")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Profile of romosozumab and its potential in the management of osteoporosis
Authors Lim SY , Bolster MB
Received 13 November 2016
Accepted for publication 28 February 2017
Published 13 April 2017 Volume 2017:11 Pages 1221—1231
DOI https://doi.org/10.2147/DDDT.S127568
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr James Janetka
Video abstract presented by Dr Sian Yik Lim.
Views: 6298
Sian Yik Lim,1 Marcy B Bolster2
1Straub Bone & Joint Center, Straub Medical Center, Honolulu, HI, 2Division of Rheumatology, Allergy & Immunology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
Abstract: Increased understanding of bone biology has led to the discovery of several unique signaling pathways that regulate bone formation and resorption. The Wnt signaling pathway plays a significant role in skeletal development, adult skeletal homeostasis, and bone remodeling. Sclerostin is an inhibitor of the Wnt signaling pathway. Romosozumab, a humanized monoclonal antibody that binds to sclerostin, prevents sclerostin from exerting this inhibitory effect. Therefore, in the presence of romosozumab, the Wnt signaling pathway is activated leading to bone formation and bone mineral density gain. Clinical studies of romosozumab have shown that this agent is one of the most potent bone anabolic agents in development to date. Romosozumab does not act solely as an anabolic agent, but rather, it has effects on increasing bone formation as well as reducing bone resorption. In the clinical studies, patients tolerated romosozumab well with no major safety signals reported. In a Phase III study, romosozumab as compared to placebo has been shown to reduce vertebral fractures by 73% after 1 year of treatment. Sequential therapy with romosozumab for 1 year followed by denosumab in the second year reduced vertebral fractures by 75% as compared to the group that received placebo for 1 year and denosumab in the second year. Romosozumab holds significant potential, by a novel mechanism of action, to expand our ability to treat osteoporosis. More studies are needed to determine the ideal setting in which romosozumab may be used to optimize osteoporosis treatment.
Keywords: romosozumab, sclerostin, osteoporosis, bone mineral density
Introduction
Osteoporosis is a skeletal disorder in which bone strength is decreased leading to an increased risk of fracture.1 Osteoporotic fractures are associated with significant disease burden, health care cost, morbidity, and mortality.1,2 Several medications have been developed to treat osteoporosis, including estrogen, raloxifene, bisphosphonates, denosumab, and teriparatide. All of these medications are antiresorptive agents except teriparatide. Developing osteoanabolic medications is needed for a more effective, individualized, and targeted approach in osteoporosis treatment.3 Other anabolic medications, including abaloparatide (Radius Health, Inc., Waltham, MA, USA) and romosozumab (AMG 785, Amgen Inc., Thousand Oaks, CA, USA), are currently under development. In this article, we discuss the profile of romosozumab and its potential role in osteoporosis treatment.
The Wnt signaling pathway and bone health
The term “Wnt” originates from the acronym between wingless (Wg) in Drosophila and Int1 in the mouse. Secreted Wnt glycoproteins are involved in the regulation of cell-to-cell communication during embryogenesis and adult tissue homeostasis.4 Wnt proteins act as ligands binding to a Frizzled family receptor subsequently activating Wnt signaling pathways.5 Well-characterized Wnt signaling pathways include the canonical Wnt-β-catenin pathway (involving β-catenin) and the noncanonical pathways (not involving β-catenin). The noncanonical pathways include the noncanonical planar cell polarity pathway and the noncanonical Wnt–calcium pathway.5
