")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 7
Profile of neratinib and its potential in the treatment of breast cancer
Authors Feldinger K, Kong A
Received 16 February 2015
Accepted for publication 25 March 2015
Published 9 June 2015 Volume 2015:7 Pages 147—162
DOI https://doi.org/10.2147/BCTT.S54414
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Katharina Feldinger,1 Anthony Kong,2
1Department of Oncology, The Weatherall Institute of Molecular Medicine, University of Oxford, Oxford, 2The Robert Aitkin Institute, School of Cancer Sciences, University of Birmingham, Birmingham, UK
Abstract: The HER (ErbB) receptor tyrosine kinase receptors are implicated in many cancers and several anti-HER treatments are now approved. In recent years, a new group of compounds that bind irreversibly to the adenosine triphosphate binding pocket of HER receptors have been developed. One of these compounds, neratinib, has passed preclinical phases and is currently undergoing various clinical trials. This manuscript reviews the preclinical as well as clinical data on neratinib. As a pan-HER inhibitor, this irreversible tyrosine kinase inhibitor binds and inhibits the tyrosine kinase activity of epidermal growth factor receptors, EGFR (or HER1), HER2 and HER4, which leads to reduced phosphorylation and activation of downstream signaling pathways. Neratinib has been shown to be effective against HER2-overexpressing or mutant tumors in vitro and in vivo. Neratinib is currently being investigated in various clinical trials in breast cancers and other solid tumors, including those with HER2 mutation. Earlier studies have already shown promising clinical activity for neratinib. However, more translational research is required to investigate biomarkers that could help to predict response and resistance for selection of appropriate patients for treatment with neratinib, either as monotherapy or in combination with other drug(s).
Keywords: neratinib, HKI 272, pan-HER inhibitor, irreversible tyrosine kinase inhibitor, HER (ErbB), breast cancer
Introduction
The family of HER (ErbB) receptor tyrosine kinases consists of four members, ie, epidermal growth factor receptors [EGFR (HER1 or ErbB1), HER2 (ErbB2, neu), HER3 (ErbB3), and HER4 (ErbB4)].1 Overexpression, mutation, or aberrant activity of these receptors has been implicated in various types of cancer. HER2 is overexpressed in approximately 15%–20% of all breast cancers1 and is correlated with poor prognosis.2 HER receptors comprise an extracellular domain, a single transmembrane domain, and an intracellular tyrosine kinase domain.3 A disintegrin and metalloproteinases (ADAMs) shed the ligands that are needed for HER member activation. Eleven ligands are known to bind to the different receptors of the family, as shown in Figure 1;3 however, HER2 does not have a known ligand.4 Ligand binding induces a conformational change in HER receptors 1, 3, and 4, which exposes the dimerization domain. This facilitates homodimerization or heterodimerization and transphosphorylation of the tyrosine kinase domains.5 Subsequently, downstream signaling pathways, most prominently the phosphatidylinositide 3-kinase and mitogen-activated protein kinase pathways, are activated and promote survival and proliferation.6–8 HER2 adopts a constant “open” conformation, with the dimerization domain being always available.9 It was shown to be the preferred dimerization partner within the HER receptor network and it can also form potent homodimers.10
