")
Back to Journals » OncoTargets and Therapy » Volume 7
Profile of selumetinib and its potential in the treatment of melanoma
Received 4 April 2014
Accepted for publication 1 July 2014
Published 19 September 2014 Volume 2014:7 Pages 1631—1639
DOI https://doi.org/10.2147/OTT.S51596
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Dae Won Kim, Sapna P Patel
Department of Melanoma Medical Oncology, University of Texas MD Anderson Cancer Center, Houston, TX, USA
Abstract: The mitogen-activated protein kinase (MAPK) pathway is a critical oncogenic driver signal in a number of malignancies. The discovery of activating mutations in the MAPK pathway has led to the development of MAPK pathway inhibitors. Selumetinib is a potent and selective inhibitor of MEK1 and MEK2, which are essential downstream molecules in the MAPK pathway. Several preclinical and clinical studies have demonstrated the promising antitumor activity of selumetinib. In this review, we discuss the MAPK pathway in melanoma and summarized data from preclinical and clinical studies of selumetinib for advanced melanoma.
Keywords: selumetinib, MEK inhibitor, melanoma, uveal melanoma
Introduction
Melanoma is the most aggressive skin cancer, and both the incidence of melanoma and melanoma-related deaths have increased over the last several decades in the United States, with more than 76,000 new cases and more than 9,000 deaths predicted in 2013.1 A majority of patients with melanoma are diagnosed early with local disease, and surgical resection is the mainstay of treatment for localized melanoma with excellent outcome. In contrast, patients with metastatic melanoma have poor outcomes, with a 6–8 month median survival.2 Furthermore, the treatment for advanced melanoma is quite challenging. Until 2011, only two drugs, dacarbazine and recombinant human high-dose interleukin 2, were approved for the treatment of metastatic melanoma by the US Food and Drug Administration (FDA). Unfortunately, both drugs have a low response rate (5%–15%), and high-dose interleukin 2 also is associated with significant systemic toxicities.3
The rapid technical advancement in molecular biology and tumor immunology led to new treatment approaches such as targeted therapy and immunotherapy. Ipilimumab, a monoclonal anticytotoxic T-lymphocyte-associated antigen 4 antibody, was approved by the FDA in 2011 on the basis of significant improvement in progression-free survival (PFS) and overall survival (OS) compared with gp100 vaccine treatment.4 Recently, mitogen-activated protein kinase (MAPK) pathway inhibitors, including selective BRAF inhibitors (dabrafenib and vemurafenib) and a selective MEK inhibitor (trametinib), have shown significant improvement of PFS and OS over chemotherapy in patients with BRAF mutant melanoma, which led to the approval of these drugs.5–7 Despite these promising new agents, the majority of metastatic melanomas are not curable. Therefore, effective and novel approaches are still desperately needed for metastatic melanoma.
MAPK pathway
One of the biggest milestones in melanoma research is the identification of BRAF gene mutations in melanoma.8 BRAF is one of the key molecules in the MAPK pathway, which is frequently dysregulated in human cancer.9 MAPK pathway, composed of RAS-RAF-MEK-ERK, is associated with cell proliferation and growth (Figure 1). Up to 75% of melanomas have dysregulated MAPK signaling pathway via BRAF (50%) or NRAS mutations (15%–25%), which results in unregulated cell proliferation and growth.8,10
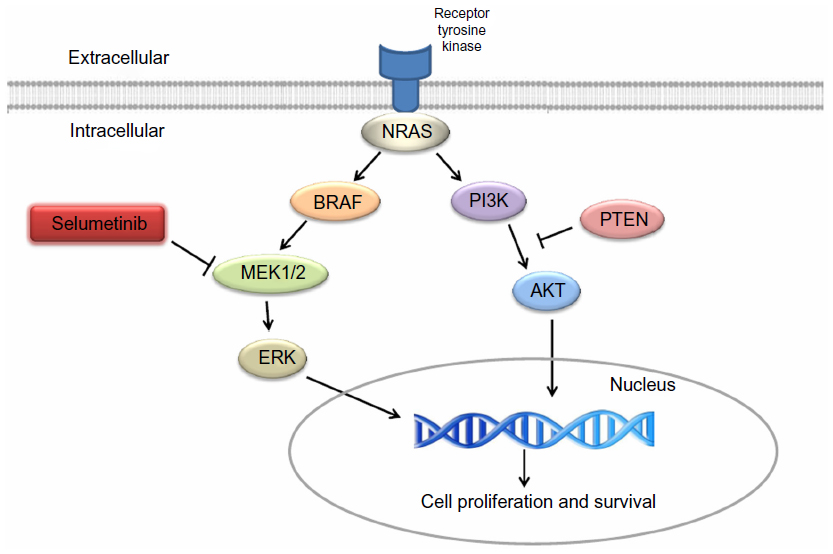 | Figure 1 Melanoma signaling pathway. |
MEK inhibition
Dysregulation of MAPK pathway in the majority of melanoma and other malignancies, including colon, pancreatic, and non-small-cell lung cancer, makes MEK an attractive therapeutic target as one of the main downstream molecules of MAPK pathway. Initial preclinical studies of selective MEK inhibitors have been shown to have promising antitumor activities in BRAF and NRAS mutant tumor cell lines.11,12 Solit et al showed complete abrogation of BRAF mutant melanoma growth and partial inhibition of NRAS mutant melanoma growth in murine xenografts with selective MEK inhibitors such as CI-1040 and PD0325901.12 However, clinical development of these MEK inhibitors, including CI-1040 and PD0325901, was discontinued because of a lack of clinical activity of CI-1040 and severe and frequent neurologic, musculoskeletal, and ocular toxicities of PD0325901. At this time, trametinib and selumetinib have been extensively studied clinically as MEK inhibitors in advanced solid tumors.
