, an intravenous combination of sodium PA and sodium benzoate (Ammonul; Valeant Pharmaceuticals North America LLC, Bridgewater, NJ, USA) was approved by the FDA.26 One very significant drawback to PA therapy was the awful odor caused by any spilled medicine and by the sweat and urine of the patients. To overcome this disadvantage, the use of PB, which is a metabolic precursor of PA with a less disagreeable odor, was proposed. Therefore, in 1983, a further amendment permitted the replacement of PA by PB. In 1987, the first PB-derivative salt (sodium PB [NaPB]) was introduced experimentally as a new investigational drug. Finally, the use of NaPB to replace the combined therapy25 was approved as a safe, well-tolerated monotherapy orphan drug for the treatment of UCDs.27 Therefore, the marketing of NaPB (Ammonaps and Buphenyl) for UCD was approved in the US by the FDA in 1996 and in Europe by the European Medicines Agency (EMA) in 1999.28 In addition, there has been a recently developed taste-masked formulation of NaPB granules (Pheburane)29 to overcome the considerable issues that taste has on adherence to therapy. Furthermore, there are other investigational agents being developed for the treatment of UCDs, such as glycerol PB (HPN-100; Hyperion Therapeutics, San Francisco, CA, USA).30,31
Common adverse effects of ammonia-scavenging drugs
Commonly reported adverse effects of combined therapy with sodium benzoate–sodium PA include disorders affecting the respiratory, blood, lymphatic, and nervous systems or metabolism and nutrition and vomiting.26 With regard to PA/PB administration, although it is well tolerated in most cases, hepatotoxicity side effects associated with interactions between these drugs and liver cytochrome P450 have been described.32 Furthermore, it should be mentioned that PA/PB acts as a histone deacetylase inhibitor and has been investigated for use in the treatment of a number of malignant disorders. Therefore, PA/PB administration may cause serious adverse effects, eg, liver injury.33,34
Levels of glutamine in the blood and various tissues in UCDs are generally elevated. PA/PB therapy can induce depletion of glutamine, so its levels should be monitored regularly. Good control is considered to be 800−;1,000 μmol/L of glutamine in the blood. Glutamine depletion may activate branched-chain amino acid (BCAA) catabolism and cause depletion of BCAAs,35–37 which may increase the risk of endogenous protein catabolism,38 suggesting a potential need for BCAA supplementation to PA/PB therapy. The main source of amino nitrogen for synthesis of glutamate (precursor of glutamine) is BCAAs.16 On the other hand, glutamine depletion may exert adverse effects on the body, such as impaired immunoresponse and gut integrity.16
NaPB represents an improvement on PA therapy for UCDs. However, it still requires the ingestion of up to 40 large capsules a day, has a disagreeable taste, and sickens some individuals. Indeed, taste disturbance and vomiting have been reported as “frequent” unpleasant adverse events associated with the drug’s oral ingestion in the summary of product characteristics of NaPB (Ammonaps).39 Moreover, dysgeusia/ageusia can also compromise the flavor of food, which is an additional problem in a patient with UCD, whose feeding is already very difficult due to dietary restrictions and need for supplementation. In fact, this adds a significant burden to the stress of families, who are faced with the daily challenge of getting their children to take medicine for this life-threatening disease.40 This well-known notoriously aversive taste of NaPB40 has been confirmed by electronic tongue and real-time dissolution in vitro studies, as well as by studies in healthy volunteers.41 Due to the unacceptability of the NaPB formulation(s) on the market, some patients can either not take the drug (even if reformulated) or change its administration, eg, via nasogastric tubing or gastrotomy. There are common practical attempts used by parents and medical staff to dilute the drug or try to obscure its taste by mixing or sprinkling it with foods or drinks. However, these approaches may lead to impaired efficacy, due to incomplete drug dosing if the mixture is not fully consumed, and may also cause the child to develop aversion to the foods used in the masking process. As a consequence, there is an ineffectiveness of the treatment that can lead to irreversible brain damage secondary to hyperammonemia. In this line of research, several studies have shown that at the recommended doses for the treatment of UCDs (250–500 mg/kg/day, maximum 12 g/day), NaPB decreases appetite, disturbs taste, and causes disagreeable body odor in approximately 5% of patients.28,40 Furthermore, chronic treatment with PB causes menstrual dysfunction/amenorrhea in about 25% of postpubertal females.28
Sodium phenylbutyrate granules