The canonical Wnt-β-catenin pathway plays a significant role in skeletal development, adult skeletal homeostasis, and bone remodeling.6 In this pathway, without the Wnt ligand binding to Frizzled family receptor, a scaffolding protein known as axin assembles a destruction complex, which phosphorylates β-catenin. Phosphorylated β-catenin is subsequently ubiquitinated and degraded by a proteasome (Figure 1).7,8 β-Catenin does not enter the nucleus of the cell, and Wnt-responsive genes are not activated, leading to decreased bone formation and increased bone resorption. When Wnt ligand binds to a specific Frizzled family receptor and an LDL-receptor-related protein (LRP) coreceptor (LRP-5 or LRP-6 coreceptor), this leads to a series of cellular changes that inhibit the function of the destruction complex. Unphosphorylated β-catenin is not degraded; therefore, β-catenin accumulates within the cell. The β-catenin then enters the nucleus of the cell and binds to the T-cell factor transcription factor in which Wnt-responsive genes are activated (Figure 1A).4,7,8
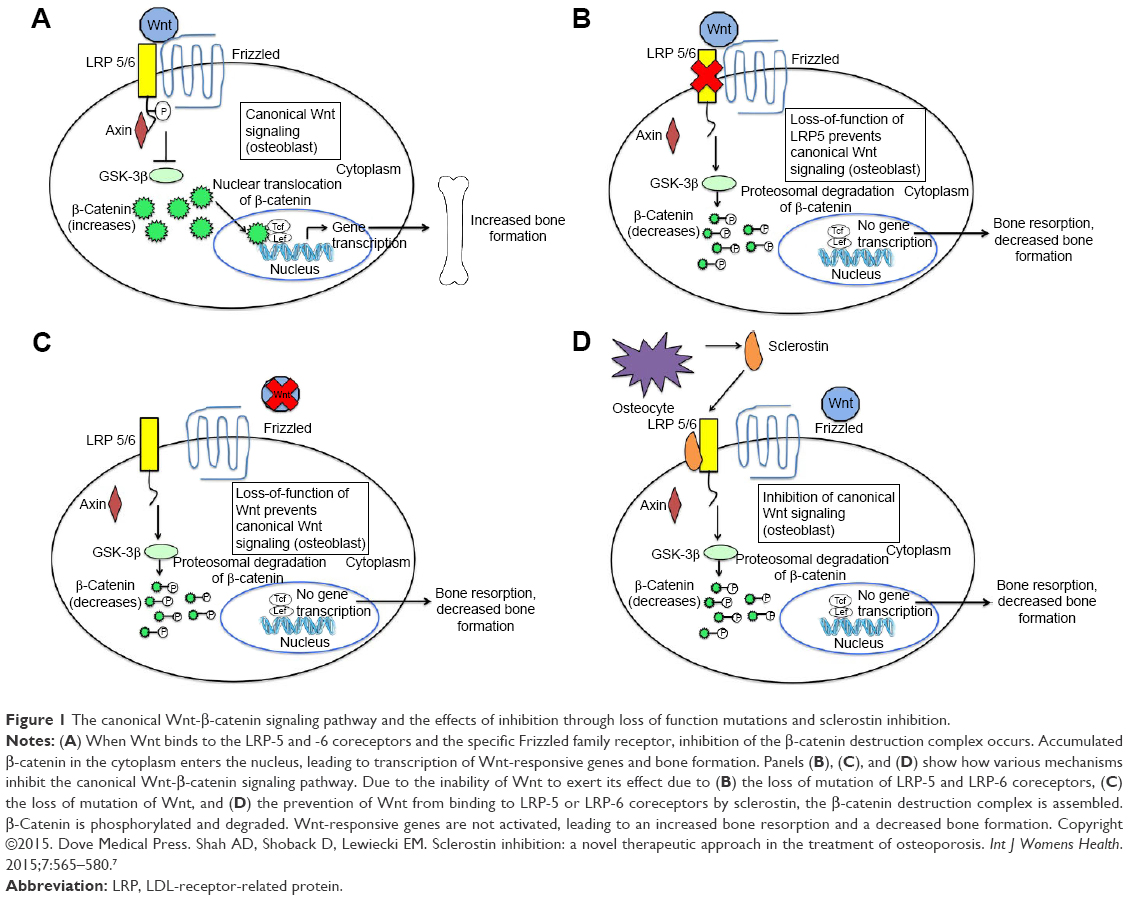 | Figure 1 The canonical Wnt-β-catenin signaling pathway and the effects of inhibition through loss of function mutations and sclerostin inhibition. |
Activation of the canonical Wnt-β-catenin pathway leads to differentiation of osteoblast precursors and osteogenesis by osteoblasts. Furthermore, increased β-catenin levels result in increased expression of osteoprotegerin. Osteoprotegerin binds to RANKL as a decoy receptor, thus preventing the binding of RANKL to RANK. In the presence of RANKL to RANK binding, osteoclast activation and differentiation occur. Thus, in the presence of increased osteoprotegerin expression, there is less binding of RANKL to RANK and therefore reduced osteoclastogenesis and bone resorption.6 Therefore, the net effect of activation of this pathway is increased bone mass. Mutation in the canonical Wnt-β-catenin signaling pathway provides significant insight into the importance of this pathway. In 2002, Little et al9 described a family found to have increased bone mass but who were normal phenotypically. Investigators identified a gain in the function of LRP-5 mutation in that family. Conversely, patients with loss of function of LRP-5 mutation have a unique phenotype of blindness, reduced bone mass, skeletal deformities, and fragility fractures during childhood, a condition known as osteoporosis–pseudoglioma syndrome.10
Sclerostin
Sclerostin is a glycoprotein produced by osteocytes. The SOST gene located on chromosome 17q12-q21 codes for sclerostin’s secretion.11 Sclerostin is a key inhibitor of the canonical Wnt signaling pathway. Sclerostin binds to LRP-5/6 and prevents Wnt from binding to the Frizzled family receptor and LRP coreceptors, therefore leading to downregulation of the canonical Wnt signaling pathway.12 Therefore, sclerostin leads to inhibition of osteoblast differentiation and function and thus decreased bone formation.13,14 SOST gene expression, and therefore sclerostin production, is mostly limited to skeletal tissue.15 Therefore, targeting sclerostin in drug development is an attractive treatment strategy because theoretically the effects of such a targeted medicine would be restricted to the skeletal system with limited risk of the drug affecting other organ systems.
Osteocytes are one of the most abundant cell types within bone tissue.16 They function as mechanosensors and also secrete sclerostin.12 The complex interaction between osteocytes, sclerostin, and the Wnt pathway mediates the coupling of mechanical stress on bone to the anabolic response. In rodents, it has been shown that hindlimb unloading induces high sclerostin levels, which downregulates the canonical Wnt signaling pathway in osteoblasts and osteocytes, leading to decreased bone formation; conversely, increased ulnar loading causes low sclerostin levels, activation of the canonical Wnt signaling pathway, and therefore bone formation.11,17 The former mechanism may have relevance in the pathogenesis of osteoporosis of disuse where prolonged immobility leads to decreased bone mass.6
