")
Back to Journals » Cancer Management and Research » Volume 12
Profile of Quizartinib for the Treatment of Adult Patients with Relapsed/Refractory FLT3-ITD-Positive Acute Myeloid Leukemia: Evidence to Date
Authors Fletcher L , Joshi SK , Traer E
Received 19 September 2019
Accepted for publication 5 December 2019
Published 8 January 2020 Volume 2020:12 Pages 151—163
DOI https://doi.org/10.2147/CMAR.S196568
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Eileen O'Reilly
Luke Fletcher,1,2 Sunil K Joshi,1–3 Elie Traer1,2
1Division of Hematology and Medical Oncology, Oregon Health & Science University, Portland, OR 97239, USA; 2Knight Cancer Institute, Oregon Health & Science University, Portland, OR 97239, USA; 3School of Medicine, Oregon Health & Science University, Portland, OR 97239, USA
Correspondence: Elie Traer
Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Mail Code: KR-HEM, Portland, OR 97239, USA
Tel +1 503 494 3553
Fax +1 503 494 3465
Email [email protected]
Abstract: Acute myeloid leukemia (AML) is a clonal hematologic neoplasm characterized by rapid, uncontrolled cell growth of immature myeloid cells (blasts). There are numerous genetic abnormalities in AML, many of which are prognostic, but an increasing number are targets for drug therapy. One of the most common genetic abnormalities in AML are activating mutations in the FMS-like tyrosine kinase 3 receptor (FLT3). As a receptor tyrosine kinase, FLT3 was the first targetable genetic abnormality in AML. The first generation of FLT3 inhibitors were broad-spectrum kinase inhibitors that inhibited FLT3 among other proteins. Although clinically active, first-generation FLT3 inhibitors had limited success as single agents. This led to the development of a second generation of more selective FLT3 inhibitors. This review focuses on quizartinib, a potent second-generation FLT3 inhibitor. We discuss the clinical trial development, mechanisms of resistance, and the recent FDA decision to deny approval for quizartinib as a single agent in relapsed/refractory AML.
Keywords: FLT3, quizartinib, AML, resistance, clinical trials, QuANTUM
Introduction
AML is a deadly disease, and identification of targetable molecular drivers in AML is an active area of investigation. FLT3, a cell-surface, membrane-bound class III receptor tyrosine kinase, plays a seminal role in normal hematopoiesis.1 FLT3 is composed of five immunoglobulin-like extracellular domains, a transmembrane domain, a juxtamembrane domain, and a tyrosine-kinase domain consisting of two lobes that are connected by a kinase-insert domain1 (Figure 1A). FLT3 is mutated in ~35% of AML patients and is one of the most common genetic abnormalities observed in AML.2,3
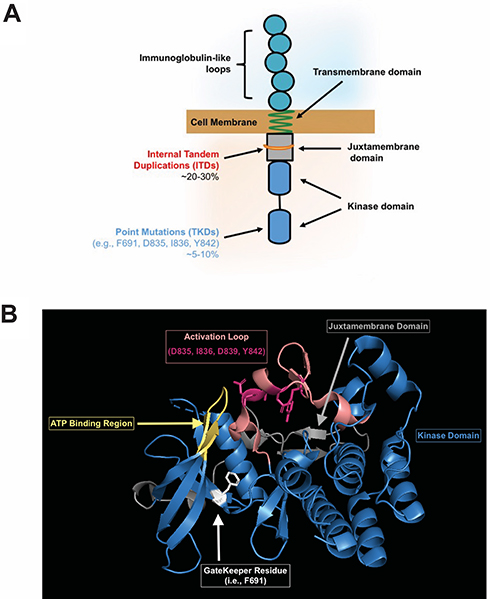 |
Figure 1 (A) Representative FLT3 receptor indicating location of internal tandem duplication (ITD) and tyrosine kinase domain (TKD) mutations. (B) The crystal structure of the FLT3 kinase domain is shown in blue (Griffith, J. et al (2004) Mol Cell 13: 169–178) with the ATP binding site in yellow (where FLT3 inhibitors bind). The activation loop (salmon) with most commonly mutated residues (magenta), and the F691 gatekeeper residue (white) are highlighted to show the close interaction of these domains. The juxtamembrane domain is also shown (gray) in relation to the kinase domain. |
There are two types of FLT3 mutations. FLT3 internal tandem duplication (ITD) mutations are more common, present in ~25% of the AML patients. FLT3-ITD mutations occur in the juxtamembrane region of the protein, interfering with the auto-inhibitory function of the domain, which leads to kinase activation. FLT3-ITD kinase activity causes uncontrolled cell proliferation, survival, and differentiation through pathways such as PI3K/AKT, MAPK/ERK, and JAK/STAT.4 FLT3-ITD mutations are a poor risk marker and associated with increased rates of relapse and reduced overall survival.5–8 The negative impact of FLT3-ITD has been further refined by the allelic ratio of FLT3-ITD, with a low allelic ratio of ITD (FLT3-ITD/FLT3 wild type <0.5) demonstrating improved outcomes in comparison to a high ITD allelic ratio.9 That being said, the impact of allelic ratio on prognosis was recently reported to be nullified by the clinical use of FLT3 inhibitors,10 so it is unclear if this distinction will remain in the future. FLT3 also has point mutations in the activation loop of the tyrosine kinase domain (FLT3-TKDs), usually around the D835 residue.11 FLT3-TKDs are present in ~5% of the AML patients but are not associated with worse outcome. FLT3-TKDs also lead to activation of the FLT3 receptor in cell line models, but are not as broadly activating of downstream kinases as FLT3-ITDs12 (Figure 1).
Due to its role in driving leukemia cell growth, there has been a concerted effort to develop clinically useful FLT3 inhibitors. The first-generation FLT3 inhibitors were multitargeted kinase inhibitors that were also noted to have FLT3 activity. These include midostaurin (protein kinase C inhibitor),13 sunitinib (VEGFR inhibitor),14 sorafenib (RAF inhibitor),15 and ponatinib (BCR-ABL inhibitor).16 These drugs were tested in patients with FLT3-mutated AML and had clinical activity, but their use was limited as single agents due to short duration of response. The second-generation FLT3 inhibitors were rationally designed to be more selective and potent FLT3 inhibitors with fewer off-targets. The second generation of FLT3 inhibitors includes quizartinib, gilteritinib, and crenolanib.
FLT3 inhibitors can be further separated into type 1 and type 2 inhibitors based upon their interaction with the kinase domain of the FLT3 receptor. Normally, FLT3 ligand binding induces the kinase domain to switch from an inactive to active conformation and drive downstream signaling. Type 1 inhibitors interact with the active conformation and are able to inhibit both FLT3-ITD and FLT3-TKD mutations. Type 2 inhibitors interact with the inactive conformation and, thus prevent activation. Since FLT3-TKD mutations favor the active conformation of FLT3, type 2 inhibitors do not inhibit most FLT3-TKD mutations.17 This difference in type 1 and type 2 inhibitors impacts resistance patterns between the two types of FLT3 inhibitors. FLT3-ITD patients treated with type 2 inhibitors frequently acquire FLT3-TKD mutations as a mechanism of resistance, whereas FLT3-ITD patients treated with type 1 inhibitors tend to have other mechanisms of resistance. Of the first-generation FLT3 inhibitors, midostaurin, lestaurtinib, and sunitinib are type 1 inhibitors, while sorafenib, ponatinib, and tandutinib are type 2 inhibitors. Of the second-generation, crenolanib and gilteritinib are type 1 inhibitors, and quizartinib is a type 2 inhibitor.