As mentioned, dietary restriction combined with NaPB is one of the main therapeutic modalities used in chronic treatment for UCDs, and has been available for over three decades.25 Due to the palatability issues associated with NaPB, there has been recent interest in the development of new formulations of this drug to overcome the serious issue of its foul, bitter taste, which may affect patient adherence to the treatment. Over the years, attempts to mask this bitter and extremely unpleasant taste in food and drink have failed, causing these children to be undertreated, with resultant poor metabolic control. The development of a taste-masked granule formulation of NaPB that can be swallowed before taste receptors are stimulated is an example of a therapeutic victory achieved through technological solutions.41,42 NaPB granules (Pheburane) are a new taste-masked and odor-free formulation of NaPB that has been developed by Lucane Pharma (Paris, France).29 NaPB granules are small spherical sugar cores coated with NaPB and ethyl cellulose in two separate layers. Ethyl cellulose is a well-known taste-masking agent for active substances.41 This taste-masked granule formulation of NaPB begins to release NaPB after a lag of approximately 10 seconds, followed by slow release over several minutes.29 In contrast, the market-licensed NaPB releases the active drug fully and immediately, leading to immediate release of the foul taste. As such, the taste-masked granule formulation of NaPB creates a window of opportunity to swallow the formulation completely before its taste and odor become apparent.
The profile of NaPB granules has been described in detail by the EMA.29 This document provides a full description of several aspects of this drug, including therapeutic indications, posology, method of administration, contraindications, and special warnings/precautions for use. In addition, it includes the safety profile of NaPB granules with a tabulated list of reported adverse reactions by system-organ class and frequency. In clinical trials with NaPB, 56% of the patients experienced at least one adverse event, and 78% of these were considered not related to NaPB. Adverse reactions mainly involved the reproductive and gastrointestinal system.29 The following common adverse reactions were reported: blood and lymphatic system disorders (anemia, thrombocytopenia, leukopenia, leukocytosis, thrombocytosis), metabolism and nutrition disorders (metabolic acidosis, alkalosis, decreased appetite), gastrointestinal disorders (abdominal pain, vomiting, nausea, constipation, dysgeusia), skin and subcutaneous tissue disorders (rash, abnormal skin odor), and renal and urinary disorders (renal tubular acidosis).29 From investigations, decreased blood potassium, albumin, total protein, and phosphate and increased blood alkaline phosphatase, transaminases, bilirubin, uric acid, chloride, phosphate, sodium, and weight were reported.29 The EMA profile also includes the pharmacological (pharmacodynamic and pharmacokinetic) properties of NaPB granules.29 It should be pointed out that NaPB granules should be administered orally; therefore, administration requires the cooperation of the patient.29
To date, two clinical trials have shown the results of the use of taste-masked NaPB granules in patients with UCD under a French-cohort temporary utilization authorization (ATU) protocol.43,44 In September 2012, the French medicines agency granted a cohort-ATU protocol for NaPB granules, allowing its use in UCD patients not able to tolerate the marketed product due to its unpalatability. NaPB granules were granted market authorization in the EU on July 31, 2013, and thus the last date for inclusion in the cohort-ATU protocol was October 31, 2013. In 2014, Kibleur et al published the results of the first French nationwide 1-year cohort study on 25 patients, of whom 21 were children.43 The aim of this study was to describe a nationwide system for premarketing follow-up of NaPB granules in France and to analyze safety and efficacy in this cohort of patients treated with UCDs. Most patients joined the study due to major issues with the taste of the marketed formulations of NaPB, as indicated by the results from patients’ surveys for assessment of palatability and ease or difficulty of administration of the available marketed drug. After just one dose of NaPB granules, taste and global acceptability evaluations indicated a dramatic increase in acceptability, a decrease in perceived bitterness, and an inverse correlation between general acceptability and bitterness compared with after one dose of the licensed marketed product.43 In addition, no patient in the ATU protocol reported any adverse event related to vomiting following administration of NaPB granules compared to when receiving marketed NaPB prior to entry into the cohort-ATU protocol.43 The absence of such vomiting reflex following drug intake with NaPB granules obviates the need for redosing and thus the risk of overdose or the risk of underdosing in those cases when the dose was not adequately readministered. Moreover, no additional measure, such as reformulation into capsules, was required to administer NaPB granules, which were taken orally in all patients in the ATU follow-up.43 This represents a dramatic improvement in care for these patients. The most important clinical outcome was the change in episodes of hyperammonemia. In the cohort-ATU protocol, the number of hyperammonemic episodes decreased from 20, as reported in ten licensed NaPB-treated patients in the previous 6 months, to 0 in the same patients treated with NaPB granules over a period of 3–11 months.43 Although the cohort-ATU protocol was not designed to collect any measurement of compliance or quality of life (QoL), analysis of the data indicated that the QoL of UCD patients and their families improved greatly with NaPB granules, as assessed on ease of administration and meaningfully reduced incidence of adverse events, notably vomiting and dysgeusia, which impair patients’ well-being and/or their acceptance/compliance with pharmaceutical treatments.43