Distinguishing genetic alterations that result in modified levels or activity of sclerostin have helped to recognize the important role that sclerostin inhibition could have in treating osteoporosis. Clues to the possible utility of inhibiting sclerostin in the treatment of osteoporosis came from two rare autosomal recessive genetic conditions of sclerosteosis and van Buchem’s disease. Each of these disorders relates directly to the SOST gene and thus to the expression of sclerostin. Patients with sclerosteosis have homozygous mutations in the SOST gene,18 while patients with van Buchem’s disease have a homozygous mutation, resulting in deletion of a region in distant regulatory elements involved in the transcription of the SOST gene.19 The following two conditions are distinguished by the severity of the symptoms: sclerosteosis being the most severe form and van Buchem being the milder form of the disorder. Due to these genetic abnormalities, patients with these diseases have decreased production of biologically active sclerostin. In the setting of reduced sclerostin activity, the Wnt signaling pathway goes uninhibited due to the inability of Wnt to bind to the specific Frizzled family receptor. Patients with these conditions have high bone mass characterized by progressive generalized osteosclerosis occurring more prominently in the skull and also involving the mandible, ribs, clavicles, and long bones.18,20 These patients manifest facial distortion and cranial nerve entrapment due to narrowing of the foramina of the cranial nerves, but they have a very low risk of fracture due to increased bone mass.18,20
SOST-knockout mice have been found to have a high bone mass phenotype with increased bone mineral density (BMD), bone formation, and improved biomechanical properties of bone strength.21 Conversely, transgenic mice overexpressing the SOST gene have been found to have a low bone mass phenotype characterized by low bone mass and decreased bone strength.22 These encouraging findings led to the idea of developing a monoclonal antibody that inhibits sclerostin (a Wnt pathway inhibitor) as a way to increase bone mass and likewise treat osteoporosis (Figure 2).7,23
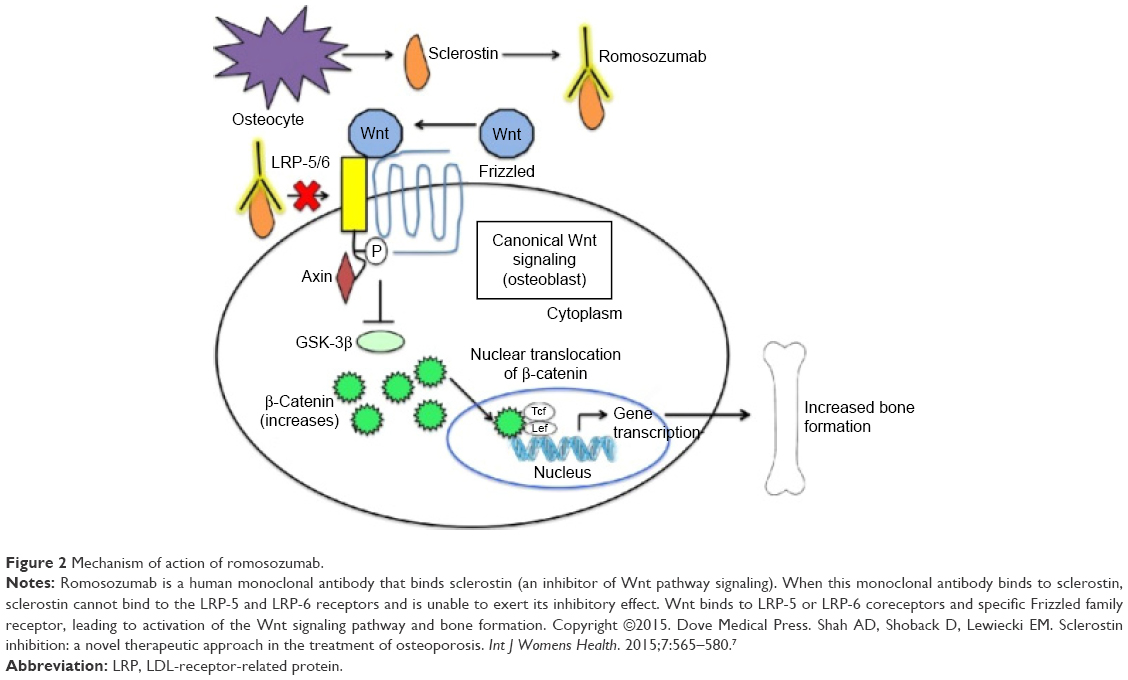 | Figure 2 Mechanism of action of romosozumab. |
Antisclerostin antibodies in animal studies
Several animal models have shown the effect of antisclerostin antibodies on bone formation. Ovariectomized rats at age 6 months, left untreated for 12 months to allow the development of significant estrogen deficiency-induced bone loss, were treated with murine antisclerostin antibodies, and this led to marked increases in bone formation on trabecular, periosteal, endocortical, and intracortical surfaces.24 The antisclerostin antibody treatment resulted in a reversal of estrogen deficiency-induced bone loss with subsequent significant increases in bone mass and bone strength. Interestingly, treatment with antisclerostin antibodies led to an increase in the osteoblast surface and mineralizing surface, while osteoclast surface was decreased, suggesting that there was increased bone formation and decreased bone resorption with the use of sclerostin antibodies.24 Furthermore, SOST-knockout mice had similarly increased bone formation and decreased bone resorption.21
Treatment of cynomolgus monkeys with antisclerostin antibodies (two monthly doses of antisclerostin antibodies over 8 weeks) led to increased BMD and improved bone strength.25 No increase in the bone resorption marker, serum collagen type 1 cross-linked C-telopeptide (CTX), was noted. The increase in bone formation markers with no change in bone resorption markers likely reflects a unique effect of antisclerostin antibodies, which involves bone “modeling” (direct activation of bone formation on quiescent surfaces without prior activation of resorption). This effect of antisclerostin antibodies differs from the effect of teriparatide, another anabolic agent, where bone “remodeling” (osteoblast-mediated bone formation follows osteoclast-mediated bone resorption) is thought to occur.
With long-term administration of antisclerostin antibody, there is an initial transcriptional response activating the canonical Wnt signaling pathway. With long-term treatment, additional regulatory pathways in the osteocyte are activated. With continued antisclerostin antibody administration, the interactions between Wnt and p53/c-Myc may be key in limiting osteoprogenitor populations, self-regulating bone formation.26
Phase I, Phase II, and Phase III trials of romosozumab