Selumetinib
Preclinical studies
Selumetinib (AZD6244, ARRY-142886; AstraZeneca, PLC, London, United Kingdom) is a highly selective, ATP-uncompetitive allosteric inhibitor of both MEK1 and MEK2 and has the empirical formula of C17H15BrCLFN4O3 (Figure 2). The half-maximal inhibitory concentration of selumetinib is 14.1 nmol/L against purified MEK1 in vitro, whereas no inhibition of 40 other serine/threonine and tyrosine kinases was observed at concentrations up to 10 μmol/L.13
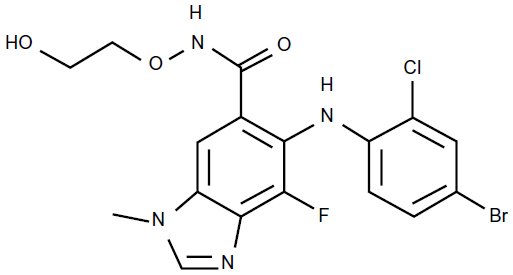 | Figure 2 Chemical structure of selumetinib. |
NRAS/KRAS/BRAF mutant cancer cell lines
Although a majority of melanomas have dysregulated MAPK pathway via BRAF and NRAS mutations, 90% of pancreatic cancers, 50% of colorectal cancers, and 25% of non-small-cell lung cancers also have constitutively activated MAPK pathway via KRAS mutations.14–16 Therefore, MAPK pathway inhibition by selumetinib was evaluated in other malignancies, including colorectal, pancreatic, non-small-cell lung, and hepatocellular cancer, as well as melanoma.
Phosphorylation of ERK1/2, downstream molecules of MEK, was effectively inhibited with selumetinib in melanoma, colorectal cancer, pancreatic cancer, non-small-cell lung cancer, and hepatocellular cancer cell lines harboring BRAF, NRAS, or KRAS mutations.13,17–19 However, growth suppression and the apoptotic effect of selumetinib varied among the cancer cell lines.17,18
Selumetinib-based combination treatment
To overcome the inconsistent efficacy of selumetinib and enhance its antitumor activity, several combinatorial approaches have been studied. The combined treatment with selumetinib and cytotoxic chemotherapy such as irinotecan, docetaxel, temozolomide, and doxorubicin resulted in significant enhanced antitumor efficacy by both cell cycle arrest and apoptosis in mice xenografts of BRAF mutant melanoma, as well as KRAS mutant colon cancer, non-small-cell lung cancer, pancreatic cancer, and hepatocellular carcinoma.17,18,20,21 Gopal et al have reported that the PI3K/AKT pathway plays a critical role in the antitumor efficacy of selumetinib in BRAF mutant melanoma, and inhibition of PI3K/AKT pathway results in synergistic antitumor activity with selumetinib.22 Because it has been reported that activation of Wnt/β-catenin signaling pathway inhibits tumor growth in mouse models of melanoma,23 the combinatorial effect of Wnt/β-catenin activation and selumetinib was examined.24 The combination of selumetinib and WNT3A, a ligand of Wnt/β-catenin pathway, induced apoptosis of melanoma cell lines harboring BRAF or NRAS mutations by degradation of AXIN1, a negative regulator of Wnt/β-catenin signaling.24 Apoptosis-resistant BRAF and NRAS mutant melanoma cell lines were able to sensitize to selumetinib by knock-down of AXIN1 expression with siRNA in the study.24
Recently, histone deacetylases have been reported to be up-regulated in cancer cells and lead to suppression of tumor suppressor gene expression such as p53 by a posttranslational modification.25 The combination of selumetinib and vorinostat, a histone deacetylase inhibitor, was evaluated in KRAS mutant colorectal cancer.26 The treatment with selumetinib plus vorinostat induced a synergistic antiproliferative activity against KRAS mutant colorectal cancer cell lines by the mechanism of apoptosis, cell-cycle arrest, and reduced cellular migration and VEGF-A secretion in vitro and in vivo.26
Uveal melanoma
Although uveal melanomas rarely have BRAF and NRAS mutations, the MAPK pathway is constitutively activated by GNAQ or GNA11 mutations that occur in approximately 80% of primary uveal melanomas.27,28 Because direct pharmacologic targeting of GNAQ/GNA11 mutations is not feasible, inhibition of the key downstream effectors of MAPK pathway has been studied. In vitro studies have demonstrated that selumetinib monotherapy is not effective in inducing antitumor activity in uveal melanoma cell lines because of reciprocal activation of the c-JUN or PI3K/AKT pathway, which is associated with cell proliferation, tumor cell invasion, and drug resistance.29,30 The combination of either c-Jun inhibition or PI3K/AKT inhibitors with selumetinib resulted in the significant induction of apoptosis in uveal melanoma cell lines.29,30 Another preclinical study demonstrated that the combination of selumetinib and a mammalian target of rapamycin (mTOR) inhibitor (AZD8055) which is downstream of PI3K/AKT pathway, induces tumor regression in BRAF mutant uveal melanoma cell