In January 2016, Kibleur and Guffon reported the results of further follow-up of part of the original ATU-cohort protocol.44 The aim of this study was to describe the status of patients with UCDs at the latest long-term clinical follow-up of treatment with NaPB granules. Patients from the original ATU cohort were followed up every 6–12 months at one reference center. Long-term follow-up data supported the previously observed improvements in drug tolerability.43 Moreover, follow-up data also confirmed the protective effect of NaPB granules against metabolic decompensations. Indeed, in the long term, improved biochemical control, as assessed by an absence of episodes of clinical decompensation, reduction in plasma ammonia and glutamine levels, and improved neurocognitive and height:weight status, were observed in the group of patients treated with NaPB granules.44 This observed improved clinical status after long-term administration of NaPB granules may reflect efficacy and improved compliance with this tasteless formulation of NaPB, as well as indicating an improvement in patients’ and their families’ QoL, as expected with tasteless products.31,45 It is worth mentioning that the safety of NaPB granules was also confirmed from experience in other patients in Sweden and in Turkey who received the drug under a named patient program prior to marketing approval.43,46
In 2015, Uçar et al reported on 1-year usage of NaPB granules in a case of late-onset ASL deficiency on a named patient program in Turkey.46 The family reported the child’s refusal to eat or drink due to the taste and smell of NaPB, as well as their unsuccessful attempts to mask the taste of the drug in food. The consequences of the child not taking the medication added a significant burden to the stress already experienced by the parents, as indicated by their QoL score. For these reasons, the tasteless and odor-free formulation NaPB granules was prescribed. After 1 month of treatment, the patient became fully compliant with the drug. In addition, no hyperammonemia episodes occurred over 1 year of treatment. Moreover, a decrease in family stress and anxiety was observed, with QoL measurement indicating a significant improvement.46
Other future alternatives: glycerol phenylbutyrate
In recent years, great effort has gone into developing new derivatives of ammonia-scavenging drugs with fewer adverse effects, such as palatability issues, intake of high numbers of capsules, or high sodium content. It should be pointed out that the maximum approved daily dose of NaPB (20 g) contains approximately 2,400 mg of sodium, which exceeds the daily allowance of 2,300 mg/day for the general population and 1,500 mg/day for individuals with hypertension and certain other sodium-retaining states, as recommended by the US Department of Health and Human Services in the 2005 Dietary Guidelines for Americans.47
In this line of research, the use of the glycerol PB, which consists of three molecules of PB and a glycerol backbone, has been recommended as an option to replace NaPB. In fact, glycerol PB (HPN-100; Hyperion Therapeutics) has been approved for treatment of UCD. Glycerol PB was shown to be far more tolerable when administered to patients than NaPB, since this drug has the advantage of being available as a tasteless, odorless liquid that can be given in doses of a few teaspoons a day. Moreover, its pharmacokinetic properties are characterized by slower, more effective release of the active metabolite (PA) when compared with the unconjugated PB. In this regard, Lee et al demonstrated that glycerol PB was as effective as NaPB in delivering PB to the body.30 Furthermore, several clinical trials have shown that glycerol PB was effective in maintaining ammonia levels in a desirable range, and the rate of hyperammonemia episodes and associated complications were reduced. In particular, the conclusions from a pivotal Phase III study of glycerol PB for UCD and from short- and long-term ammonia-control and neurocognitive outcomes from 91 UCD patients in four clinical trials showed that glycerol PB exhibited favorable pharmacokinetics and ammonia control relative to NaPB, and that long-term glycerol PB treatment in pediatric patients was associated with improved executive function.31 Finally, it has been demonstrated that glycerol PB is well tolerated and has fewer gastrointestinal complications than sodium benzoate or NaPB treatment.48
Conclusion
For over 40 years, ammonia-scavenging drugs have been used in the treatment of UCDs. Benzoate, PA/PB, and NaPB target liver-nitrogen metabolism by offering an alternative pathway for nitrogen disposal through the urinary excretion of hippurate and phenylacetylglutamine. Their safety and efficacy in the treatment of UCDs is well established,18 and they are usually well tolerated among patients. However, in recent years the identification of novel ammonia-scavenging derivatives, which display improved actions and exhibit fewer overall adverse effects, has been a hot topic in research.