Romosozumab is a humanized antisclerostin monoclonal antibody. In the following sections, we summarize the Phase I, II, and III trials of romosozumab. All published trials at the time of this review are summarized in Table 1.27–30 Through these trials, we have learned that romosozumab treatment leads to a significant gain in bone density. A total of 12 months of romosozumab treatment leads to a bone density gain at the lumbar spine of 11.3%–13.3%, the total hip of 4.1%–6.9%, and the femoral neck of 3.7%–5.9%.27,30 Subcutaneous romosozumab 210 mg monthly led to the greatest BMD gain among the studied doses without an increased incidence of adverse effects.30 The gains in BMD with subcutaneous romosozumab 210 mg monthly were significantly greater as compared to patients treated with teriparatide or alendronate.30 Studies using bone turnover markers point to a distinct mechanism of action of romosozumab where there occurs unique coupling of the bone remodeling process: an increase in bone formation and a decrease in bone resorption.27–30 In a Phase III study, romosozumab as compared to placebo has been shown to reduce vertebral fractures by 73% after 1 year of treatment. Sequential therapy with romosozumab for 1 year followed by denosumab in the second year reduced vertebral fractures by 75% as compared to the group that received placebo for 1 year followed by denosumab in the second year.30
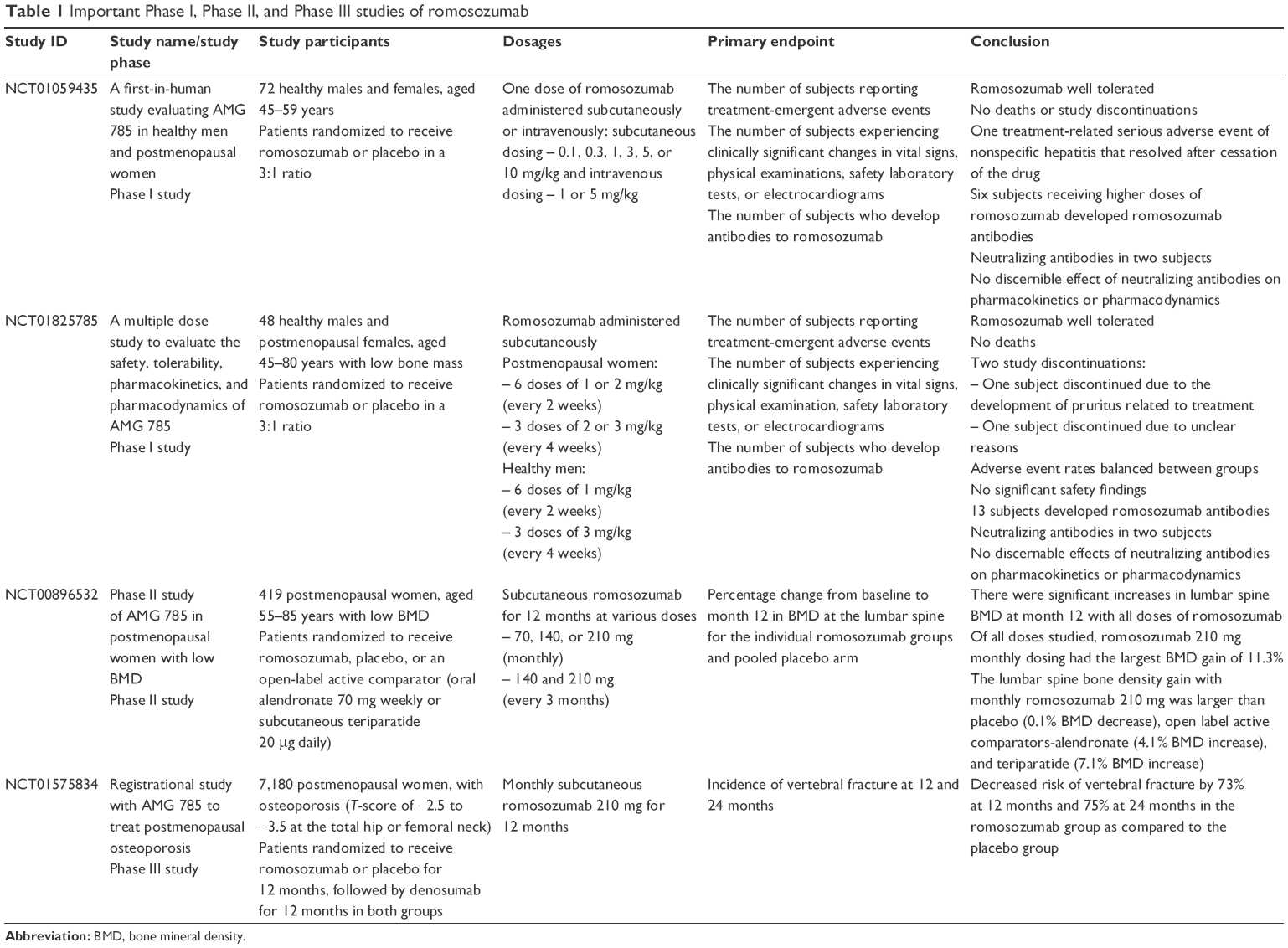 | Table 1 Important Phase I, Phase II, and Phase III studies of romosozumab |
Phase I trials: pharmacokinetics, pharmacodynamics, and safety of romosozumab
Romosozumab is an IgG2 monoclonal antibody generated by humanizing a mouse sclerostin monoclonal antibody. Romosozumab neutralizes the activity of human, monkey, and rat sclerostin and has a high binding affinity for human sclerostin with a pKd of 11.2–12.2.31
There are no specific studies published about the absorption, distribution, and excretion of romosozumab; however, it is likely similar to other monoclonal antibodies.32 When subcutaneously administered, systemic absorption of monoclonal antibodies occurs via the lymphatic vessels.33 Because of their large molecular size, monoclonal antibodies distribute from the blood compartment to the peripheral tissue by convection or through endocytosis/pinocytosis via endothelial cells.33 For monoclonal antibodies, the role of hepatic and renal excretion in elimination is minor. Elimination of monoclonal antibodies happens via protein catabolism, occurring through several mechanisms including less specific processes of proteolysis by the liver and reticuloendothelial system and nonspecific endocytosis. More specific elimination occurs at the target cell, a process involving endocytosis and intracellular degradation within the target cell. Target-mediated elimination has small capacity and hence is susceptible to saturation. Because of this, many but not all monoclonal antibodies exhibit nonlinear elimination pharmacokinetics.33 At low serum concentrations, rapid saturable target-mediated elimination regulates the elimination rate of the antibody. However, at higher serum concentrations, when target-mediated elimination is saturated, elimination of antibody protein occurs more slowly via nonspecific endocytosis and other processes.33,34
Two pivotal Phase I trials of romosozumab assessed the safety, pharmacokinetics, and pharmacodynamics of this agent (Table 1). The first study was a placebo-controlled, randomized study of 72 healthy subjects where patients received a single dose of romosozumab subcutaneously (0.1, 0.3, 1, 3, 5, or 10 mg/kg), intravenously (1 or 5 mg/kg), or placebo.29 Patient follow-up ranged from 29 to 85 days depending on the dose of romosozumab administration.
There was a subsequent randomized, double-blind, placebo-controlled study of multiple doses of romosozumab in 48 healthy postmenopausal women and men. The postmenopausal women received six doses of 1 or 2 mg/kg every 2 weeks or three doses of 2 or 3 mg/kg once every 4 weeks or placebo. The healthy men received six doses of 1 mg/kg every 2 weeks or 3 mg/kg once every 4 weeks or placebo.28 The study involved 3 months of treatment followed by 3 months of follow-up after treatment.