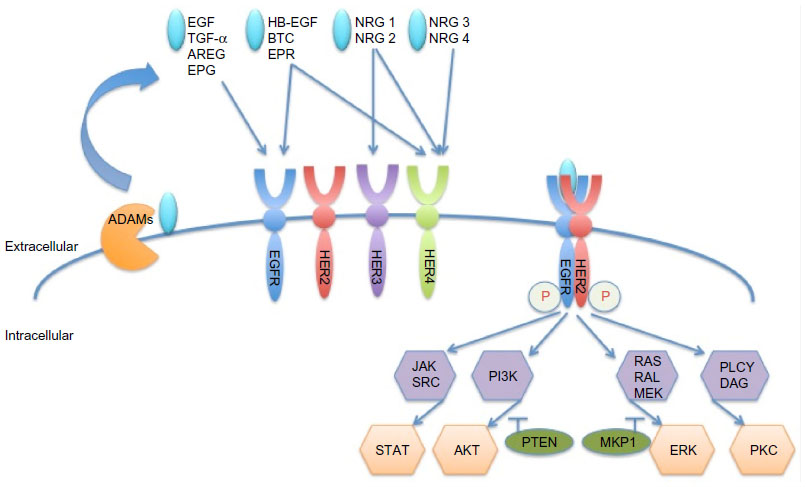 | Figure 1 HER member family and activation. |
Development of neratinib to target HER family kinase activity
Neratinib (HKI-272, Puma Biotechnology Inc., Los Angeles, CA, USA) is an oral tyrosine kinase inhibitor (TKI). The scientific approach behind the development of this and other similar compounds was to design small molecules that could bind to the tyrosine kinase domain and inhibit its interaction with adenosine triphosphate (ATP) in order to prevent receptor phosphorylation. A selection of compounds with relevant references is listed in Figure 2 and Table 1, although a direct comparison of IC50 values is difficult due to different assay conditions. Earlier compounds such as erlotinib (Tarceva®) and gefitinib (Iressa®), which are approved in non-small cell lung cancer (NSCLC), or lapatinib (Tyverb), which is licensed for HER2-positive breast cancer, were designed as reversible compounds that directly compete with ATP for binding. However, considering the high endogenous ATP levels within the cell (mM range) and that drug resistance is a common problem with these reversible inhibitors, there was a need to develop a more potent inhibitor that could have enhanced and sustained antitumor activity.11
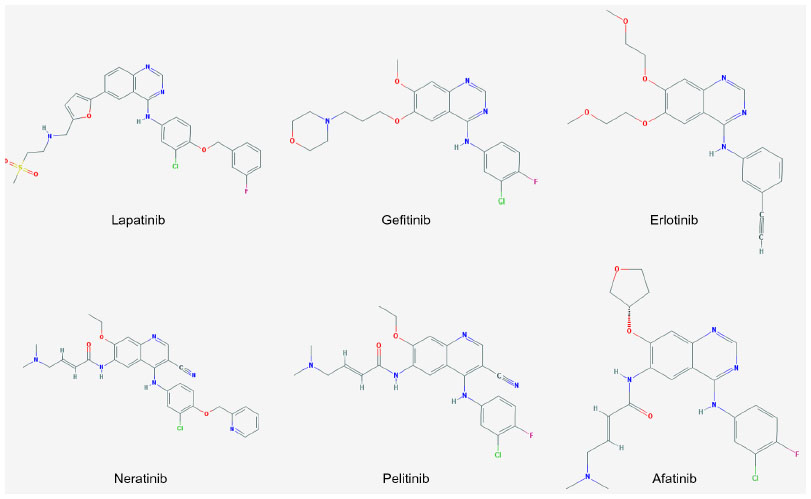 | Figure 2 Chemical structure of tyrosine kinase inhibitors. |
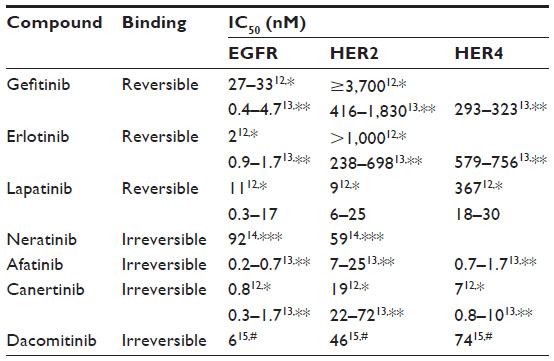 | Table 1 Comparison of various tyrosine kinase inhibitors |
A variety of irreversible inhibitors of quinazoline derivatives has been developed, including dacomitinib and afatinib, which showed a high affinity and efficacy in inhibiting autophosphorylation of EGFR.22 Neratinib is a compound comprising a quinolone core with the same reactive substituent as afatinib, but with a lower affinity and biochemical potency.22 The effectiveness of these compounds is based on a Michael acceptor moiety, which is designed to form a covalent bond with the conserved cysteine residue at the lip of the EGFR ATP binding site.23–25 Neratinib interacts with cysteine residues Cys-773 and Cys-805 in EGFR and HER2, respectively, in the ATP binding pocket.11 Due to this selective binding, a higher specificity of the compound is achieved. It was shown that neratinib did not significantly inhibit serine-threonine kinases such as Akt in cell-free assays. Tyrosine kinases tested included c-met, Kdr (vascular endothelial growth factor receptor 2), and Src, and either no effect or a 14-fold and 24-fold decreased efficacy, respectively, was found in comparison with HER2.14