It is well known that due to NaPB’s unpleasant taste and odor, many patients with UCDs have reported that they could not take the drug. As such, patients’ nonacceptance of the marketed NaPB product led to the optimization and development of novel pharmacological derivatives of this drug, such as NaPB granules (Pheburane), or other new derivatives with distinct pharmacological characteristics, such as glycerol PB.30
With this review, we have shared reported experience in the use of NaPB granules with the aim of informing clinicians about this new option for treatment in UCDs.43,44,46 Firstly, the new granule formulation of NaPB has been shown to be effectively taste-masked.41,42 Indeed, according to patient surveys, taste and global acceptability are improved. Secondly, dysgeusia or vomiting was not reported by patients following NaPB-granule intake compared to marketed NaPB formulations. Therefore, fewer adverse effects due to over/underdosing might be expected with NaPB granules. Thirdly, and probably the most important clinical outcome, a decrease in episodes of hyperammonemia was observed in a cohort of NaPB granule-treated patients. Finally, this taste-masked formulation of NaPB also improved compliance in treatment and QoL for UCD patients and their families.
Finally, considering that new aspects of NaPB usage are emerging that may indicate further therapeutic activity, the development of a new NaPB formulation that is far more palatable is extremely important. In this regard, it has been shown that PB enhances pyruvate dehydrogenase-complex enzymatic activity in vitro and in vivo by increasing the proportion of unphosphorylated enzymes through inhibition of pyruvate dehydrogenase kinase. These findings suggest the potential use of PB for treatment of patients with pyruvate dehydrogenase-complex deficiency and other forms of primary and secondary lactic acidosis.49 Moreover, PB has also been used as chemical chaperone in the frame of other diseases, eg, in patients with rare biliary diseases associated with defects in the BSEP/ABCB11 transporter.