Quizartinib Preclinical Investigations
Quizartinib (AC220, Daiichi Sankyo) was originally developed by Ambit Biosciences, which was acquired by Daiichi Sankyo in 2014. Quizartinib is a potent second generation, type 2 FLT3 inhibitor with additional inhibitory activity against KIT and PDGFR.18 Quizartinib was identified in 2009 out of a series of compounds and found to have good efficacy and tolerability in xenograft models.19 It is an extremely potent FLT3 inhibitor, with activity in the low nanomolar range in cell culture assays, and animal models at doses as low as 1 mg/kg.20 From this promising pre-clinical data, quizartinib was taken into clinical trials21 (Table 1).
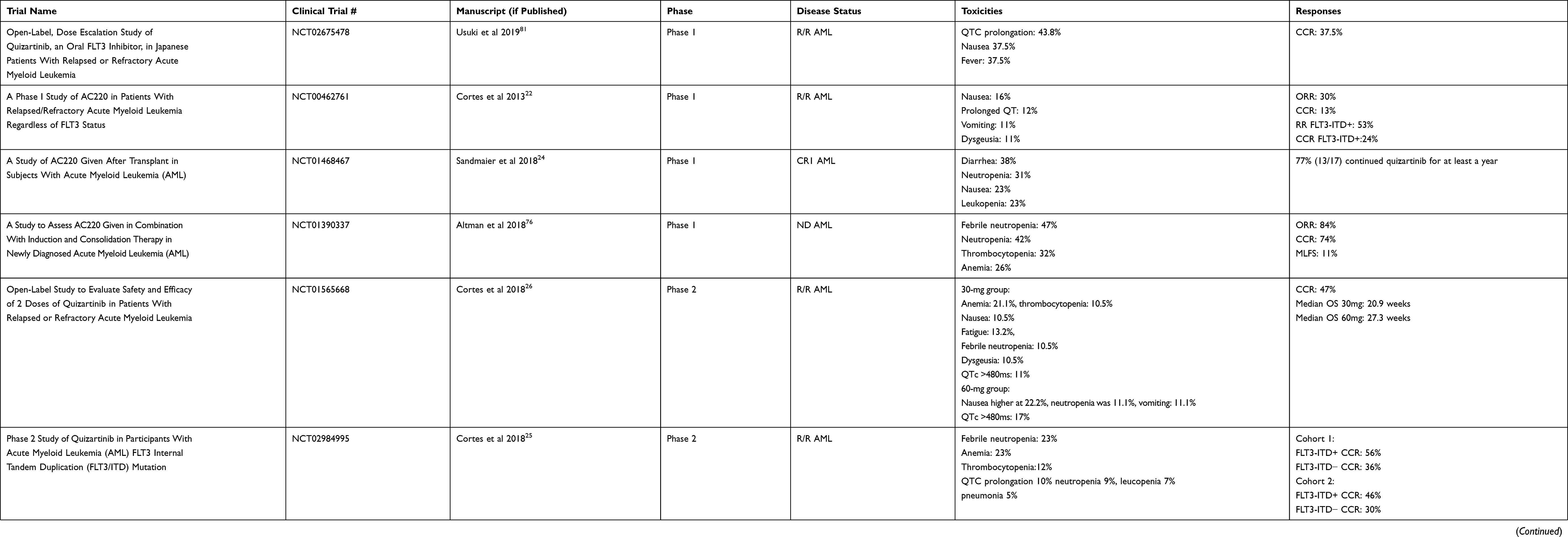 |
Table 1 Summary of Completed Quizartinib Trials |
Phase 1 Clinical Trials
In an American and Republic of Georgia phase 1 safety study, quizartinib was tested in relapsed/refractory patients regardless of FLT3 mutation status. Seventy-six patients received escalating doses of quizartinib monotherapy. The maximum tolerated dose was found to be 200 mg daily with the main dose-limiting toxicity at 200–300 mg being grade 3–4 QTc prolongation at 16% (4/25).22 The response rate was 30% (23/76) with 13% (10/76) composite complete remission22 (CRc). CRc includes complete remission (CR, defined by the International Working Group as <5% blasts in the bone marrow, neutrophils >1000/mm3, platelets >100K/mm3, absence of circulating blasts/extramedullary disease, and transfusion independence23) and CR with incomplete count recovery (CRi if neutrophils <1000/mm3 and platelets <100K/mm3, or CRp if only platelets <100K/mm3). Of those that obtained a CRc, three had incomplete platelet recovery and five had incomplete neutrophil recovery. Of note, in patients with FLT3-ITD, 53% responded (9/17), with five CRcs.22
Due to the high risk of relapse after allogeneic transplant in patients with FLT3-ITD, a phase 1 study was undertaken to determine optimal dosing to potentially prevent relapse after transplant. A 3+3 design enrolled 13 patients and identified a dose of 60 mg daily.24 Seventy-seven percent (10/13) continued quizartinib for at least 1 year with nine patients (69%) alive >50 weeks and four patients (31%) were still alive >2 years.24 Main toxicities included neutropenia (4/17, 31%), nausea (3/17, 23%), and leukopenia (3/17, 23%).24 Instead of a standalone trial, maintenance therapy was included in the QuANTUM-R Phase 3 trial (see below).
Phase 2 Trials
Building off the initial phase 1 results, a single-arm multi-center trial was performed with quizartinib monotherapy. It contained two cohorts, one cohort enrolling ages >60 years old with relapsed/refractory AML within 1 year of initial induction treatment and without allogeneic transplant, and a second cohort 18–85 years of age with relapsed/refractory AML after at least one salvage regimen or allogeneic transplant (>100 days post-transplant). Patients were considered to be FLT3-ITD+ if allelic frequency was >10%, but were not required to have FLT3-ITD mutations for the trial. Patients were started on 200 mg daily but due to QTc prolongation in the first patient, dosing was reduced to 135 mg for men and 90 mg for women, since women were noted to be more susceptible to QTc prolongation.25 A total of 333 patients were enrolled. In cohort 1, 56% (63/112) FLT3-ITD+ patients and 36% (16/44) of FLT3-ITD− patients achieved CRc(including <10% FLT3-ITD variant allele frequency and one-third of patients were low positive); however, most of these were CRi/CRp (FLT3-ITD+ 60/63, ITD− 14/16). Only three FLT3-ITD+ and two FLT3-ITD− patients achieved CR.25 In cohort 2, 62/136 (46%) FLT3-ITD+ patients achieved CRc and 12/40 (30%) FLT3-ITD− patients achieved CRc. In cohort 2, only five FLT3-ITD+ patients and one FLT3-ITD− patient achieved CR.25 The most common adverse events included cytopenias (neutropenia, anemia, thrombocytopenia), infections (febrile neutropenia and pneumonia), and QTc prolongation. One patient developed torsade de pointes while on study, and 5% of the patients (18/333) died secondary to an adverse event considered to be treatment related.25
Due to the QTc prolongation issues and cytopenias, a second randomized phase 2b trial was done to evaluate lower doses of quizartinib. Patients >18 years of age with either relapse after allogeneic transplant or relapsed/refractory AML after at least one salvage regimen were eligible for treatment. In contrast to the initial phase 2, only FLT3-ITD+ patients (defined at >10% allelic frequency) were included. Patients were randomized to 30 mg (n=38) or 60 mg (n=36) per day with the option to escalate by 30 mg if lack of response. Primary endpoints were CRc rate and QTcF >480 (QT corrected by Fridericia’s formula). The CRc rate was 47% for patients started at 30 or 60 mg per day. Eleven percent of the patients who started on 30 mg and 17% of the patients started on 60 mg developed a QTcF >480.26 In the 60 mg group, there were higher transplant rates (42% vs 32%), higher median overall survival (27.3 weeks vs 20.9 weeks), survivors living beyond 1 year (5 vs 1), and longer duration of CRc (9.1 weeks vs 4.2 weeks) when compared to the 30 mg daily group.26 This was only numerically higher as the study was not powered to statistically compare these groups. Of note, 61% (23/38) of the patients at 30 mg required dose escalation to 60 mg per day, with four achieving CRc after dose increase. Common toxicities in the 30 mg per day group were cytopenias (anemia 21.1%, thrombocytopenia 10.5%), nausea 10.5%, fatigue 13.2%, febrile neutropenia 10.5%, and dysgeusia 10.5%. In the 60 mg per day group, nausea was more frequent at 22.2%, absolute neutropenia was 11.1%, and vomiting was 11.1%.26 The remainder of the toxicities were similar.