Neratinib was developed as a modification of EKB-569 (pelitinib), an irreversible EGFR inhibitor developed by Wyeth Pharmaceuticals.11 The chemical name is 2-butenamide, N-[4- [[3-chloro-4-(2-pyridinylmethoxy) phenyl] amino]-3-cyano-7-ethoxy-6-quinolinyl]-4-(dimethylamino)-, (2E)-, (2Z) -2-butenedioate (1:1). In comparison with EKB-569, neratinib has a lipophilic 2-pyridinylmethyl moiety at the para-position of the aniline and a lipophilic chlorine atom at the meta-position, which is thought to improve its activity against HER2.14,24 However, as the cysteine residue required for binding is conserved in three of the HER receptors, ie, EGFR, HER2, and HER4, neratinib is a pan-HER inhibitor.11 Neratinib inhibits EGFR and HER2 at IC50 values of 92 nM and 59 nM, respectively; specific IC50 data for HER4 has not been published.14 However, the compound is similar to afatinib, so activity against HER4 can be expected and has indeed been shown previously.26,27
Efficacy of neratinib in preclinical models
As neratinib binds irreversibly to the ATP binding pocket of HER member receptor tyrosine kinases, it was hypothesized that after binding a prolonged bioavailability in the blood would not be needed. Therefore, the efficacy of the compound would only depend on receptor turnover.11 Acute treatment with low doses of neratinib led to a decrease of HER2 and EGFR phosphorylation in HER2-overexpressing BT474 and EGFR-amplified A431 cells, respectively, which persisted even after the removal of the drug. However, the time point chosen was only 5 hours.14 Neratinib was shown to decrease tumor growth in EGFR and HER2-expressing cell lines in vitro, which was correlated with a decrease in cyclin D1 and an increased level of p27, leading to G1-S cell cycle arrest and an increased sub-G1 population, indicating apoptosis.14
HER2-positive breast cancer
Despite reasonable IC50 values with regard to both EGFR and HER2, several studies have shown that neratinib is more effective in HER2-positive breast cancers relative to other breast cancer subtypes. In a study using a panel of cell lines, we showed that the response to neratinib was correlated with baseline HER2 and phosphorylated HER2 levels, but not with EGFR levels in vitro.26 Interestingly, neratinib decreased the activation of phosphatidylinositide 3-kinase and mitogen-activated protein kinase downstream pathways, and the activity against AKT and ERK could be correlated with efficacy.14,26,28 Other biomarker candidates that have been investigated in vitro include upregulation of RB1CC1, HER3, FOXO3a, and NR3C1 as well as downregulation of CCND1 mRNA, all of which have been correlated with response to neratinib.29 In vivo, neratinib was shown to be more effective in decreasing xenograft tumor growth of EGFR-amplified (A431) and HER2-overexpressing (BT474 and SKOV3) cell lines compared with low EGFR and HER2-expressing cell lines (MCF-7 and MX-1) in athymic (nude) mice.14 However, the decrease in tumor growth in the EGFR-dependent tumor model was less than that seen in HER-2-dependent tumors at comparable doses.14
Dual targeting of HER2-positive breast cancers and overcoming treatment resistance
Considerable effort has been made to target HER2 signaling. The first treatment approved by the US Food and Drug Administration was the monoclonal antibody trastuzumab (Herceptin®), which targets domain IV of the extracellular domain of HER2.9 Trastuzumab was approved in 1998 and has been shown to be effective, particularly in combination with chemotherapeutic agents, in the treatment of various stages of HER2-positive breast cancer.30–33 However, de novo or acquired resistance to trastuzumab is frequent and is still a subject of extensive research.