Author contributions
LPQ, DRS, and LAE conceptualized and designed the review and drafted the initial manuscript. ML drafted the manuscript. All authors contributed toward data analysis, drafting and critically revising the paper, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Sumar ML, Dasouki MJ, Schofield PJ, et al. Physical and linkage mapping of human carbamyl phosphate synthetase I (CPS1) and reassignment from 2p to 2q35. Cytogenet Cell Genet. 1995;71(3):266–267. | ||
Choi JH, Lee BH, Kim JH, et al. Clinical outcomes and the mutation spectrum of the OTC gene in patients with ornithine transcarbamylase deficiency. J Hum Genet. 2015;60(9):501–507. | ||
Engel K, Höhne W, Häberle J. Mutations and polymorphisms in the human argininosuccinate synthetase (ASS1) gene. Hum Mutat. 2009;30(3):300–307. | ||
Erez A, Nagamani SC, Lee B. Argininosuccinate lyase deficiency: argininosuccinic aciduria and beyond. Am J Med Genet C Semin Med Genet. 2011;157:45–53. | ||
Sin YY, Baron G, Schulze A, Funk CD. Arginase-1 deficiency. J Mol Med (Berl). 2015;93(12):1287–1296. | ||
Brusilow SW, Horwich AL. Urea cycle enzymes. In: Driver CR, Beaded AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001:1909–1963. | ||
Häberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. | ||
Sumar ML, Koelker S, Freedenberg D, et al. The incidence of urea cycle disorders. Mol Genet Metab. 2013;110(1–2):179–180. | ||
Msall M, Batshaw ML, Suss R, Brusilow SW, Mellits ED. Neurologic outcome in children with inborn errors of urea synthesis: outcome of urea-cycle enzymopathies. N Engl J Med. 1984;310(23):1500–1505. | ||
Bachmann C. Outcome and survival of 88 patients with urea cycle disorders: a retrospective evaluation. Eur J Pediatr. 2003;162(6):410–416. | ||
Nassogne MC, Héron B, Touati G, Rabier D, Saudubray JM. Urea cycle defects: management and outcome. J Inherit Metab Dis. 2005;28(3):407–414. | ||
Martín-Hernández E, Aldámiz-Echevarría L, Castejón-Ponce E, et al. Urea cycle disorders in Spain: an observational, cross-sectional and multicentric study of 104 cases. Orphanet J Rare Dis. 2014;9:187. | ||
Kölker S, Cazorla AG, Valayannopoulos V, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders – part 1: the initial presentation. J Inherit Metab Dis. 2015;38(6):1041–1057. | ||
Kölker S, Valayannopoulos V, Burlina AB, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders – part 2: the evolving clinical phenotype. J Inherit Metab Dis. 2015;38(6):1059–1074. | ||
Krivitzky L, Babikian T, Lee HS, Thomas NH, Burk-Paull KL, Batshaw ML. Intellectual, adaptive, and behavioral functioning in children with urea cycle disorders. Pediatr Res. 2009;66(1):96–101. | ||
Holecek M. Evidence of a vicious cycle in glutamine synthesis and breakdown in pathogenesis of hepatic encephalopathy: therapeutic perspectives. Metab Brain Dis. 2014;29(1):9–17. | ||
Berry GT, Steiner RD. Long-term management of patients with urea cycle disorders. J Pediatr. 2001;138(1 Suppl):S56–S61. | ||
Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr. 1996;43:127–170. | ||
Urea Cycle Disorders Conference Group. Consensus statement from a conference for the management of patients with urea cycle disorders. J Pediatr. 2001;138(1 Suppl):S1–S5. | ||
Carbaglu [prescribing information]. Lebanon (NJ): Recordati Rare Diseases; 2015. | ||
Picca S, Dionisi-Vici C, Abeni D, et al. Extracorporeal dialysis in neonatal hyperammonemia: modalities and prognostic indicators. Pediatr Nephrol. 2001;16(11):862–867. | ||
Bachmann C. Outcome and survival of 88 patients with urea cycle disorders: a retrospective evaluation. Eur J Pediatr. 2003;162(6):410–416. | ||
Lewis HB. Studies in the synthesis of hippuric acid in the animal organisms – II: the synthesis and rate of elimination of hippuric acid after benzoate ingestion in man. J Biol Chem. 1914;18(2):225–231. | ||
Batshaw ML, Brusilow S, Waber L, et al. Treatment of inborn errors of urea synthesis: activation of alternative pathways of waste nitrogen synthesis and excretion. N Engl J Med. 1982;306(23):1387–1392. | ||
Brusilow S, Tinker J, Batshaw ML. Amino acid acylation: a mechanism of nitrogen excretion in inborn errors of urea synthesis. Science. 1980;207(4431):659–661. | ||