lines but not GNAQ mutant cell lines.31 Unfortunately, these cell lines are likely contaminants of cutaneous melanoma cell lines because BRAF mutations are not present in uveal melanoma. The resistance to selumetinib plus AZD8055 in GNAQ mutant uveal melanoma cell lines is associated with expression of the prosurvival protein MCL1.31 Interestingly, there is a discrepancy between the two preclinical uveal melanoma studies using MEK inhibitors and PI3K/AKT/mTOR inhibitors. One study has shown that the combination of a MEK inhibitor (trametinib) with a PI3K inhibitor (GSK2126468) induced significant antitumor activity with a GNAQ mutant melanoma cell line,30 whereas the other study demonstrated that concurrent treatment of a MEK inhibitor (selumetinib) and mTOR inhibitor (AZD8055) does not result in any significant apoptosis or tumor growth suppression with the same GNAQ mutant cell line.31 The discrepancy of the studies may result from the different activity of each inhibitor.
Drug development
Pharmacokinetics
Initially, selumetinib was formulated as a freebase oral suspension with a median half-life of 8 hours and 100 mg twice a day of 50% maximum tolerated dose (MTD).32 Subsequently, a solid oral capsule formulation incorporating a hydrogen sulfate (Hyd-Sulfate) salt was developed to facilitate patient compliance and convenience. In a phase 1 clinical study, MTD of selumetinib with Hyd-Sulfate capsule was established as 75 mg twice a day.33 When MTD of Hyd-Sulfate capsule (75 mg twice a day) was compared with that of freebase suspension (100 mg twice a day), the maximum plasma concentration (Cmax) and the area under the concentration-time curve from 0–24 hours of Hyd-Sulfate capsule were 1,316 ng/mL and 4,454 ng × hour/mL, respectively, and for freebase suspension they were 523 ng/mL and 2,260 ng × hour/mL, respectively.33 The estimated oral bioavailability of the Hyd-Sulfate capsule relative to the freebase suspension was 263% (90% confidence interval, 241%–322%),33 which suggests a better pharmacokinetic profile of the Hyd-Sulfate capsule than the freebase formulation.
A food effect study with 75 mg twice a day of the Hyd-Sulfate formulation has demonstrated that high-fat meals reduced Cmax and area under the curve for selumetinib by 62% and 19%, respectively, and delayed the rate of absorption of selumetinib by 2.5 hours in comparison with fasting.34 Therefore, it is recommended that selumetinib should be taken on an empty stomach with water either 2 hours before eating or 1 hour after.
Pharmacodynamics
Because inhibition of ERK phosphorylation has been used as a pharmacodynamic biomarker of MEK inhibitor activity,35 the level of inhibition of ERK phosphorylation was measured in circulating lymphocytes and tumor samples before and after selumetinib treatment in phase 1 studies. Up to 100% inhibition of ERK phosphorylation was seen 1 hour after the first dose of selumetinib, and up to 90% of inhibition (geometric mean, 79%) was observed on day 15 and day 22 of selumetinib treatment in circulating lymphocytes.32,33 In tumor biopsy samples, selumetinib treatment induced strong inhibition of ERK phosphorylation, on average, by 79% by immunohistochemistry.32 However, Ki67, a proliferation marker, was not reduced consistently in tumor samples after selumetinib treatment.32
Early phase 1 studies of selumetinib
The first phase 1 study of selumetinib used freebase formulation for 57 previously treated patients with advanced solid tumors.32 This trial established MTD of freebase formulation of selumetinib to be 100 mg twice a day and demonstrated significant inhibition of ERK phosphorylation in peripheral blood mononuclear cells and tumor samples after selumetinib treatment, as described earlier.32 Nine of 57 patients with advanced solid tumors including metastatic melanoma and medullary thyroid cancer had long-term disease stabilization. However, no objective response was observed.32
In a phase 1 study of selumetinib Hyd-Sulfate oral capsule, 75 mg twice a day was established as the MTD dose, as previously described.33 One complete response of BRAF V600E mutant metastatic melanoma (2.9%) and 16 stable disease (45.7%) were observed among 35 patients with advanced solid tumors who received 75 mg twice a day of selumetinib.33 Because this study demonstrated a better pharmacokinetic profile of the Hyd-Sulfate formulation than the freebase formulation, as discussed earlier, subsequent studies have used the Hyd-Sulfate formulation of selumetinib.