Previous reports identified phosphatase and tensin homolog (PTEN) deficiency,34,35 phosphatidylinositide 3-kinase (PI3K) mutation,36 loss of p27,37 and reduced binding of the antibody due to HER2 cleavage38 or allosteric hindrance39,40 as mechanisms involved in trastuzumab resistance. Interestingly, it was also reported that treatment with trastuzumab led to an increased activation of other receptor tyrosine kinases, including the insulin-like growth factor 1 receptor (IGFR1)38 and c-Met.41 In addition, we have previously shown that trastuzumab induced an increase of ADAM10 and ADAM17 expression, which enhanced the release of the HER ligands betacellulin and heregulin (neuregulin), resulting in activation of HER receptors as well as downstream signaling pathways during prolonged treatment with trastuzumab.42,43 A ligand-dependent mechanism has also been reported in vitro with regard to lapatinib resistance.44,45
As shown in Figure 3, neratinib targets tyrosine kinase activity at the intracellular domain of the HER receptors (EGFR, HER2, and HER4), whereas trastuzumab targets the extracellular domain of HER2. This difference in mechanism of action would explain the improved response when both drugs are combined, since trastuzumab can induce ligand-dependent activation of HER receptors,42,43 whereas neratinib inhibits the phosphorylation and activity of HER receptors. Indeed, it has been shown that neratinib can overcome trastuzumab resistance in vitro.26 It was reported that neratinib was able to suppress ligand-stimulated HER2–HER3 dimers and could effectively disrupt preformed HER2–HER3 dimers in non-HER2-overexpressing MCF7 cells more effectively than lapatinib.46 Additionally, both TKIs decreased internalization of the HER2 receptor from the cell membrane, leading to increased trastuzumab-induced antibody-dependent cellular cytotoxicity.46 It has been shown that during trastuzumab treatment and resistance, HER4 cleavage and its translocation to the nucleus was increased; and nuclear HER4 was associated with a poor prognosis.27 However, cotreatment with neratinib decreased trastuzumab-induced nuclear HER4 in vitro and in BT474 xenograft models.27 In the same in vivo model, it was shown that trastuzumab and neratinib treatment in combination was more effective than either treatment alone.26
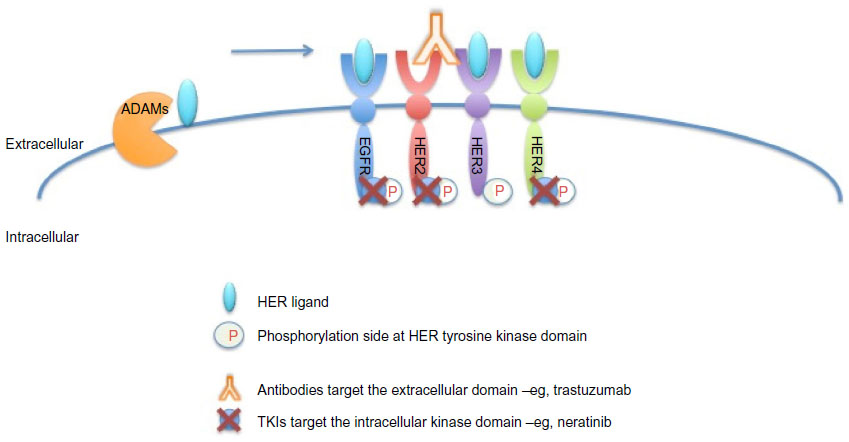 | Figure 3 Sites of interaction of trastuzumab and neratinib with HER receptors. |
Additionally, neratinib was also shown to enhance response when added to mammalian target of rapamycin/phosphatidylinositide 3-kinase inhibitors47 and the PARP inhibitor olaparib.48 These results, as well as those of the study from Seyhan et al28 point to the potential added benefit of using neratinib as a combinational treatment option alongside other targeted treatments as well as conventional chemotherapeutics.