US Food and Drug Administration. Drug approval package: Ammonul (sodium phenylacetate and sodium benzoate) injection. 2007. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2005/020645s000TOC.cfm. Accessed August 5, 2017. | ||
Brusilow SW. Phenylacetylglutamine may replace urea as a vehicle for waste nitrogen excretion. Pediatr Res. 1991;29(2):147–150. | ||
Batshaw ML, MacArthur RB, Tuchman M. Alternative pathway therapy for urea cycle disorders: twenty years later. J Pediatr. 2001;138(1 Suppl):S46–S55. | ||
European Medicines Agency. Summary of product characteristics: Pheburane. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002500/WC500147373.pdf. Accessed August 5, 2017. | ||
Lee B, Rhead W, Diaz GA, et al. Phase 2 comparison of a novel ammonia scavenging agent with sodium phenylbutyrate in patients with urea cycle disorders: safety, pharmacokinetics and ammonia control. Mol Genet Metab. 2010;100(3):221–228. | ||
Diaz GA, Krivitzky LS, Mokhtarani M, et al. Ammonia control and neurocognitive outcome among urea cycle disorder patients treated with glycerol phenylbutyrate. Hepatology. 2013;57(6):2171–2179. | ||
Shneider BL, Vockley J. Possible phenylacetate hepatotoxicity during 4-phenylbutyrate therapy of Byler disease. J Pediatr Gastroenterol Nutr. 2016;62(3):424–428. | ||
Iannitti T, Palmieri B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D. 2011;11(3):227–249. | ||
Holecek M, Vodenicarovova M. Phenylbutyrate exerts adverse effects on liver regeneration and amino acid concentrations in partially hepatectomized rats. Int J Exp Pathol. 2016;97(3):278–284. | ||
Rodney S, Boneh A. Amino acid profiles in patients with urea cycle disorders at admission to hospital due to metabolic decompensation. JIMD Rep. 2013;9:97–104. | ||
Holecek M, Kandar R, Sispera L, Kovarik M. Acute hyperammonemia activates branched-chain amino acid catabolism and decreases their extracellular concentrations: different sensitivity of red and white muscle. Amino Acids. 2011;40(2):575–584. | ||
Holecek M, Sprongl L, Tichý M. Effect of hyperammonemia on leucine and protein metabolism in rats. Metabolism. 2000;49(10):1330–1334. | ||
Burrage LC, Jain M, Gandolfo L, Lee BH. Sodium phenylbutyrate decreases plasma branched-chain amino acids in patients with urea cycle disorders. Mol Genet Metab. 2014;113(1–2):131–135. | ||
Ammonaps 940 mg/g granules [summary of product characteristics]. Stockholm: Swedish Orphan Biovitrum AB; 2010. | ||
Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr. 1996;43:127–170. | ||
Guffon N, Kibleur Y, Copalu W, Tissen C, Breitkreutz J. Developing a new formulation of sodium phenylbutyrate. Arch Dis Child. 2012;97(12):1081–1085. | ||
Rieder M. How sweet it isn’t: a new formulation of sodium phenylbutyrate and the challenge of palatability for medicines for children. Arch Dis Child. 2012;97(12):1080. | ||
Kibleur Y, Dobbelaere D, Barth M, Brassier A, Guffon N. Results from a nationwide cohort temporary utilization authorization (ATU) survey of patients in France treated with Pheburane (sodium phenylbutyrate) taste-masked granules. Paediatr Drugs. 2014;16(5):407–415. | ||
Kibleur Y, Guffon N. Long-term follow-up on a cohort temporary utilization authorization (ATU) survey of patients treated with Pheburane (sodium phenylbutyrate) taste-masked granules. Paediatr Drugs. 2016;18(2):139–144. | ||
Cederbaum S, LeMons C, Batshaw ML. Alternative pathway or diversion therapy for urea cycle disorders now and in the future. Mol Genet Metab. 2010;100(3):219–220. | ||
Uçar SK, Ozbaran B, Altinok YA, et al. One year experience of Pheburane (sodium phenylbutyrate) treatment in a patient with argininosuccinate lyase deficiency. JIMD Rep. 2015;19:31–33. | ||
US Department of Health and Human Services. Dietary Guidelines for Americans 2005. Washington: US Government Printing Office; 2005. | ||
Oishi K, Diaz G. Glycerol phenylbutyrate for the chronic management of urea cycle disorders. Expert Rev Endocrinol Metab. 2014;9(5):427–434. | ||
Ferriero R, Manco G, Lamantea E, et al. Phenylbutyrate therapy for pyruvate dehydrogenase complex deficiency and lactic acidosis. Sci Transl Med. 2013;5(175):175ra31. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.