PHASE 3 Clinical Trials
QuANTUM-R was a randomized, controlled, multicenter phase 3 trial to determine the efficacy of quizartinib monotherapy in relapsed/refractory FLT3-ITD AML.27 Patients were randomized 2:1 to quizartinib 60 mg daily vs investigator’s choice of chemotherapy. There was a 30 mg lead-in of quizartinib for the first 15 days, which was then increased to 60 mg if the mean QTcF was <450 ms. The options for chemotherapy included a low-intensity salvage regimen (low dose cytarabine) and higher intensity regimens (mitoxantrone, etoposide, cytarabine [MEC] or fludarabine, cytarabine, granulocyte colony-stimulating factor, idarubicin [FLAG-IDA]). Patients were able to move to allogeneic transplant when appropriate and could restart quizartinib as maintenance after transplant. The primary endpoint was overall survival. A total of 367 patients were enrolled (245 quizartinib, 122 chemotherapy) over a span of just under 3.5 years. Quizartinib was associated with a 24% reduction in risk of death compared to chemotherapy (hazard ratio 0.76, 95% CI 0.58–0.98; p=0.02) while overall survival was improved from 4.7 months in the chemotherapy arm to 6.2 months in the quizartinib arm.27 The CRc rate was 48.2%, and similar to previous trials most of these were CRi or CRp (108/118). In terms of allogeneic transplantation, 78 (32%) patients on quizartinib proceeded transplant while only 14 (11%) of 122 patients in the chemotherapy arm proceeded to transplant.27 The most common side effect in the quizartinib group were infections: 19% sepsis/septic shock, 12% pneumonia. Grade 3 QTc prolongation was seen in 3–4% of the patients on quizartinib. There were no grade 4 events. Based on this data, quizartinib was submitted for FDA approval as a single agent for relapsed/refractory disease. However, despite the positive results, quizartinib approval was rejected by the FDA, which we discuss in further detail below.
Challenging Toxicities of Quizartinib
One of the notable toxicities of quizartinib is prolonged cytopenias. As noted above, in phase 2 and phase 3 studies most patients achieved CRi or CRp, with only a few CRs (10/118 in phase 3 QuANTUM-R).27 For comparison, in the phase 3 ADMIRAL trial with the FLT3 inhibitor gilteritinib,28 52/134 patients achieved CR. One potential explanation for this disparity is that quizartinib also inhibits KIT, which is important for both myeloid and erythroid progenitor cell function.29 The impact of inhibition of both FLT3 and KIT inhibition as an explanation for more profound cytopenias is supported by in vitro bone marrow progenitor cell assays.30
The other notable toxicity of quizartinib is QT prolongation via potassium channelblockade.31 This was noted in the initial phase 1 and 2 trials, with subsequent dose adjustments to 135 mg for men and 90 mg for women as discussed above. The concern about QT prolongation led to the additional phase 2b study with 30 and 60 mg doses before opening the phase 3 QuANTUM trial. In the FDA review of the QuANTUM-R study, there was a concern that four deaths were possibly related to QTc prolongation, either from direct cardiac toxicity in form of myocardial infarction, cardiac failure, or fatal subdural hematoma from fall related to cardiac event.31 Furthermore, in the QuANTUM-First trial, the Oncologic Drugs Advisory Committee meeting noted five cardiac deaths to date in the quizartinib arm vs none in the placebo arm (2 cardiac arrest, 1 sudden death, 1 ventricular fibrillation, 1 ventricular dysfunction).32
In comparison, midostaurin does not prolong the QT interval but interacts with many anti-emetic and antifungal medications, so the recent phase 3 RATIFY clinical trial included close monitoring with ECGs and dose adjustments for prolonged QTc.33 This was similar for gilteritinib, which also had a low rate of QTc prolongation (4.9%) in the phase 3 ADMIRAL trial.28 This issue did not impact FDA approval for either of these drugs, but monitoring of QTc and dose reductions are recommended in the package inserts.
Resistance to Quizartinib
Despite the initial clinical efficacy of FLT3 inhibitors such as quizartinib, resistance to single agents develops after months of therapy, which limits their use in the clinic. Although clinical resistance eventually develops with all FLT3 inhibitors, there are differences between each drug with respect to the development of resistance mutations and off-target effects. Resistance to quizartinib can be broadly categorized into intrinsic and extrinsic. In the context of quizartinib, tumor intrinsic mechanisms involve (i) secondary point mutations in the FLT3 receptor, (ii) expansion of pre-existing subclones with additional gene mutations, and (iii) activation of alternative signaling pathways. Extrinsic mechanisms involve crosstalk between leukemia cells and cells of the bone marrow microenvironment that modulate quizartinib response.
Intrinsic Resistance Mechanisms
The most common tumor intrinsic mechanism is the accumulation of secondary point mutations in the TKD of FLT3 that confer quizartinib resistance. In 2012, shortly after the release of the interim analysis for the phase 2 trial of quizartinib monotherapy of 53 patients with relapsed/refractory FLT3-ITD AML, point mutations at three residues within the kinase domain of the FLT3 receptor were reported to confer resistance to quizartinib.34 These residues consist of the “gatekeeper” residue (i.e., F691) and residues within the activation loop of FLT3 (i.e., D835, Y842). The binding of quizartinib to the crystal structure of the FLT3 kinase domain was modeled and suggested that substitutions of F691 with non-aromatic residues could hinder the π-π stacking interaction needed to stabilize the benzo-imidazol-thiazol ring of quizartinib.34 In contrast, replacement of residues D835 or Y842 resulted in a loss of hydrogen bonding between these residues and S838, which is critical to maintain the inactive confirmation of FLT3 needed for quizartinib to bind to FLT334 (Figure 1B).