We have recently shown that neuregulin (NRG1) can counteract the response to lapatinib, which was improved by the combination of lapatinib with pertuzumab, an antibody binding to the HER2 dimerization domain.45 It would thus be interesting to test whether NRG1 mediates resistance to neratinib and whether pertuzumab could also enhance the response to neratinib. In addition, since the combination of trastuzumab with new agent(s), including a heat shock protein 90 inhibitor,49 was shown to be additional or synergistic, it will be interesting to test the combination of neratinib with these new agents. However, more data will be needed to guide the sequence and/or combinations of different compounds, which could benefit patients.
Neratinib for CNS metastasis in HER2-positive breast cancer
A major problem in the treatment of patients with HER2-positive breast cancer is the high incidence of central nervous system (CNS) metastases. Although clinical studies suggest that patients with CNS metastases who receive trastuzumab have an increased overall survival compared with patients who do not, it is well understood that penetration of trastuzumab across the blood–brain barrier is inefficient.50 Therefore, there is a growing interest in using TKIs to target CNS disease in patients with HER2-positive breast cancer. One obvious advantage of these small-molecule compounds is their size. The blood–brain barrier is at least in part regulated by ATP-binding cassette (ABC) transporters, which contribute to multidrug resistance by actively extruding the drug compounds. Recent studies have shown that lapatinib is a substrate as well as an inhibitor of ABCB1, potentially explaining why brain concentrations of lapatinib tend to be lower.51 However, in contrast with lapatinib, neratinib has been shown to reverse ABCB1-mediated chemoresistance52 and it is unaltered by the presence of the ABCG2 transporter.53 Moreover, neratinib could inhibit the function of this transporter at higher doses, leading to an increased level of ABCG2 substrate drugs within cells.53 At the 2014 American Society of Clinical Oncology annual meeting, the preliminary data from a clinical trial investigating the efficacy of neratinib in HER2-positive breast cancer patients presenting with brain metastasis (ClinicalTrials.gov identifier NCT01494662, see Table 2) suggested that neratinib could help to control disease in some HER2-positive breast cancer patients presenting with brain metastasis; however, the CNS overall response rate was only 7.5%.54
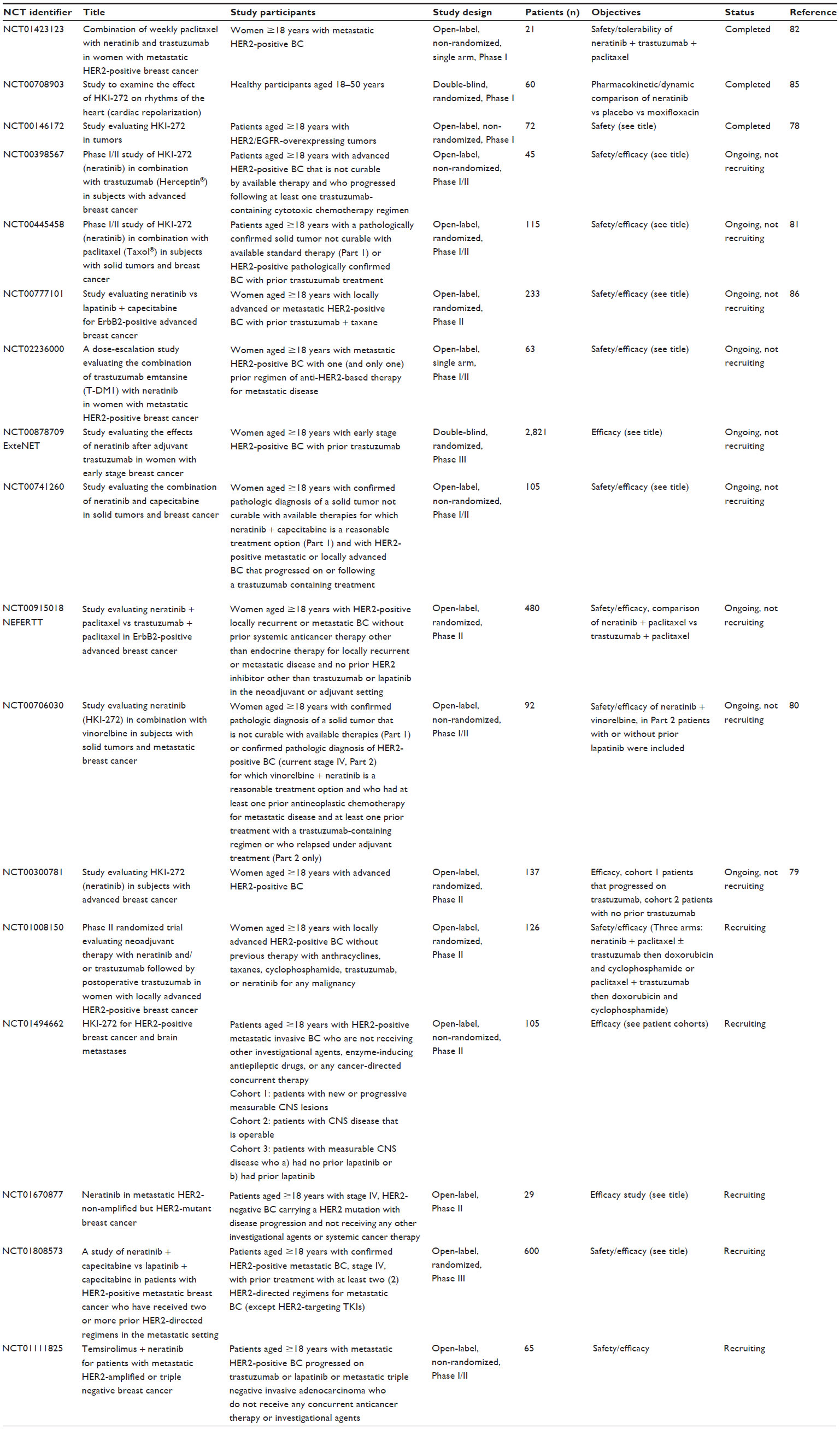 | Table 2 Clinical trials involving neratinib in breast cancer |
HER2-negative breast cancer