In a phase 1 open-label, randomized food effect study mentioned previously, two patients (7.1%) with metastatic melanoma had partial response.34
Because preclinical studies have shown synergistic antitumor activity of selumetinib in combination with cytotoxic or molecularly targeted drugs,17 selumetinib-based combination therapies have been evaluated in the clinical setting. In one phase 1 study of selumetinib at 75 mg twice a day in combination with one of four drugs (dacarbazine, docetaxel, erlotinib, or temsirolimus) involving 18 patients with metastatic melanoma, five objective clinical responses (28%) were observed and all responders had BRAF mutant melanoma.36 The choice of the combination was at the discretion of the local investigator. In the study, a median time to progression of patients with BRAF mutant melanoma was 51 weeks, and for wild BRAF it was 12 weeks (hazard ratio [HR], 0.22; 95% confidence interval [CI], 0.06–0.82; P=0.02).36 There is an ongoing phase 1 study of the combination of selumetinib and cediranib maleate, a vascular endothelial growth factor receptor inhibitor (AZD2171) in patients with advanced solid tumors, including melanoma, that is currently recruiting participants (NCT01364051).
Later-stage clinical trials
In a randomized open-label phase 2 study, efficacy of selumetinib in freebase formulation was compared with temozolomide in patients with chemotherapy-naive advanced melanoma.37 The primary outcome of the study was PFS. Two hundred patients were randomized to either selumetinib (100 mg twice daily in 28-day cycles) or temozolomide (200 mg/m2 daily for 5 days, followed by 23 days off treatment). Objective response was observed in 6 patients (5.8%) receiving selumetinib and 9 patients (9.4%) in the temozolomide group. Among patients with BRAF-positive melanoma, objective response rate was similar between the two treatment groups (11.1% for selumetinib and 10.7% for temozolomide). Five of the six responders in the selumetinib group had BRAF mutant melanoma. Median PFS was not different significantly between selumetinib and temozolomide (78 and 80 days, respectively).
Because MEK inhibition has better antitumor activity in melanoma harboring BRAF mutations than wild BRAF,12,13 clinical efficacy of Hyd-Sulfate selumetinib was evaluated in selected patients with BRAF mutant melanoma in another phase 2 study.38 In addition, all patients were stratified by activation of PI3K/AKT pathway based on phosphorylated-AKT (pAKT) expression (high versus low) in the study, as PI3K/AKT pathway is one of the critical regulators of selumetinib efficacy in BRAF mutant melanoma.22 The primary endpoint of the study was clinical response, and three objective clinical responses were observed in a total of 15 patients.38 All three responders had low pAKT expression in the study. The estimated median PFS and OS were 2.2 and 8 months in the high pAKT cohort and 7.1 and 18 months in the low pAKT cohort without statistical significance.38
In a double-blind randomized phase 2 study, the combination of Hyd-Sulfate selumetinib and dacarbazine was evaluated as a first-line treatment for BRAF mutant metastatic melanoma.39 Patients with metastatic melanoma harboring BRAF mutations were randomly assigned to either selumetinib combined with dacarbazine or to dacarbazine alone, and the primary objective was to compare overall survival analyzed by intention to treat between the two groups. The objective response rate was 40% (18/45 patients) in the selumetinib plus dacarbazine group and 26% (12/46 patients) in the dacarbazine-alone group.39 Although OS did not differ significantly between selumetinib in combination with dacarbazine and dacarbazine alone (median, 13.9 and 10.5 months, respectively), PFS was significantly improved in the selumetinib plus dacarbazine group compared with dacarbazine alone, with a median of 5.6 and 3.0 months, respectively (HR, 0.63; 80% CI, 0.47–0.84; P=0.021).39
Because uveal melanomas also have activated MAPK pathway, inhibition of MAPK pathway using MEK inhibitors has been studied in uveal melanoma. In a randomized open-label phase 2 study of freebase selumetinib versus temozolomide in patients with advanced melanoma including uveal melanoma, none of seven patients with uveal melanoma in the selumetinib group achieved clinical response.37 Hyd-Sulfate selumetinib was studied in a randomized phase 2 study specifically for metastatic uveal melanoma (NCT01143402). In the study, patients were randomized to receive either selumetinib or temozolomide, and all patients were stratified by GNAQ and GNA11 mutations. The primary endpoint of the study was PFS. Patients in the temozolomide group with disease progression were allowed to crossover to receive selumetinib. At interim analysis, the median PFS duration was 15.9 weeks, and the median OS duration was 10.8 months in the selumetinib group (n=47), regardless of mutation status, whereas the temozolomide group (n=49) had a median PFS and OS of 7.0 weeks and 9.4 months, respectively. The HR for PFS was 0.46 (95% CI, 0.30–0.71; P=0.0005), and for OS it was 0.79 (95% CI, 0.46–1.37; P=0.4).40
There are several ongoing phase 2 clinical studies using selumetinib in Hyd-Sulfate formulation. One double-blind randomized phase 2 study comparing the efficacy of docetaxel plus selumetinib with docetaxel in wild BRAF melanoma is recruiting patients in the United Kingdom (NCT01256359).