These observations were further confirmed with the first cocrystal structure of quizartinib bound to the FLT3 kinase domain. Importantly, this structure demonstrated that quizartinib binding to FLT3 relies on edge-to-face aromatic interactions mediated by the gatekeeper residue, F691, and activation loop residues.35 Only disruptions that strongly hinder this interaction enabled quizartinib resistance.35 A more recent study that performed extensive atomistic molecular dynamics simulations of the FLT3-quizartinib complex further suggests that once the active state of FLT3 is adopted due to the TKD mutations, the transition to the FLT3 inactive state is less likely due to the reaction kinetics.36 However, in a follow-up study, it was reported that not all D835 mutations facilitate quizartinib resistance.37 Specifically, bulky hydrophobic substitutions (i.e., D835Y/V/I/F) at this residue produced a resistant phenotype as these mutations prohibited hydrogen bonding between the activation loop and the S838 region of FLT3 and sterically hindered the binding of quizartinib.37 In aggregate, these early studies demonstrated that TKD mutations provide a survival mechanism by enabling the FLT3 receptor to shift from an inactive to active confirmation, precluding the binding of quizartinib. Likewise, these mutations also promote resistance to other type 2 FLT3 inhibitors such as sorafenib and ponatinib.37
More recently, single-cell analysis of FLT3-ITD primary AML cells suggests that mutational resistance to quizartinib is more complex than initially thought.38 While FLT3-ITD AML cells can acquire de novo FLT3-TKD mutations following treatment with quizartinib, this model alone does not accurately depict what is observed clinically. In an analysis of 15 patients treated with quizartinib, FLT3-TKD mutations were detected in 14 at resistance. Interestingly, the FLT3-TKD mutations were often found on the native FLT3 allele rather than the FLT3-ITD allele, and there were subpopulations that were resistant to quizartinib that did not contain FLT3-TKD mutations.38 Thus, in some patients, both FLT3-dependent and -independent resistant mechanisms can coexist, highlighting the underlying clonal heterogeneity that contributes to development of quizartinib resistance.
Apart from resistance mutations in the FLT3 receptor, upregulation of receptor tyrosine kinase AXL provides another route to quizartinib resistance. Previous studies have shown that increased expression of AXL is associated with worse progression-free and overall survival for patients with AML.39–41 Mechanistically, upregulation of AXL has been shown to mediate phosphorylation of FLT3.42 Inhibition of AXL via inhibitor or siRNA suppresses cell growth, induces apoptosis, and restores myeloid differentiation in vivo.42 Upon treatment with quizartinib, it was shown that expression of AXL increases in AML cell lines and in patients after treatment with quizartinib, supporting the idea that AXL upregulation may enable quizartinib resistance in the setting of leukemia.43 Inhibition of AXL with a small-molecule inhibitor TP-0903 restored sensitivity to quizartinib, corroborating its role in mediating resistance. The FLT3 inhibitor gilteritinib is also an AXL inhibitor,44 and it has been suggested that AXL inhibition delays the development of resistance. Apart from AXL’s role in promoting tumor-intrinsic resistance, a recent study has shown that marrow stromal cells support increased phosphorylation of STAT5, which in turn leads to increased AXL activity that drives quizartinib resistance both in vitro and in vivo.45 The study showed that the hypoxic marrow microenvironment further contributed to increased AXL activity, and thereby, aids quizartinib resistance in an extrinsic manner.45
Extrinsic Resistance Mechanisms
Many studies,46–51 including our own,52–54 have shown that the bone marrow microenvironment contributes significantly to the development of drug resistance in the setting of AML. Leukemia cells circulating in the peripheral blood are rapidly cleared by FLT3 inhibitors while leukemia cells within the microenvironment respond more slowly and a small number of leukemia cells persist despite treatment.50,55,56 Survival of these residual cells leads to the development of resistance and eventual relapse.57,58 This tumor-permissive microenvironment consists of a collection of mesenchymal stromal cells, immune cells, and hematopoietic cells that signal to residual leukemia cells.
Marrow stromal cells produce a number of growth factors, cytokines, and adhesion molecules within the AML microenvironment that provide the necessary cues for leukemia cells to survive initial therapy and eventually become resistant.46,50,53,54 One such factor is FLT3 ligand (FL), which is secreted by stromal cells.1 FL binds to the FLT3 receptor and in turn leads to restoration of FLT3 and downstream MAPK signaling, allowing FLT3-ITD AML cells to survive.59 The addition of exogenous FL to leukemia cell lines in vitro protects cells and increases the IC50 for FLT3 inhibition by activating the MAPK pathway.56 In agreement with this model, FL expression also increases in patients treated with FLT3 inhibitors.56,60 The addition of a MAPK inhibitor is able to abrogate stromal-mediated resistance and restore sensitivity to quizartinib.56 Other groups have found that AKT is also activated by marrow stromal cells. AKT inhibitors have been shown to have synergy with quizartinib and lead to increased cell death in FLT3-ITD+ cell lines such as MOLM14 and MV4-11, and overcomes the protective effects of bone marrow stromal cells in vitro.61
Previous work from our laboratory has shown that fibroblast growth factor 2 (FGF2) is secreted by marrow stromal cells and can protect FLT3-ITD AML cells from quizartinib.54,62 Addition of FGF2 leads to increased survival of FLT3-ITD AML cell lines and primary cells in vitro. In patients treated with quizartinib, expression of FGF2 in marrow stromal cells increased significantly during treatment and peaked just prior to resistance. FGF2 binds FGFR1 on AML cells, leading to downstream RAS/MAPK signaling, quizartinib resistance, and eventually relapse. Combined inhibition of FLT3 and FGFR signaling overcame FGF2-mediated protection of these AML cells.54,62 In comparison to FL resistance, FGF2 activates an accessory pathway through FGFR for survival, yet both ligand-mediated resistance mechanisms converge on the downstream MAPK pathway to drive resistance. In a separate but similar finding, a genome-wide CRISPR screen identified that loss of SPRY3, an intracellular inhibitor of FGF signaling, and GSK3, a canonical Wnt signaling antagonist, can also induce quizartinib resistance.63 Deletion of these genes in the FLT3-ITD AML cell line MV4-11, conferred quizartinib resistance as evidenced by increased cell viability and increased downstream MAPK and Wnt signaling.63 These findings were further confirmed in quizartinib-resistant AML patient samples.