There is an interest in assessing whether neratinib could also be effective in breast tumors with lower HER2 expression, eg, immunohistochemistry (IHC) 2+ but fluorescence in situ hybridization (FISH)-negative patients. These will be those tumors that do not fulfill the current criteria for HER2-positive breast cancer. HER2 testing and its criteria have not always been consistent. Initially, the US Food and Drug Administration guidelines for HER2 testing recommended that if ≥10% of the tumor cells show strong (3+) staining by IHC or if the HER2/chromosome 17 ratio at FISH is ≥2.0, the tumor is classified as HER2-positive.55 The threshold recommendations were updated by the American Society of Clinical Oncology/College of American Pathologists to ≥30% of tumor cells with 3+ staining and a ratio of ≥2.2 for IHC and FISH, respectively, in 2007.56 However, the latest recommendation states thresholds similar to the initial guidelines.57 To be eligible for HER2 targeting treatments, breast cancer patients have to be classified as HER2-positive, depending on the local criteria used. However, IHC and FISH testing can be equivocal, and in two trials, patients initially classified as HER2-positive were found to be HER2-negative upon retesting. However, some of these negative patients still responded to treatment with trastuzumab.58,59
As the original HER2 positivity threshold was based on trastuzumab response, it is unknown whether the threshold should be different for TKIs such as lapatinib and neratinib, since there have been studies suggesting that breast cancer with a lower HER2 level may benefit from TKIs. For example, one study showed that patients with low HER2 by IHC may be more sensitive to lapatinib than to trastuzumab.60 The I-SPY2 trial suggested that HER2-negative patients may benefit from the addition of neratinib to chemotherapy (see below).61
Triple-negative breast cancer
Triple negative breast cancer (TNBC) represents a group of heterogeneous breast cancers that lack expression of the estrogen receptor and progesterone receptor as well as HER2 overexpression or gene amplification. Patients with TNBC tend to have a poor prognosis and very aggressive breast cancer with a lack of targeted therapy options. EGFR is frequently overexpressed in TNBC, and thus there is an interest in targeting this subtype of cancer using EGFR inhibitors.62 It was reported that ADAM17 was expressed at significantly higher levels in TNBC than in non-TNBC, and an ADAM17 inhibitor could inhibit cell proliferation by a decrease of the release of the EGFR ligands and could also enhance the response of TNBC cells to neratinib.63 However, despite the promising in vitro data, TNBC patients did not have a statistically significant improvement in pathological complete response (pCR) when treated with neratinib and chemotherapy in comparison with chemotherapy alone in the I-SPY 2 trial (see below).64 Moreover, a few trials involving cetuximab, an EGFR-targeting monoclonal antibody, have shown a poor response rate in patients with TNBC.65,66