Romosozumab was found to demonstrate nonlinear pharmacokinetics similar to other monoclonal antibody treatments: clearance of romosozumab decreased as the dose of romosozumab increased.29 With single doses of romosozumab, serum concentrations declined in a biphasic manner after maximum concentration with half-lives of 11–18 days and then 6–7 days subsequently.29 After administration of a single dose of romosozumab, serum levels of romosozumab peaked within the first week.28
Patient development of antibodies directed against therapeutic monoclonal antibodies may affect the medication pharmacokinetics and result in reduced efficacy.33 Among the 54 patients who received single-dose romosozumab, six (11%) patients in the higher-dose groups developed antibodies against romosozumab. Only two of the patients had neutralizing antibodies, and there was no discernable effect of these antibodies on the pharmacokinetics and pharmacodynamics of romosozumab.29 In 36 patients who received multiple doses of romosozumab, two patients developed neutralizing antibodies, while ten patients developed nonneutralizing antibodies. There were no apparent effects of any of these antibodies on pharmacokinetics and pharmacodynamics. There was one patient involved in a prior romosozumab study who was found to have preexisting neutralizing antibodies against romosozumab. In this particular patient, serum concentrations of romosozumab declined rapidly after the first dose of romosozumab and were unmeasurable, despite receiving subsequent doses of romosozumab.28
As with the treatment with antisclerostin antibodies in animal studies, Phase I clinical studies in which humans were administered romosozumab showed that romosozumab treatment led to a rapid increase in the bone formation markers of serum type I amino-terminal propeptide (P1NP), osteocalcin, and bone-specific alkaline phosphatase (BSAP). There was a decrease in the bone resorption marker, serum CTX, confirming the notion of both increased bone formation and decreased bone resorption with the use of antisclerostin antibodies resulting in a large anabolic window, a period where romosozumab’s effects are mainly osteoanabolic. Treatment with a single dose of romosozumab led to increases in BMD, 5.3% in the lumbar spine and 2.8% in the hip at 85 days, as compared to placebo.29 In patients treated for 3 months with multiple doses of romosozumab, there was an increase in lumbar spine BMD at 6 months.28 Aside from injection site reactions, the study subjects tolerated romosozumab at all doses. These encouraging results in Phase I studies led to Phase II studies evaluating the efficacy of romosozumab for the treatment of osteoporosis.28,29
Phase II trials: efficacy and safety of romosozumab
A Phase II randomized, placebo-controlled, parallel group, eight-group study evaluated the effectiveness and safety of romosozumab in postmenopausal women with low bone density (Table 1).30 The study included 419 postmenopausal women aged 55–85 years, with BMD T-scores <−2.0 and >−3.5. A total of 383 (91%) patients completed the study. The mean patient T-scores were as follows: lumbar spine −2.29, total hip −1.53, and femoral neck −1.93. Patients were randomized to receive romosozumab monthly (doses 70, 140, and 210 mg) or every 3 months (doses 140 and 210 mg), placebo, or open-label comparator group (oral alendronate 70 mg weekly or subcutaneous teriparatide 20 μg daily). The primary endpoint of the study was percentage change from baseline of lumbar spine BMD at 12 months in patients who received romosozumab as compared to the pooled placebo group.
At 12 months, pooled romosozumab group participants achieved a statistically significant increase in BMD at the lumbar spine, total hip, and femoral neck as compared to the pooled placebo group participants, regardless of romosozumab dose and frequency. Romosozumab 210 mg administered subcutaneously monthly was associated with the highest gain in BMD at 12 months (11.3% in the lumbar spine, 4.1% in the total hip, and 3.7% in the femoral neck) among the doses evaluated. The BMD gain on romosozumab 210 mg subcutaneously monthly was larger when compared to active comparators, such as subcutaneous teriparatide 20 μg daily and oral alendronate 70 mg weekly.30 A total of 12 months treatment with romosozumab led to gains in trabecular and cortical compartments in the spine and hip as assessed by quantitative computed tomography (QCT).35
Bone formation markers, such as serum P1NP, showed a marked transitory increase that peaked at 1 month after initiating treatment. After peaking, serum P1NP returned to baseline or dropped below baseline at months 2–9 depending on the romosozumab dose. Serum CTX (a bone resorption marker) decreased the most in the first week but remained below baseline up to month 12 of treatment (Figure 3).30,36 Romosozumab seems to cause a rapid initial gain in bone formation and also a more prolonged decrease in bone resorption, leading to a significant increase in BMD, although the full mechanism of this remains unclear.30
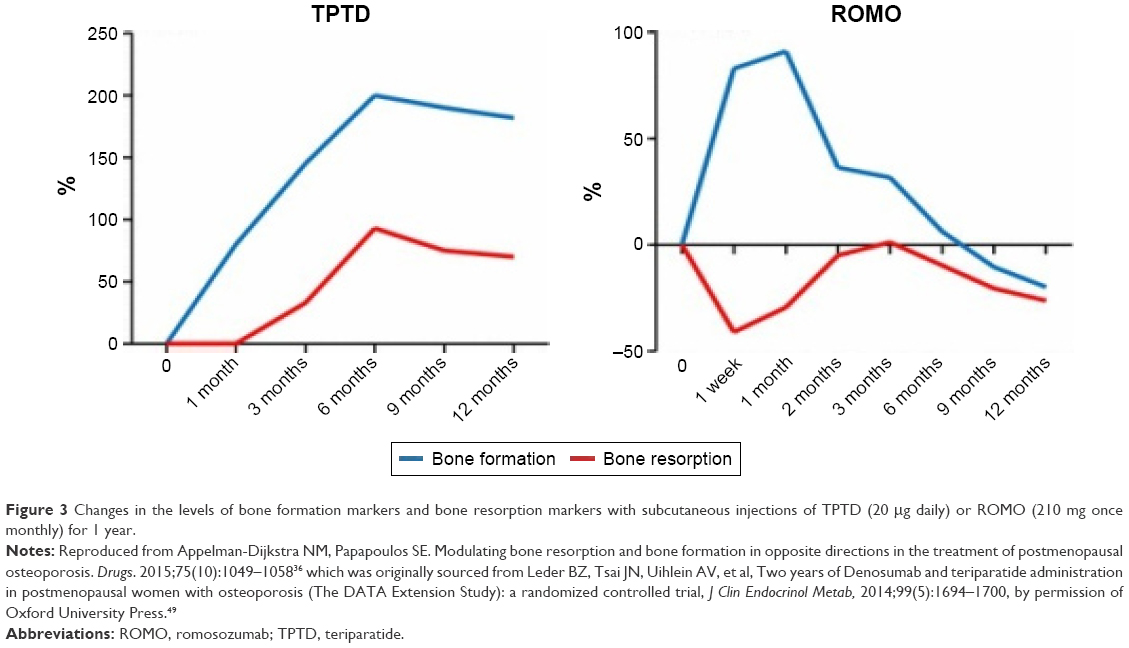 | Figure 3 Changes in the levels of bone formation markers and bone resorption markers with subcutaneous injections of TPTD (20 μg daily) or ROMO (210 mg once monthly) for 1 year. |
There were no significant differences in the percentage of serious adverse events between all groups. However, there were more injection site reactions with romosozumab treatment. The injection site reactions include pain, hematoma, erythema, discomfort, hemorrhage, or rash at the injection site. Binding antibodies were detected in 20% of patients receiving romosozumab, but only 3% of them were romosozumab-neutralizing antibodies. There was no relationship between romosozumab-neutralizing antibodies and measures of efficacy.