In another phase 2 study, the efficacy of the combination of selumetinib and an AKT inhibitor (MK2206) is being evaluated in patients with stage III or IV melanoma who failed prior therapy with BRAF inhibitors (NCT01519427).
The results of the phase 2 studies and ongoing clinical trials of selumetinib are summarized in Tables 1 and 2, respectively.
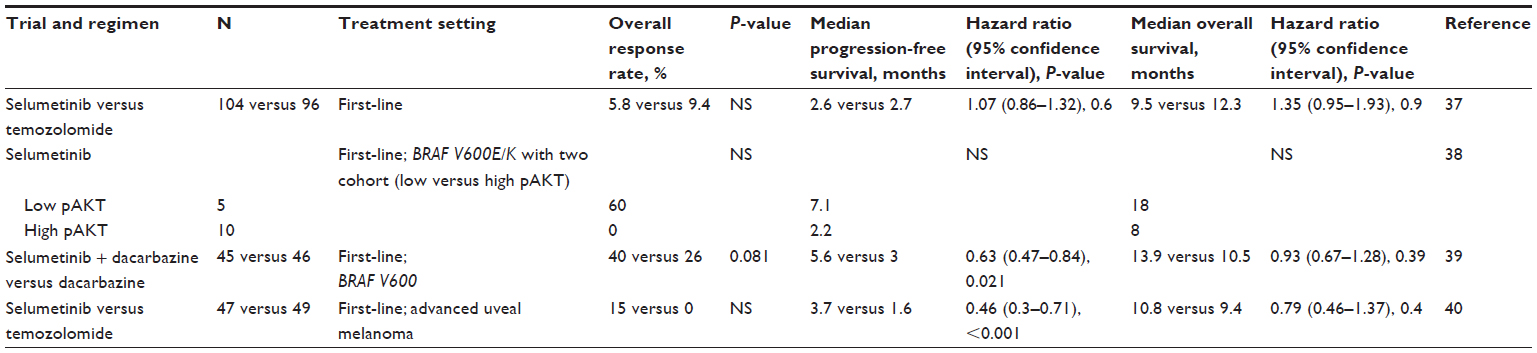 | Table 1 Clinical efficacy data of randomized phase 2 studies of selumetinib in melanoma |
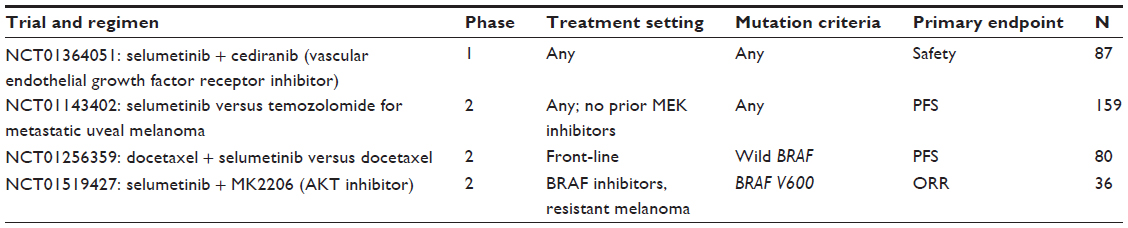 | Table 2 Ongoing clinical trials of selumetinib |
Selumetinib in specific populations
BRAF-positive melanoma
Several preclinical and clinical studies have demonstrated that selumetinib has better antitumor activity in BRAF-positive melanoma than in BRAF-negative melanoma,13,36,37 as discussed previously. Two BRAF inhibitors such as vemurafenib and dabrafenib have been approved by the FDA after demonstrating a survival benefit for patients with BRAF V600E/K mutant melanoma.5,6 The combination of a BRAF inhibitor with a MEK inhibitor has shown better toxicity profiles and clinical benefit than monotherapy in patients with a BRAF V600E/K mutation.41 In a phase 1/2 study of the combination of dabrafenib and trametinib versus single-agent dabrafenib in patients with BRAF V600 mutation, the response rates and median PFS duration were significantly better in the combination group than the dabrafenib-alone group (response rate, 76% versus 54%; P=0.03; the median PFS:9.4 versus 5.8 months; HR, 0.39; 95% CI, 0.25–0.62; P<0.001).41 Another phase 1 study of the combination of vemurafenib and GDC-0973, a selective MEK inhibitor, showed that all 25 evaluable BRAF inhibitor-naive patients had tumor reduction with the combined therapy.42 At this time, several phase 3 studies of the combination of BRAF inhibitors and MEK inhibitors are underway.