This indicates that TNBC tumors are heterogeneous and may be driven by several different oncogenes other than EGFR. Therefore, targeting EGFR may only be suitable for a small subset of patients. Thus, the data so far show that the mechanisms of response and resistance to neratinib and other EGFR inhibitors in TNBCs are not well understood. There is therefore a need for further investigation of relevant predictive biomarkers for use of EGFR inhibitors in TNBC.
Role of neratinib in treatment of tumors with EGFR, HER2, HER3, or HER4 somatic mutations
EGFR
Previously, erlotinib and gefitinib have been found to be particularly effective against NSCLCs with EGFR mutations, including L858R EGFR mutation.67 However, secondary resistance is common due to an alternative T790M mutation, which causes increased affinity for ATP. However, this can be overcome by irreversible TKIs like afatinib, as they are not in a competitive equilibrium with ATP.68 Two trials have demonstrated the clinical benefit of afatinib in patients with lung cancer who had progressed on erlotinib or gefitinib.69,70 The recent results from the LUX-Lung 3 trial showed superiority of afatinib with regard to response rate and progression-free survival over cisplatin and pemetrexed as first-line agents in treatment-naïve patients with advanced adenocarcinoma of the lung and EGFR mutation.71 Since then, afatinib has been approved for use in NSCLC with EGFR mutation. Since neratinib is a similar compound to afatinib as an irreversible TKI, there is an interest in assessing whether neratinib could be effective in solid tumors (including breast cancer) with EGFR mutations or amplification, and this is the basis for the Basket Study (NCT01953926, see below).
HER2
Similarly, neratinib could also be a treatment option for patients who carry a mutation in the HER2 protein. A recent publication evaluated data from eight breast cancer genome sequencing projects and identified a total of 13 somatic HER2 mutations, including seven activating mutations.72 This analysis showed that approximately 1.6% of all newly diagnosed breast cancers may harbor a HER2 mutation, and most of these patients do not have HER2 gene amplification or overexpression.72 This percentage might be higher for patients who have relapsed. Interestingly, all functionally characterized mutations were sensitive to neratinib, including those that rendered cells resistant to lapatinib.72 This provides the rationale for testing neratinib in patients with HER2 mutations, including those with breast cancer.
HER3
Different studies have shown a 2%–4% prevalence of HER3 mutations in breast cancer with evidence for occurrence in other cancer types as well.73 A majority of these somatic mutations were identified in the extracellular region. Although not by themselves, in combination with HER2, these mutations promoted oncogenic signaling and enhanced colony formation and in vivo growth. However, several HER3 or HER2 targeting agents, including trastuzumab and lapatinib, were effective in targeting these mutants,73 indicating that neratinib may also have an effect.