In the abovementioned trial, patients continued their assigned treatment for an additional year. Patients were then randomized to receive denosumab treatment or placebo for the third year. In the second year on romosozumab, there was a continued gain of BMD in the spine and hip, but the magnitude of increase in the second year was smaller than that which occurred in the first year. After the first 2 years of romosozumab therapy, patients who switched to denosumab had a continued increase in BMD. Notably, the magnitude of gain was almost the same with BMD increases in the second year of romosozumab treatment but not as large as in the first year of therapy. Patients who stopped romosozumab at year 2 and did not receive further denosumab treatment had their bone density and bone turnover markers return to close to baseline values. After 3 years of treatment, there were no differences noted in regard to adverse events between treatment and placebo groups.37
Phase III trials: efficacy, effectiveness, and safety of romosozumab
Several Phase III trials of romosozumab are currently underway or have been recently completed with encouraging results. One of the studies is the randomized, open-label, international multicenter STRUCTURE study (STudy evaluating effect of RomosozUmab Compared with Teriparatide in postmenopaUsal women with osteoporosis at high risk for fracture pReviously treated with bisphosphonatE therapy).38 This study aimed to assess the effect of a 12-month treatment with either romosozumab or teriparatide on BMD after bisphosphonate treatment. The study included 436 postmenopausal women aged 55–85 years with osteoporosis (T-score ≤−2.5 at the lumbar spine, femoral neck, or total hip) who had taken an oral bisphosphonate for >3 years before screening and, specifically, had taken weekly alendronate 1 year before screening. Patients also had to have a history of a vertebral fracture or a nonvertebral fracture after the age of 50 years. In the study, the mean patient T-scores were as follows: lumbar spine −2.2, total hip −2.9, and femoral neck −2.5. Patients, all of whom had previously received bisphosphonate treatment, were randomized to receive subcutaneous romosozumab or teriparatide. The primary endpoint of the study was total hip BMD on Dual-energy X-ray absorptiometry at month 12.
Romosozumab significantly increased total hip BMD (2.9%) and was superior to teriparatide (−0.5%). Romosozumab also led to superior gains in lumbar spine BMD as compared to teriparatide (9.8% in patients on romosozumab and 3.5% in patients on teriparatide). It is interesting to note that on QCT assessments, romosozumab treatment led to gains in BMD in the cortical and integral compartments of the hip and improved the estimated hip strength (as opposed to teriparatide where there was a decrease in the estimated hip strength). Adverse effects in both treatment arms were well balanced. Therefore, romosozumab seems to be a good treatment option for patients who are transitioning from bisphosphonate therapy because it is well tolerated and also leads to BMD gains in the hip and spine.
Currently, there are two large Phase III fracture studies exploring the utility of romosozumab in the treatment of osteoporosis. The first study (ClinicalTrial.gov identifier: NCT01631214) compares romosozumab in the first year followed by 2 years of alendronate therapy.39
The second study, recently published, a Phase III placebo-controlled FRActure study in postmenopausal woMen with ostEoporosis (FRAME) (ClinicalTrials.gov identifier: NCT01575834), is a multicenter, international, randomized, double-blind, placebo-controlled, parallel group study that compares the 1-year treatment of romosozumab followed by denosumab with 1-year treatment with placebo followed by denosumab.27 The study included 7,180 postmenopausal women aged 55–90 years, with a total hip or femoral neck BMD T-score of −2.5 to −3.5. The first year of the trial was completed by 6,390 patients (89.1%), while 6,026 patients (83.9%) completed the second year. The mean patient T-scores were as follows: lumbar spine −2.72, total hip −2.47, and femoral neck −2.75. Patients were randomized to receive romosozumab 210 mg monthly or placebo for the first year, followed by subcutaneous denosumab every 6 months for the second year. The primary endpoints for this study were vertebral fracture reduction at 12 and 24 months.
At the end of 12 months of romosozumab treatment, in the study by Cosman et al, vertebral fractures were reduced by 73% (the incidence of vertebral fracture in the romosozumab group was 0.5% as compared to 1.8% in the placebo group). The romosozumab treatment group also had a 63% reduction in clinical fractures (composite of nonvertebral fracture and symptomatic vertebral fracture) as compared to the placebo group. At 24 months, the incidence of vertebral fractures was reduced by 75% in patients who received romosozumab in the first year and denosumab in the second year (vertebral fracture incidence 0.6%) as compared to the group who received placebo in the first year and denosumab in the second year (vertebral fracture incidence 2.5%). There was no significant difference in nonvertebral fracture incidence at 12 and 24 months between the two groups.27 One possible explanation for the lower than expected nonvertebral fracture incidence in the placebo group was attributed to low nonvertebral fracture incidence in patients enrolled from the Latin America region (Colombia, Brazil, Argentina, Dominican Republic, and Mexico). Patients from the Latin America region consisted of 42.7% of patients enrolled in the study.