NRAS-positive melanoma
NRAS is upstream of MAPK pathway, and point mutation of the NRAS gene both is the second most common mutation in melanoma (15%–25%) and is associated with progression of melanoma.43 Because direct pharmacologic targeting of mutated NRAS has not been clinically tolerable, inhibition of key downstream effectors has been extensively studied. In a phase 2 study of MEK162, 6 (20%) of 30 patients with metastatic melanoma harboring NRAS mutations had an objective clinical response.44 However, other clinical studies of MEK inhibitors using trametinib or selumetinib in patients with NRAS mutant melanoma failed to show clinical activity.37,45 One of the possible explanations for the failed anticancer activity is that NRAS mutations activate multiple pathways including CDK4 driven cell-cycle progression and PI3K/AKT signaling in addition to MAPK pathway.46,47 Therefore, selumetinib plus other signaling pathway blockade such as CDK4 inhibitors and PI3K/AKT inhibitors may be a reasonable strategy for NRAS mutant melanoma.
GNAQ/GNA11-positive melanoma
In contrast to cutaneous melanoma, uveal melanomas do not harbor BRAF or NRAS mutations. Instead, point mutations of GNAQ or GNA11 have been reported in more than 80% of uveal melanomas, and the mutations are associated with activation of MAPK pathway and progression of uveal melanoma.27–29 As described earlier, a dedicated uveal melanoma trial comparing selumetinib with temozolomide has been completed. Despite the promising result of the study showing improvement in PFS with no improvement in OS in selumetinib-treated patients, combination therapy may be more effective than selumetinib single-agent therapy in GNAQ/GNA11 mutant uveal melanoma. To that end, a randomized phase 3 registration study of selumetinib in combination with dacarbazine versus placebo plus dacarbazine for patients with metastatic uveal melanoma is ongoing (NCT01974752).
Drug resistance
Although MAPK pathway inhibitors such as BRAF inhibitors and MEK inhibitors have demonstrated significant improvement of the clinical response rate, PFS, and OS in comparison with chemotherapy in patients with advanced melanoma harboring BRAF mutations, most responders develop a resistance to the therapy within a year.5–7 The resistance mechanisms are divided into MEK-dependent resistance and MEK-independent resistance, in general. MEK-dependent resistance is associated with reactivation of down-regulated p-ERK or p-MEK by MAPK pathway inhibitors at the time of disease progression.48 MEK-dependent resistance has been explained by the acquisition of new NRAS or MEK mutation,48 overexpression of COT-1 (serine/threonine kinase protein),49 elevated CRAF kinase protein expression,50 and alternate splicing of the BRAF gene.51 In particular, a MEK1 mutation was identified as acquired resistance to selumetinib therapy in melanoma cell lines and tumor samples of patients treated with selumetinib.52 The MEK-dependent resistance can be overcome by more potent inhibition of the MAPK pathway, such as the combination of BRAF inhibitors and MEK inhibitors41 and ERK inhibitors.
In contrast, MEK-independent resistance can develop through overexpression of MITF, which is a transcription factor and regulates melanogenesis,53 activation of PI3K/AKT pathway by loss of PTEN,48 overexpression of receptor tyrosine kinases,54,55 and secretion of hepatocyte growth factor from tumor microenvironment.56 In non-small-cell lung cancer cell lines, selumetinib resistance was mediated by a high level of AKT activity, and the resistance was reversed by AKT inhibition.57 Because MEK-independent resistance is not sensitive to MEK inhibitor monotherapy, the combined inhibition of MEK inhibitors and AKT inhibitors or MEK inhibitors plus histone deacetylase inhibitors suppressing MITF may reverse the resistance. At this time, a phase 2 study of the combination of selumetinib and an AKT inhibitor (MK2206) is being evaluated in patients with metastatic melanoma who failed prior therapy with BRAF inhibitors (NCT01519427).