Although discussed separately, it should be noted that extrinsic and intrinsic resistance mechanisms are not distinct, but interrelated. As previously mentioned, AXL expression can be increased in AML cells during treatment with quizartinib through intrinsic and extrinsic mechanisms, and others have shown increased GAS6 expression in the marrow microenvironment (ligand for AXL), that may also influence resistance.64 Likewise, FL- or FGF2-mediated resistance to quizartinib can lead to acquisition of resistance mutations over time in FLT3-ITD AML cell lines and patients treated with quizartinib, suggesting that extrinsic mechanisms of resistance mediate early resistance, which then leads to acquisition and outgrowth of intrinsic resistance mutations.54 Further characterization of the unique features of the leukemia microenvironment may define targets in the microenvironment for future clinical trials. For example, the finding that increased FGF2 expression in leukemia stromal cells can be blocked by FGFR inhibitors suggests a strategy to target the leukemia-permissive microenvironment that protects leukemia cells.53
Quizartinib FDA Review
Based upon promising initial clinical trial results, quizartinib was granted FDA breakthrough designation in 2018.65 However, in May 2019 the Oncologic Drugs Advisory Committee (ODAC) voted 8 to 3 against approval of the drug.32 This decision raised doubts as to whether quizartinib would obtain FDA approval. In June 2019, the FDA rejected approval for quizartinib for relapsed/refractory AML. Of note, this decision came just 3 days after quizartinib was approved for use in Japan.65 The QuANTUM-R phase 3 results, although positive, were greeted with skepticism by the FDA. First, while there was a significant overall survival benefit, the median survival was only extended 6 weeks (6.2 vs 4.7 months). In addition to this, there was no difference in event-free survival, which raised questions about what led to the improvement in overall survival. One specific criticism was that 23% of the patients randomized to chemotherapy did not receive treatment while only 2% of those randomized to quizartinib did not receive treatment, a concerning drop-out rate for the chemotherapy arm. Another concern was that only 11% of the patients on the chemotherapy arm underwent allogeneic stem cell transplant whereas 32% of the patients treated with quizartinib made it to transplant.27 Proponents of quizartinib have argued that this effect size is due to the activity of the drug, but the lower rate of patients who received allogeneic transplant in the chemotherapy arm raised questions as to whether this affected the difference in survival between the two groups. Although the unique toxicities of QTc prolongation and myelosuppression were noted, this did not appear to be a major factor in the rejection decision.
FDA-Approved FLT3 Inhibitors
Development of quizartinib took place concurrently with many other FLT3 inhibitors. Two other FLT3 inhibitors were recently approved by the FDA and are discussed for comparison. The first is midostaurin (PKC-412, Novartis), a multi-kinase inhibitor with activity against both FLT3-ITD and FLT3-TKD (type 1 inhibitor). As a first-generation FLT3 inhibitor, the clinical development of midostaurin has been ongoing for many years. The initial activity was shown in relapsed/refractory disease with 70% of the patients having a 50% reduction in peripheral blasts while 30% had a similar reduction in the bone marrow, including one CR with hypocellularity.66 Although it had clinical activity, the duration of response was limited as a single agent, so combination approaches were prioritized over monotherapy. Midostaurin was combined with induction chemotherapy and found to be safe and well tolerated.67 The Randomized AML Trial in FLT3 (RATIFY) study was a phase 3 trial of midostaurin in combination with 7+3 induction chemotherapy. A total of 717 patients with either FLT3-ITD or -TKD mutations were randomized to receive standard 7+3 induction (daunorubicin + cytarabine) and consolidation chemotherapy with or without midostaurin (360 midostaurin, 357 placebo). Patients were able to move to allogeneic transplantation when clinically feasible during consolidation. Patients in the midostaurin arm had a significantly increased median overall survival of 74.7 months vs 25.6 months (hazard ratio for death 0.78; P = 0.009).33 Based on this positive outcome, it was approved by the FDA for combination therapy at 50 mg orally twice a day on days 8–21 with standard induction chemotherapy and consolidation chemotherapy for newly diagnosed AML.68 In comparison to quizartinib development, the decision to test midostaurin in combination with chemotherapy started relatively early, which was related in part to the limited efficacy of midostaurin as a single agent. However, the positive results of the RATIFY trial resulted in midostaurin being the first FDA-approved FLT3 inhibitor for AML. Whether or not more potent type 1 and type 2 second- generation FLT3 inhibitors (including quizartinib, gilteritinib, and crenolanib) will be more effective than midostaurin in combination with chemotherapy is of great interest and currently under investigation.
The second FDA approved FLT3 inhibitor is gilteritinib (ASP2215, Astellas). Gilteritinib is a potent type 1 inhibitor of both FLT3-ITD and FLT3-TKD and also has activity against AXL.44 Initial safety and efficacy were determined in a phase 1/2 trial of 252 patients with relapsed or refractory FLT3-mutated AML. The recommended phase 2 dose was 120 mg orally daily after the initial dose escalation.69 In the full analysis set, 40% of the patients (100/249) had at least some response, with 30% (75/249) achieving CRc and the other 10% (25/249) achieving partial remission.69 Most common grade 3–4 adverse events included cytopenias and infectious complications (sepsis, febrile neutropenia, pneumonia). Other side effects included liver function abnormalities, diarrhea, nausea/vomiting, stomatitis, fatigue, and myalgias/arthralgias. These promising early results led to the ADMIRAL Trial, a phase 3 randomized trial of gilteritinib vs salvage chemotherapy in FLT3+ relapsed/refractory AML.28 Gilteritinib was given orally at a dose of 120 mg daily until unacceptable toxicity or lack of clinical benefit. After a median follow-up of 4.6 months (range: 2.8 to 15.8), 29 patients achieved CRc (21%, 95% CI: 14.5, 28.8).70 Based upon the interim results of the ADMIRAL trial, gilteritinib was FDA approved for relapsed/refractory AML in November 2018, and the full results of the ADMIRAL trial were presented in April 201970,71 which demonstrated an increase in median overall survival compared to salvage chemotherapy. In comparison to quizartinib, the ADMIRAL trial had more durable responses with a median overall survival of 9.6 vs. 5.6 months for chemotherapy (P<0.001) compared to the median overall survival with quizartinib 6.2 vs. 4.7 months with chemotherapy (p=0.02). The major caveat to comparing these trials is that the quizartinib trial limited the patient population to primary refractory AML and relapsed AML within 6 months of first CRc, essentially a higher risk population. This is reflected in the shorter duration of response to chemotherapy in the QuANTUM-R trial. That being said, the frequent development of FLT3-TKD resistance mutations with quizartinib and potentially the impact of AXL inhibition still appear to lead to more durable responses with gilteritinib.
In addition to this approval, gilteritinib is currently being investigated in multiple other settings in AML: including in combination with standard induction chemotherapy;72 in newly diagnosed not fit for standard induction as standalone treatment or in combination with hypomethylating agents;73,74 and as maintenance therapy after completion of consolidation chemotherapy or as post-transplant maintenance to prevent relapse.75
Future Directions
Quizartinib is approved in Japan for relapsed/refractory FLT3-mutated AML, and there are still a number of ongoing clinical trials with quizartinib in the US (see Table 2), so it may be that quizartinib will eventually obtain FDA approval as part of combination treatment. This would be more akin to the approval of midostaurin in combination with induction and consolidation chemotherapy, as opposed to the approval of gilteritinib as a single agent in relapsed/refractory AML. In the phase 1 dose escalation study of quizartinib with induction chemotherapy, 40 mg per day of quizartinib given on days 4–17 with standard 7+3 induction chemotherapy was identified as the maximal tolerated dose.76 Preliminary efficacy from the trial showed a 74% (14/19) CRc, and the phase 3 randomized QuANTUM-First trial is ongoing to evaluate efficacy.77 The results of induction chemotherapy trials with more potent second-generation FLT3 inhibitors such as quizartinib will be interesting to compare with results from the midostaurin RATIFY trial since many have speculated that some of midostaurin’s clinical success is partly attributable to off-target effects. The other path to FDA approval for quizartinib in the near future may also be in combination with other drugs such as hypomethylating agents or venetoclax as these combinations are currently in clinical investigation (see Table 2).