Consistent with the findings in Phase I and Phase II studies, there were significant gains in BMD by 12 months in the lumbar spine (13.3%), total hip (6.9%), and femoral neck (5.9%). With romosozumab treatment, serum P1NP (bone formation marker) increased rapidly and returned to baseline by 9 months of treatment. Serum CTX (bone resorption marker) decreased early during treatment and remained low during the 12 months of treatment. The change in bone turnover markers is consistent with prior studies and suggests that both increased bone formation and decreased bone resorption comprise a unique mechanism of action of romosozumab, one of the most potent osteoanabolic agents developed to date. Severe adverse events of hypersensitivity reactions to romosozumab were rare. There were mild injection site reactions in 5.2% of patients treated with romosozumab. There were two cases of osteonecrosis of the jaw and one case of atypical femoral fracture reported in the romosozumab treatment group.
Potential complications of romosozumab treatment
One of the concerns of inhibition of sclerostin is the development of bony overgrowth and skeletal deformities as seen in individuals with sclerosteosis and van Buchem’s disease. Patients with both conditions have homozygous mutations, leading to decreased levels of sclerostin. Reassuringly, heterozygous carriers of these mutations have moderate levels of sclerostin, increased bone formation, and bone mass but none of the sequelae of bony overgrowth.40–42 In a large study of 3,321 patients on romosozumab, the frequency of hyperostosis was balanced between the romosozumab treatment group and the placebo group.27 Therefore, inhibition of sclerostin with romosozumab, especially over a limited period, would be less likely to lead to musculoskeletal sequela as seen in sclerosteosis and van Buchem’s disease.
Another concern related to pharmacological sclerostin inhibition is the possibility of the extraskeletal effects of romosozumab, given the diverse role the Wnt signaling pathway plays in development and homeostasis of adult organs and tissues.43 Based on current understanding, the secretion of sclerostin is fairly limited to osteocytes within the musculoskeletal system; therefore, theoretically, the extraskeletal effects of sclerostin should be minimal.
However, based on several studies in patients with chronic kidney disease, sclerostin may play a role in vascular physiology. Patients with chronic kidney disease who have vascular and aortic calcifications have high serum sclerostin levels; furthermore, sclerostin is expressed in vascular tissue undergoing calcification. The sclerostin expression may represent a counter-regulatory mechanism aimed at suppressing vascular calcification progression. However, more studies are needed to clarify this hypothesis.44 Notably, patients with sclerosteosis or van Buchem’s disease have not been reported to be at increased risk for cardiovascular disease.45 Furthermore, SOST-knockout mice have not been noted to develop vascular calcification.45 In the aforementioned Phase III trial, cardiovascular events were balanced between the romosozumab treatment group and the placebo group.27 Nevertheless, further evaluation of the effects of romosozumab on cardiovascular health, especially in high-risk patients, is warranted.
Future directions
Romosozumab holds significant potential to enhance our ability to treat osteoporosis in the future. Due to its potency as an osteoanabolic agent, romosozumab should be an excellent treatment option for patients with severe osteoporosis. With better ability to increase BMD at both the hip and the spine, romosozumab may help clinicians to adopt a “treat to target” approach for osteoporosis in the future.46 There have been encouraging results of the utility of sclerostin antibodies in glucocorticoid-induced osteoporosis, immobilization-induced bone loss, chronic inflammation-induced bone loss, type 2 diabetes bone loss, and bone loss associated with multiple myeloma.47 The utility of romosozumab in these conditions warrants further study.
Due to the uncertain effects of continuous bone formation on the skeletal system, there is a concern for exposing patients to prolonged romosozumab. Therefore, romosozumab may be used in sequence with antiresorptive agents, such as denosumab and bisphosphonates, to increase BMD as has been evaluated in Phase III clinical trials in the aforementioned studies. Further studies are needed to evaluate ways to optimally use this potent bone anabolic agent. It is reassuring that trials, to date, have not noted significant safety signals for romosozumab. However, these findings need to be confirmed in larger studies as Phase I and Phase II studies may not detect rare side effects due to smaller sample sizes.
Summary
Increased knowledge of bone biology has led to the discovery of several unique signaling pathways that regulate bone formation and resorption. The Wnt signaling pathway is important in bone formation, and sclerostin is an inhibitor of this pathway. Romosozumab, a monoclonal antibody that binds to sclerostin, prevents sclerostin from exerting its inhibitory effect. The Wnt signaling pathway is then activated leading to bone formation and increased BMD. Romosozumab is one of the most potent bone anabolic agents to date and holds significant potential to increase our ability to treat osteoporosis in the future. Further studies are needed to confirm its safety profile and to evaluate ways to optimize the use of this potent bone anabolic agent to treat osteoporosis.
Acknowledgment
MBB is currently receiving grants as a coinvestigator from the National Institutes of Health (# R01AR064850, 1U01AR068043, and R01HL127118) and Eli Lilly, outside the submitted work.
Disclosure
MBB is the chair of the American Board of Internal Medicine (ABIM) Rheumatology Board and the ABIM Rheumatology Board Exam Committee. No ABIM exam questions are disclosed in this article. MBB holds investments (stocks, stock options, or bond holdings) in Johnson & Johnson unrelated to this publication. SYL reports no conflicts of interest in this work.