Toxicities
Safety data have demonstrated that selumetinib overall appears to be well-tolerated. The most common adverse events were rash (63%–74% of patients) and fatigue (41%–68% of patients) in a dose-dependent manner in phase 1 studies.32,33 Selumetinib-induced skin reactions have been evaluated in patients with advanced melanoma in a phase 2 study.58 The most common acute skin reaction was papulopustular rash on the face, upper chest, and back with increased apoptotic keratinocytes and focal neutrophil infiltration histologically. In a half-side treatment experiment, one side of the face of all patients was treated with topical corticosteroid and the other side with topical antibiotics. A faster improvement of the selumetinib-associated skin reactions was observed with topical steroid than antibiotics, which suggests an inflammatory component of the rash caused by neutrophils. Diarrhea and nausea, the two most common gastrointestinal toxicities, have been reported in 48%–61% and 44%–54% of the patients, respectively.32,33 These toxicities were generally responsive to supportive care. Peripheral edema, reversible mild to moderate transaminitis, and transient and reversible blurry vision have been reported as adverse events as well. Hematologic toxicity is a rare toxicity with selumetinib. Grade 3/4 toxicities were reported up to 71% in a phase 1 study, but the majority of the toxicities were reported by only a single patient each. Grade 3/4 events reported by more than one patient were left ventricular dysfunction, nausea, vomiting, fatigue, febrile infection, pneumonia, elevation of alkaline phosphatase, elevation of γ-glutamyltransferase, hypoxia, dyspnea, abdominal pain, constipation, anemia, rash, and hypertension.32–34
Conclusion
Selumetinib is an orally available highly selective inhibitor of both MEK1 and MEK2 with a favorable toxicity profile. Preclinical data have demonstrated that MEK inhibition by selumetinib is one of the effective strategies for targeting dysregulated MAPK pathway that leads to unregulated cell proliferation and development and progression of melanoma. Clinical studies have proven the potential antitumor activity of selumetinib in a subset of melanoma patients such as BRAF mutant melanoma and uveal melanoma harboring GNAQ/GNA11 mutations. In addition, selumetinib-based combination treatment is being investigated more extensively to potentiate synergistic effects and overcome resistance. At this time, a phase 3 registration trial of selumetinib plus dacarbazine versus placebo plus dacarbazine for metastatic uveal melanoma represents the first potential indication for this drug. If successful, selumetinib with dacarbazine will be the first treatment regimen considered for approval for metastatic uveal melanoma.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | |
Lee ML, Tomsu K, Von Eschen KB. Duration of survival for disseminated malignant melanoma: results of a meta-analysis. Melanoma Res. 2000;10(1):81–92. | |
Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004;351(10):998–1012. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | |
Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. | |
Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. | |
Flaherty KT, Robert C, Hersey P, et al; METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. | |
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. | |
Pratilas CA, Solit DB. Targeting the mitogen-activated protein kinase pathway: physiological feedback and drug response. Clin Cancer Res. 2010;16(13):3329–3334. | |
Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353(20):2135–2147. | |
von Euw E, Atefi M, Attar N, et al. Antitumor effects of the investigational selective MEK inhibitor TAK733 against cutaneous and uveal melanoma cell lines. Mol Cancer. 2012;11(1):22. | |
Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439(7074):358–362. | |
Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13(5):1576–1583. | |
Balko JM, Black EP. A gene expression predictor of response to EGFR-targeted therapy stratifies progression-free survival to cetuximab in KRAS wild-type metastatic colorectal cancer. BMC Cancer. 2009;9(1):145. | |
Au HJ, Karapetis CS, O’Callaghan CJ, et al. Health-related quality of life in patients with advanced colorectal cancer treated with cetuximab: overall and KRAS-specific results of the NCIC CTG and AGITG CO.17 Trial. J Clin Oncol. 2009;27(11):1822–1828. | |
Sequist LV, Heist RS, Shaw AT, et al. Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol. 2011;22(12):2616–2624. | |
Haass NK, Sproesser K, Nguyen TK, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14(1):230–239. | |
Davies BR, Logie A, McKay JS, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6(8):2209–2219. | |
Huynh H, Soo KC, Chow PK, Tran E. Targeted inhibition of the extracellular signal-regulated kinase kinase pathway with AZD6244 (ARRY-142886) in the treatment of hepatocellular carcinoma. Mol Cancer Ther. 2007;6(1):138–146. | |
Huynh H, Chow PK, Soo KC. AZD6244 and doxorubicin induce growth suppression and apoptosis in mouse models of hepatocellular carcinoma. Mol Cancer Ther. 2007;6(9):2468–2476. | |
Holt SV, Logié A, Odedra R, et al. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br J Cancer. 2012;106(5):858–866. | |