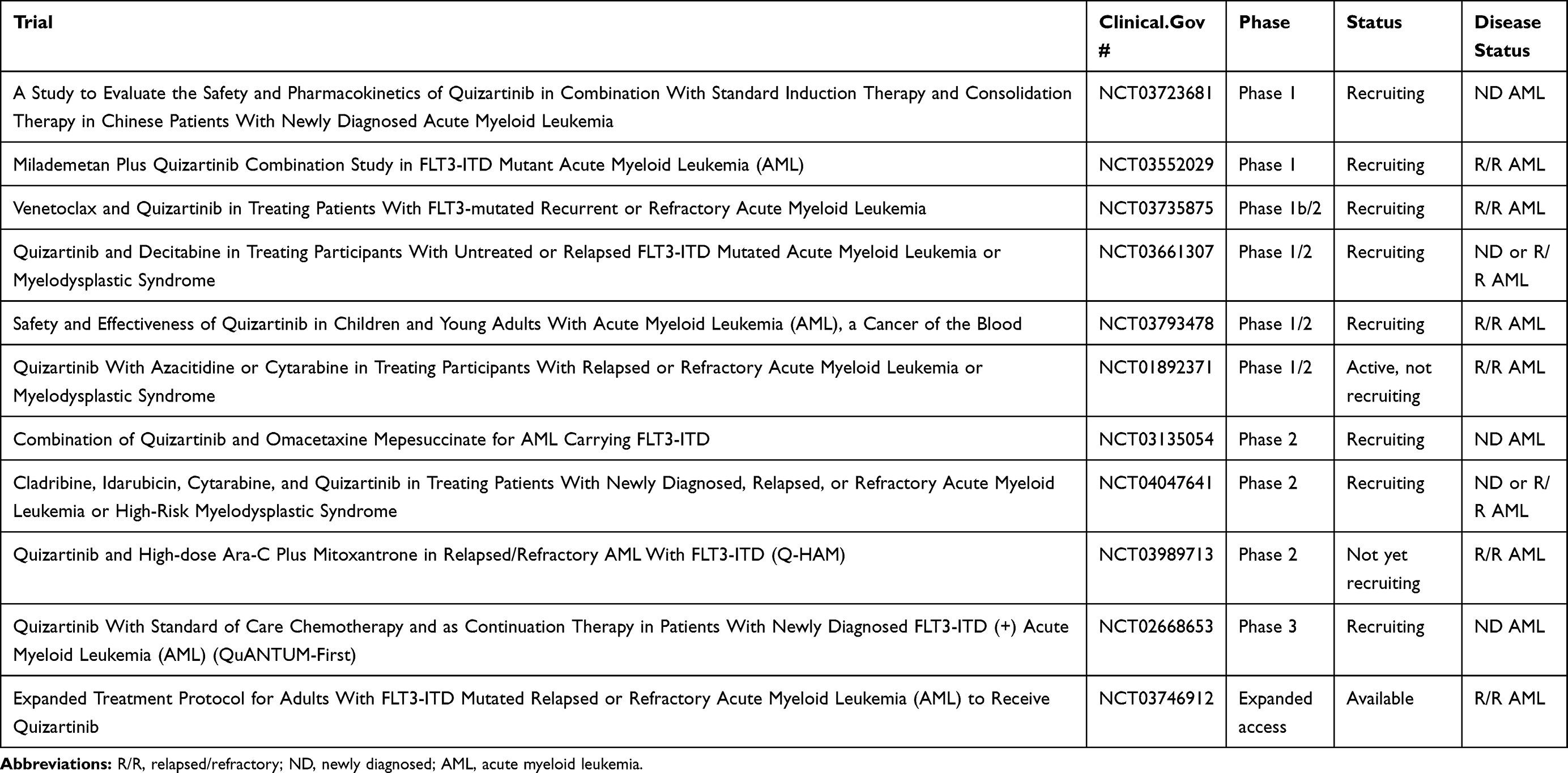 |
Table 2 Ongoing and Future Quizartinib Trials |
In addition, there are a number of pre-clinical studies with quizartinib investigating novel combinations. For example, 8-chloroadenosine is a novel RNA directed nucleoside analog with activity against both proliferative and quiescent leukemia stem cells; and was shown to be synergistic with quizartinib in cell lines and patient samples.78 In another study, quizartinib downregulated DNA repair genes such as BRCA1, BRCA2, and RAD51 leading to inhibition of double-strand DNA repair pathways. The PARP inhibitor BMN673 was able to overcome this resistance mechanism and was synergistic with quizartinib in cell line models, patient samples, and xenograft mouse models.79 And as a final example, the autophagy inhibitor TAK-165 was also shown to be synergistic with quizartinib in both cell lines and patient sample models.80
Given the surge of recently approved drugs in AML, there are a number of possible combinations with quizartinib that may improve patient responses and overcome resistance mechanisms. Many of these combinations are already in clinical trials, but of course, so are combinations with other FLT3 inhibitors. The right drug combination and timely completion of a phase 3 clinical trial will be important for quizartinib to move forward.
Disclosure
Dr. Elie Traer reports consulting honorarium from Astellas, Agios, Daiichi Sankyo, Abbvie, and ImmunoGen, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3(9):650–665. doi:10.1038/nrc1169
2. Nagel G, Weber D, Fromm E, et al. Epidemiological, genetic, and clinical characterization by age of newly diagnosed acute myeloid leukemia based on an academic population-based registry study (AMLSG BiO). Ann Hematol. 2017;96(12):1993–2003. doi:10.1007/s00277-017-3150-3
3. Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33(2):299–312. doi:10.1038/s41375-018-0357-9
4. Kim HG, Kojima K, Swindle CS, et al. FLT3-ITD cooperates with inv(16) to promote progression to acute myeloid leukemia. Blood. 2008;111(3):1567–1574. doi:10.1182/blood-2006-06-030312
5. Frohling S, Scholl C, Levine RL, et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell. 2007;12(6):501–513. doi:10.1016/j.ccr.2007.11.005
6. Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–4335. doi:10.1182/blood.V99.12.4326
7. Whitman SP, Archer KJ, Feng L, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. 2001;61(19):7233–7239.
8. Whitman SP, Maharry K, Radmacher MD, et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. Blood. 2010;116(18):3622–3626. doi:10.1182/blood-2010-05-283648
9. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447.
10. Yalniz F, Abou Dalle I, Kantarjian H, et al. Prognostic significance of baseline FLT3-ITD mutant allele level in acute myeloid leukemia treated with intensive chemotherapy with/without sorafenib. Am J Hematol. 2019. doi:10.1002/ajh.25553
11. Dicker F, Haferlach C, Sundermann J, et al. Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia. 2010;24(8):1528–1532. doi:10.1038/leu.2010.124
12. Choudhary C, Schwable J, Brandts C, et al. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood. 2005;106(1):265–273. doi:10.1182/blood-2004-07-2942
13. Fabbro D, Ruetz S, Bodis S, et al. PKC412–a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des. 2000;15(1):17–28.