References
NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA. 2001;285(6):785–795. | ||
Hernlund E, Svedbom A, Ivergard M, et al. Osteoporosis in the European Union: medical management, epidemiology and economic burden. A report prepared in collaboration with the International Osteoporosis Foundation (IOF) and the European Federation of Pharmaceutical Industry Associations (EFPIA). Arch Osteoporos. 2013;8(1–2):136. | ||
Geusens P. New insights into treatment of osteoporosis in postmenopausal women. RMD Open. 2015;1(suppl 1):e000051. | ||
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. | ||
Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4(2):68–75. | ||
Canalis E. Wnt signalling in osteoporosis: mechanisms and novel therapeutic approaches. Nat Rev Endocrinol. 2013;9(10):575–583. | ||
Shah AD, Shoback D, Lewiecki EM. Sclerostin inhibition: a novel therapeutic approach in the treatment of osteoporosis. Int J Womens Health. 2015;7:565–580. | ||
Deal C. Potential new drug targets for osteoporosis. Nat Clin Pract Rheumatol. 2009;5(1):20–27. | ||
Little RD, Carulli JP, Del Mastro RG, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70(1):11–19. | ||
Gong Y, Slee RB, Fukai N, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107(4):513–523. | ||
Sapir-Koren R, Livshits G. Osteocyte control of bone remodeling: is sclerostin a key molecular coordinator of the balanced bone resorption-formation cycles? Osteoporos Int. 2014;25(12):2685–2700. | ||
Poole KE, van Bezooijen RL, Loveridge N, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19(13):1842–1844. | ||
van Bezooijen RL, Svensson JP, Eefting D, et al. Wnt but not BMP signaling is involved in the inhibitory action of sclerostin on BMP-stimulated bone formation. J Bone Miner Res. 2007;22(1):19–28. | ||
Krause C, Korchynskyi O, de Rooij K, et al. Distinct modes of inhibition by sclerostin on bone morphogenetic protein and Wnt signaling pathways. J Biol Chem. 2010;285(53):41614–41626. | ||
van Bezooijen RL, Roelen BA, Visser A, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199(6):805–814. | ||
Aarden EM, Burger EH, Nijweide PJ. Function of osteocytes in bone. J Cell Biochem. 1994;55(3):287–299. | ||
Robling AG, Niziolek PJ, Baldridge LA, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283(9):5866–5875. | ||
Balemans W, Ebeling M, Patel N, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10(5):537–543. | ||
Loots GG, Kneissel M, Keller H, et al. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005;15(7):928–935. | ||
Moester MJ, Papapoulos SE, Lowik CW, van Bezooijen RL. Sclerostin: current knowledge and future perspectives. Calcif Tissue Int. 2010;87(2):99–107. | ||
Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860–869. | ||
Winkler DG, Sutherland MK, Geoghegan JC, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22(23):6267–6276. | ||
Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179–192. | ||
Li X, Ominsky MS, Warmington KS, et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009;24(4):578–588. | ||
Ominsky MS, Vlasseros F, Jolette J, et al. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 2010;25(5):948–959. | ||
Taylor S, Ominsky MS, Hu R, et al. Time-dependent cellular and transcriptional changes in the osteoblast lineage associated with sclerostin antibody treatment in ovariectomized rats. Bone. 2016;84:148–159. | ||
Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016;375(16):1532–1543. | ||
Padhi D, Allison M, Kivitz AJ, et al. Multiple doses of sclerostin antibody romosozumab in healthy men and postmenopausal women with low bone mass: a randomized, double-blind, placebo-controlled study. J Clin Pharmacol. 2014;54(2):168–178. | ||
Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26(1):19–26. | ||
McClung MR, Grauer A, Boonen S, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. 2014;370(5):412–420. | ||
IUPHAR BPS Guide to Pharmacology [webpage on the Internet]. Romosozumab. Available from: http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=8092. Accessed February 2, 2017. | ||
Lewiecki EM. Sclerostin: a novel target for intervention in the treatment of osteoporosis. Discov Med. 2011;12(65):263–273. | ||
Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507. | ||
Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–558. | ||
Genant HK, Engelke K, Bolognese MA, et al. Effects of romosozumab compared with teriparatide on bone density and mass at the spine and hip in postmenopausal women with low bone mass. J Bone Miner Res. 2017;32(1):181–187. | ||
Appelman-Dijkstra NM, Papapoulos SE. Modulating bone resorption and bone formation in opposite directions in the treatment of postmenopausal osteoporosis. Drugs. 2015;75(10):1049–1058. | ||
McClung M, Chines A, Brown J, et al. Effects of 2 years of treatment with romosozumab followed by 1 year of denosumab or placebo in postmenopausal women with low bone mineral density. Ann Rheum Dis. 2015;74:166–167. | ||
Langdahl B, Libanati C, Crittenden D, et al. Superior gains in bone mineral density and estimated strength at the hip for romosozumab compared with teriparatide in women with postmenopausal osteoporosis transitioning from bisphosphonate therapy: Results of a phase 3, open-label clinical trial [abstract]. Arthritis Rheumatol. 2016;68 (suppl 10). | ||
Amgen [webpage on the Internet]. Study to Determine the Efficacy and Safety of Romosozumab in the Treatment of Postmenopausal Women with Osteoporosis. Available from: https://clinicaltrials.gov/ct2/show/NCT01631214?term=NCT01631214&rank=1. Accessed November 1, 2016. | ||
Gardner JC, van Bezooijen RL, Mervis B, et al. Bone mineral density in sclerosteosis; affected individuals and gene carriers. J Clin Endocrinol Metab. 2005;90(12):6392–6395. | ||
van Lierop AH, Hamdy NA, Hamersma H, et al. Patients with sclerosteosis and disease carriers: human models of the effect of sclerostin on bone turnover. J Bone Miner Res. 2011;26(12):2804–2811. | ||
van Lierop AH, Hamdy NA, van Egmond ME, Bakker E, Dikkers FG, Papapoulos SE. Van Buchem disease: clinical, biochemical, and densitometric features of patients and disease carriers. J Bone Miner Res. 2013;28(4):848–854. | ||
Ge X, Wang X. Role of Wnt canonical pathway in hematological malignancies. J Hematol Oncol. 2010;3:33. | ||
Evenepoel P, D’Haese P, Brandenburg V. Sclerostin and DKK1: new players in renal bone and vascular disease. Kidney Int. 2015;88(2):235–240. | ||
McClung MR, Grauer A. Romosozumab in postmenopausal women with osteopenia. N Engl J Med. 2014;370(17):1664–1665. | ||
Lewiecki EM, Cummings SR, Cosman F. Treat-to-target for osteoporosis: is now the time? J Clin Endocrinol Metab. 2013;98(3):946–953. | ||
Ominsky MS, Boyce RW, Li X, Ke HZ. Effects of sclerostin antibodies in animal models of osteoporosis. Bone. 2017;96:63–75. | ||
Papapoulos SE. Anabolic bone therapies in 2014: new bone-forming treatments for osteoporosis. Nat Rev Endocrinol. 2015;11(2):69–70. | ||
Leder BZ, Tsai JN, Uihlein AV, et al. Two years of Denosumab and teriparatide administration in postmenopausal women with osteoporosis (The DATA Extension Study): a randomized controlled trial. J Clin Endocrinol Metab. 2014;99(5):1694–1700. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.