Gopal YN, Deng W, Woodman SE, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010;70(21):8736–8747. | |
Chien AJ, Moore EC, Lonsdorf AS, et al. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc Natl Acad Sci U S A. 2009;106(4):1193–1198. | |
Conrad WH, Swift RD, Biechele TL, Kulikauskas RM, Moon RT, Chien AJ. Regulating the response to targeted MEK inhibition in melanoma: enhancing apoptosis in NRAS- and BRAF-mutant melanoma cells with Wnt/β-catenin activation. Cell Cycle. 2012;11(20):3724–3730. | |
Shankar S, Srivastava RK. Histone deacetylase inhibitors: mechanisms and clinical significance in cancer: HDAC inhibitor-induced apoptosis. Adv Exp Med Biol. 2008;615:261–298. | |
Morelli MP, Tentler JJ, Kulikowski GN, et al. Preclinical activity of the rational combination of selumetinib (AZD6244) in combination with vorinostat in KRAS-mutant colorectal cancer models. Clin Cancer Res. 2012;18(4):1051–1062. | |
Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. | |
Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363(23):2191–2199. | |
Ambrosini G, Pratilas CA, Qin LX, et al. Identification of unique MEK-dependent genes in GNAQ mutant uveal melanoma involved in cell growth, tumor cell invasion, and MEK resistance. Clin Cancer Res. 2012;18(13):3552–3561. | |
Khalili JS, Yu X, Wang J, et al. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin Cancer Res. 2012;18(16):4345–4355. | |
Ho AL, Musi E, Ambrosini G, et al. Impact of combined mTOR and MEK inhibition in uveal melanoma is driven by tumor genotype. PLoS ONE. 2012;7(7):e40439. | |
Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26(13):2139–2146. | |
Banerji U, Camidge DR, Verheul HM, et al. The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res. 2010;16(5):1613–1623. | |
Leijen S, Soetekouw PM, Jeffry Evans TR, et al. A phase I, open-label, randomized crossover study to assess the effect of dosing of the MEK 1/2 inhibitor Selumetinib (AZD6244; ARRY-142866) in the presence and absence of food in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2011;68(6):1619–1628. | |
Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22(22):4456–4462. | |
Patel SP, Lazar AJ, Papadopoulos NE, et al. Clinical responses to selumetinib (AZD6244; ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma. Cancer. 2013;119(4):799–805. | |
Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18(2):555–567. | |
Catalanotti F, Solit DB, Pulitzer MP, et al. Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanoma. Clin Cancer Res. 2013;19(8):2257–2264. | |
Robert C, Dummer R, Gutzmer R, et al. Selumetinib plus dacarbazine versus placebo plus dacarbazine as first-line treatment for BRAF-mutant metastatic melanoma: a phase 2 double-blind randomised study. Lancet Oncol. 2013;14(8):733–740. | |
Carvajal R, Sossman J, Quevedo F, Milhelm M, Joshua A, Kudchadkar R. Phase II study of selumetinib vs temozolomide in patients with advanced uveal melanoma. 2014 Annual Meeting of the American Society of Clinical Oncology; May 31–June 4, 2013; Chicago, Illinois. | |
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. | |
Gonzalez R, Ribas A, Daud A, Pavlick A, Gajewski T, Puzanov I. Phase IB study of vemurafenib in combination with the MEK inhibitor, GDC-0973, in patients (pts) with unresectable or metastatic BRAFV600 mutated melanoma (BRIM7). 37th European Society for Medical Oncology Congress; September 28–October 2, 2012; Vienna, Austria. Abstract 2744. | |
Jakob JA, Bassett RL Jr, Ng CS, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118(16):4014–4023. | |
Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249–256. | |
Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):782–789. | |
Jaiswal BS, Janakiraman V, Kljavin NM, et al. Combined targeting of BRAF and CRAF or BRAF and PI3K effector pathways is required for efficacy in NRAS mutant tumors. PLoS ONE. 2009;4(5):e5717. | |
Kwong LN, Costello JC, Liu H, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18(10):1503–1510. | |
Trunzer K, Pavlick AC, Schuchter L, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31(14):1767–1774. | |
Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972. | |
Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68(12):4853–4861. | |
Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387–390. | |
Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106(48):20411–20416. | |
Johannessen CM, Johnson LA, Piccioni F, et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504(7478):138–142. | |
Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. | |
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. | |
Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–504. | |
Meng J, Peng H, Dai B, et al. High level of AKT activity is associated with resistance to MEK inhibitor AZD6244 (ARRY-142886). Cancer Biol Ther. 2009;8(21):2073–2080. | |
Schad K, Baumann Conzett K, Zipser MC, et al. Mitogen-activated protein/extracellular signal-regulated kinase kinase inhibition results in biphasic alteration of epidermal homeostasis with keratinocytic apoptosis and pigmentation disorders. Clin Cancer Res. 2010;16(3):1058–1064. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.