14. Mendel DB, Laird AD, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–337.
15. Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. doi:10.1158/0008-5472.CAN-04-1443
16. O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–412. doi:10.1016/j.ccr.2009.09.028
17. Larrosa-Garcia M, Baer MR. FLT3 Inhibitors in acute myeloid leukemia: current status and future directions. Mol Cancer Ther. 2017;16(6):991–1001. doi:10.1158/1535-7163.MCT-16-0876
18. Kampa-Schittenhelm KM, Heinrich MC, Akmut F, Dohner H, Dohner K, Schittenhelm MM. Quizartinib (AC220) is a potent second generation class III tyrosine kinase inhibitor that displays a distinct inhibition profile against mutant-FLT3, -PDGFRA and -KIT isoforms. Mol Cancer. 2013;12:19. doi:10.1186/1476-4598-12-19
19. Chao Q, Sprankle KG, Grotzfeld RM, et al. Identification of N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor. J Med Chem. 2009;52(23):7808–7816. doi:10.1021/jm9007533
20. Zarrinkar PP, Gunawardane RN, Cramer MD, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114(14):2984–2992. doi:10.1182/blood-2009-05-222034
21. Gunawardane RN, Nepomuceno RR, Rooks AM, et al. Transient exposure to quizartinib mediates sustained inhibition of FLT3 signaling while specifically inducing apoptosis in FLT3-activated leukemia cells. Mol Cancer Ther. 2013;12(4):438–447. doi:10.1158/1535-7163.MCT-12-0305
22. Cortes JE, Kantarjian H, Foran JM, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31(29):3681–3687. doi:10.1200/JCO.2013.48.8783
23. Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the international working group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. J Clin Oncol. 2003;21(24):4642–4649. doi:10.1200/JCO.2003.04.036
24. Sandmaier BM, Khaled S, Oran B, Gammon G, Trone D, Frankfurt O. Results of a phase 1 study of quizartinib as maintenance therapy in subjects with acute myeloid leukemia in remission following allogeneic hematopoietic stem cell transplant. Am J Hematol. 2018;93(2):222–231. doi:10.1002/ajh.24959
25. Cortes J, Perl AE, Dohner H, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018;19(7):889–903. doi:10.1016/S1470-2045(18)30240-7
26. Cortes JE, Tallman MS, Schiller GJ, et al. Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3-ITD-mutated, relapsed or refractory AML. Blood. 2018;132(6):598–607. doi:10.1182/blood-2018-01-821629
27. Cortes JE, Khaled S, Martinelli G, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019;20(7):984–997. doi:10.1016/S1470-2045(19)30150-0
28. le Coutre P, Ottmann OG, Giles F, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood. 2008;111(4):1834–1839. doi:10.1182/blood-2007-04-083196
29. Lyman SD, Jacobsen SE. c-kit ligand and Flt3 ligand: stem/progenitor cell factors with overlapping yet distinct activities. Blood. 1998;91(4):1101–1134.
30. Galanis A, Levis M. Inhibition of c-Kit by tyrosine kinase inhibitors. Haematologica. 2015;100(3):e77–e79. doi:10.3324/haematol.2014.117028
31. NDA 212166. Quizaritinib.
32. White D, Saunders V, Grigg A, et al. Measurement of in vivo BCR-ABL kinase inhibition to monitor imatinib-induced target blockade and predict response in chronic myeloid leukemia. J Clin Oncol. 2007;25(28):4445–4451. doi:10.1200/JCO.2006.09.9499
33. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–464. doi:10.1056/NEJMoa1614359
34. Smith CC, Wang Q, Chin CS, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260–263. doi:10.1038/nature11016
35. Smith CC, Zhang C, Lin KC, et al. Characterizing and overriding the structural mechanism of the quizartinib-resistant FLT3 “gatekeeper” F691L mutation with PLX3397. Cancer Discov. 2015;5(6):668–679. doi:10.1158/2159-8290.CD-15-0060
36. Friedman R. The molecular mechanism behind resistance of the kinase FLT3 to the inhibitor quizartinib. Proteins. 2017;85(11):2143–2152. doi:10.1002/prot.v85.11
37. Smith CC, Lin K, Stecula A, Sali A, Shah NP. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia. 2015;29(12):2390–2392. doi:10.1038/leu.2015.165
38. Smith CC, Paguirigan A, Jeschke GR, et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood. 2017;130(1):48–58. doi:10.1182/blood-2016-04-711820
39. Challier C, Uphoff CC, Janssen JW, Drexler HG. Differential expression of the ufo/axl oncogene in human leukemia-lymphoma cell lines. Leukemia. 1996;10(5):781–787.
40. Neubauer A, Fiebeler A, Graham DK, et al. Expression of axl, a transforming receptor tyrosine kinase, in normal and malignant hematopoiesis. Blood. 1994;84(6):1931–1941. doi:10.1182/blood.V84.6.1931.1931
41. Rochlitz C, Lohri A, Bacchi M, et al. Axl expression is associated with adverse prognosis and with expression of Bcl-2 and CD34 in de novo acute myeloid leukemia (AML): results from a multicenter trial of the Swiss Group for Clinical Cancer Research (SAKK). Leukemia. 1999;13(9):1352–1358. doi:10.1038/sj.leu.2401484
42. Park I-K, Mishra A, Chandler J, Whitman SP, Marcucci G, Caligiuri MA. Inhibition of the receptor tyrosine kinase Axl impedes activation of the FLT3 internal tandem duplication in human acute myeloid leukemia: implications for Axl as a potential therapeutic target. Blood. 2013;121(11):2064–2073. doi:10.1182/blood-2012-07-444018
43. Park IK, Mundy-Bosse B, Whitman SP, et al. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia. 2015;29(12):2382–2389. doi:10.1038/leu.2015.147
44. Lee LY, Hernandez D, Rajkhowa T, et al. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood. 2017;129(2):257–260. doi:10.1182/blood-2016-10-745133
45. Dumas PY, Naudin C, Martin-Lanneree S, et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5- and hypoxia- dependent up-regulation of AXL. Haematologica. 2019. doi:10.3324/haematol.2018.205385
46. Kornblau SM, Ruvolo PP, Wang RY, et al. Distinct protein signatures of acute myeloid leukemia bone marrow-derived stromal cells are prognostic for patient survival. Haematologica. 2018;103:810–821. doi:10.3324/haematol.2017.172429
47. Raaijmakers MH, Mukherjee S, Guo S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852–857. doi:10.1038/nature08851
48. Tabe Y, Konopleva M. Advances in understanding the leukaemia microenvironment. Br J Haematol. 2014;164(6):767–778. doi:10.1111/bjh.12725
49. Xia B, Tian C, Guo S, et al. c-Myc plays part in drug resistance mediated by bone marrow stromal cells in acute myeloid leukemia. Leuk Res. 2015;39(1):92–99. doi:10.1016/j.leukres.2014.11.004
50. Zeng Z, Shi YX, Samudio IJ, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113(24):6215–6224. doi:10.1182/blood-2008-05-158311
51. Spaeth EL, Labaff AM, Toole BP, Klopp A, Andreeff M, Marini FC. Mesenchymal CD44 expression contributes to the acquisition of an activated fibroblast phenotype via TWIST activation in the tumor microenvironment. Cancer Res. 2013;73(17):5347–5359. doi:10.1158/0008-5472.CAN-13-0087
52. Hosseini MM, Kurtz SE, Abdelhamed S, et al. Inhibition of interleukin-1 receptor-associated kinase-1 is a therapeutic strategy for acute myeloid leukemia subtypes. Leukemia. 2018;32:2374–2387. doi:10.1038/s41375-018-0112-2
53. Carey A, Edwards D, Eide CA, et al. Identification of interleukin-1 by functional screening as a key mediator of cellular expansion and disease progression in acute myeloid leukemia. Cell Rep. 2017;18(13):3204–3218. doi:10.1016/j.celrep.2017.03.018
54. Traer E, Martinez J, Javidi-Sharifi N, et al. FGF2 from marrow microenvironment promotes resistance to FLT3 inhibitors in acute myeloid leukemia. Cancer Res. 2016;76(22):6471–6482. doi:10.1158/0008-5472.CAN-15-3569
55. Weisberg E, Liu Q, Nelson E, et al. Using combination therapy to override stromal-mediated chemoresistance in mutant FLT3-positive AML: synergism between FLT3 inhibitors, dasatinib/multi-targeted inhibitors and JAK inhibitors. Leukemia. 2012;26(10):2233–2244. doi:10.1038/leu.2012.96
56. Yang X, Sexauer A, Levis M. Bone marrow stroma-mediated resistance to FLT3 inhibitors in FLT3-ITD AML is mediated by persistent activation of extracellular regulated kinase. Br J Haematol. 2014;164(1):61–72. doi:10.1111/bjh.2013.164.issue-1
57. Ayala F, Dewar R, Kieran M, Kalluri R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia. 2009;23(12):2233–2241. doi:10.1038/leu.2009.175
58. Hui L, Chen Y. Tumor microenvironment: sanctuary of the devil. Cancer Lett. 2015;368(1):7–13. doi:10.1016/j.canlet.2015.07.039
59. Ghiaur G, Levis M. Mechanisms of resistance to FLT3 Inhibitors and the role of the bone marrow microenvironment. Hematol Oncol Clin North Am. 2017;31(4):681–692. doi:10.1016/j.hoc.2017.04.005
60. Sato T, Yang X, Knapper S, et al. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood. 2011;117(12):3286–3293. doi:10.1182/blood-2010-01-266742
61. Weisberg E, Liu Q, Zhang X, et al. Selective Akt inhibitors synergize with tyrosine kinase inhibitors and effectively override stroma-associated cytoprotection of mutant FLT3-positive AML cells. PLoS One. 2013;8(2):e56473. doi:10.1371/journal.pone.0056473
62. Javidi-Sharifi N, Martinez J, English I, et al. FGF2-FGFR1 signaling regulates release of leukemia-protective exosomes from bone marrow stromal cells. Elife. 2019;8:e40033. doi:10.7554/eLife.40033
63. Hou P, Wu C, Wang Y, et al. A genome-wide CRISPR screen identifies genes critical for resistance to FLT3 inhibitor AC220. Cancer Res. 2017;77(16):4402–4413. doi:10.1158/0008-5472.CAN-16-1627
64. Ben-Batalla I, Schultze A, Wroblewski M, et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood. 2013;122(14):2443–2452. doi:10.1182/blood-2013-03-491431
65. FDA. Grants Breakthrough Designation to Daiichi Sankyo’s FLT3 Inhibitor Quizartinib for Relapsed/Refractory FLT3-ITD AML [Press Release]. Daiichi Sankyo; 2018.
66. Stone RM, DeAngelo DJ, Klimek V, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105(1):54–60. doi:10.1182/blood-2004-03-0891
67. Stone RM, Fischer T, Paquette R, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26(9):2061–2068. doi:10.1038/leu.2012.115
68. Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–2551. doi:10.1056/NEJMoa055104
69. Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017;18(8):1061–1075. doi:10.1016/S1470-2045(17)30416-3
70. Kantarjian H, Pasquini R, Hamerschlak N, et al. Dasatinib or high-dose imatinib for chronic-phase chronic myeloid leukemia after failure of first-line imatinib: a randomized phase 2 trial. Blood. 2007;109(12):5143–5150. doi:10.1182/blood-2006-11-056028
71. Perl AE, Martinelli G, Cortes JE, et al. Abstract CT184: gilteritinib significantly prolongs overall survival in patients with FLT3-mutated (FLT3mut+) relapsed/refractory (R/R) acute myeloid leukemia (AML): results from the Phase III ADMIRAL trial. AACR Annu Meet. 2019;79(13):CT184–CT184.
72. Pellicano F, Copland M, Jorgensen HG, Mountford J, Leber B, Holyoake TL. BMS-214662 induces mitochondrial apoptosis in chronic myeloid leukemia (CML) stem/progenitor cells, including CD34+38- cells, through activation of protein kinase Cbeta. Blood. 2009;114(19):4186–4196. doi:10.1182/blood-2009-05-219550
73. Mahon FX, Rea D, Guilhot F, et al. Discontinuation of imatinib therapy after achieving a molecular response in chronic myeloid leukemia patients. In. ASH Abstract #8592009. Blood. 2009;114:859. doi:10.1182/blood.V114.22.859.859
74. Mauro MJ. Appropriate sequencing of tyrosine kinase inhibitors in chronic myelogenous leukemia: when to change? A perspective in 2009. Curr Opin Hematol. 2009;16(2):135–139. doi:10.1097/MOH.0b013e3283257b2b
75. Kantarjian HM, Talpaz M, O’Brien S, et al. Dose escalation of imatinib mesylate can overcome resistance to standard-dose therapy in patients with chronic myelogenous leukemia. Blood. 2003;101(2):473–475. doi:10.1182/blood-2002-05-1451
76. Altman JK, Foran JM, Pratz KW, Trone D, Cortes JE, Tallman MS. Phase 1 study of quizartinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed acute myeloid leukemia. Am J Hematol. 2018;93(2):213–221. doi:10.1002/ajh.24974
77. Talpaz M, Silver RT, Druker BJ, et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood. 2002;99(6):1928–1937. doi:10.1182/blood.V99.6.1928
78. Buettner R, Nguyen LXT, Kumar B, et al. 8-chloro-adenosine activity in FLT3-ITD acute myeloid leukemia. J Cell Physiol. 2019;234:16295–16303. doi:10.1002/jcp.v234.9
79. Maifrede S, Nieborowska-Skorska M, Sullivan-Reed K, et al. Tyrosine kinase inhibitor-induced defects in DNA repair sensitize FLT3(ITD)-positive leukemia cells to PARP1 inhibitors. Blood. 2018;132(1):67–77. doi:10.1182/blood-2018-02-834895
80. Ouchida AT, Li Y, Geng J, et al. Synergistic effect of a novel autophagy inhibitor and quizartinib enhances cancer cell death. Cell Death Dis. 2018;9(2):138. doi:10.1038/s41419-017-0170-9
81. Usuki K, Handa H, Choi I, et al. Safety and pharmacokinetics of quizartinib in Japanese patients with relapsed or refractory acute myeloid leukemia in a phase 1 study. Int J Hematol. 2019;110:654–664. doi:10.1007/s12185-019-02709-8
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.