HER4
HER4 mutations have been found in 19% of patients with melanoma.74 A majority of these mutations are in the extracellular domain of HER4 and affect, for example, ligand binding. HER4 mutations were found to increase kinase activity and transformation ability.74 Interestingly, melanoma cells with a HER4 mutation were sensitive to treatment with lapatinib.74 As a result, lapatinib has been taken into clinical trials in melanoma, where patients with no more than two oncogenic somatic HER4 mutations will be offered lapatinib monotherapy (NCT01264081). Being an irreversible pan-HER inhibitor, neratinib could probably inhibit HER4 activation more effectively than lapatinib, although a head-to-head comparison trial has not been done. It will be interesting to assess whether neratinib is effective in tumors with HER4 mutations, including melanoma and breast cancer.
Proposed resistance mechanisms
Despite the encouraging results, experience has shown that single treatment with targeting agents frequently triggers resistance mechanisms. Therefore, work is underway to investigate potential counteracting pathways that come into action during treatment with neratinib.
One in vitro study used a pool-based lentiviral genome-wide functional RNA interference screen to discover genes which, if low or absent, would confer resistance to neratinib. Interestingly, the screen identified, among others, genes involved in oncogenesis, transcription factors, as well as genes known to interact with breast cancer-associated genes,75 highlighting the complexity of the cell machinery. One gene implicated was insulin-like growth factor binding protein 1, part of the insulin-like growth factor 1 pathway.75 Another group identified miR-630 deficiency as a cause of neratinib resistance, at least partly due to its regulation of the insulin-like growth factor 1 receptor.76 Recently, expression of the neuropeptide neuromedin U was associated with a poor prognosis in HER2-overexpressing breast cancer and has been shown to correspond with resistance to HER-targeting agents, including neratinib in vitro. It was suggested that neuromedin U acts through the chaperone heat shock protein 27 to aid HER2 stability.77 As with lapatinib,46 we observed a reactivation of HER3 and AKT phosphorylation after 24 hours of neratinib treatment in HER2-positive breast cancer cells, although we did not assess whether this was NRG1-dependent.26 In addition, our previous work showed that ADAM10 was upregulated in neratinib-treated breast cancer cells, albeit to a lesser extent compared with trastuzumab-treated cells;43 however, a thorough investigation was not carried out. Thus, it may be important to investigate whether HER ligand release mediated by ADAM10 and/or 17, a resistance mechanism found in trastuzumab,42,43 also mediates acquired resistance to neratinib.
Biomarkers
Biomarker studies in vitro have identified several potential biomarkers, including HER2, AKT, ERK, and other potential markers for response to neratinib (see HER2-positive breast cancer section above). However, there are not much data available from clinical trials as yet. So far, the I-SPY 2 trial has shown that neratinib in addition to standard neoadjuvant chemotherapy may be beneficial for patients with HER2-positive breast cancer, and in particular for those who have hormone receptor-negative disease (pCR rate 56% vs 33% for patients treated with standard chemotherapy alone).64 Patients with hormone receptor-positive/HER2-positive disease also had a higher pCR rate with the addition of neratinib (30% vs 17%).64 In the same study, patients were stratified according to their MammaPrint score (based on the median score of the I-SPY 1 trial). Patients with a higher score than the previous median MammaPrint score had a pCR of 47.5% if treated with a neratinib-containing regimen in comparison with 29.4% if treated with chemotherapy alone. Interestingly, 80.5% of these patients were negative for HER2. Nevertheless, patients with TNBC did not seem to benefit;64 however, this subtype of breast cancer is heterogeneous and other factors in addition to hormone receptor and HER2 status may be important. Further preclinical work as well as translational research using historical and prospective tumor samples from clinical trials will be needed to establish reliable biomarkers for neratinib response and resistance.
Clinical trials
Neratinib is currently being tested in various clinical trials for its safety and efficacy in various types of tumors, including NSCLC, colorectal, bladder and breast cancers. The trials that have been registered with Clinicaltrials.gov as of December 2014 are listed in Table 3. However, in this section, the main focus is on trials involving patients with breast cancer (Table 2).
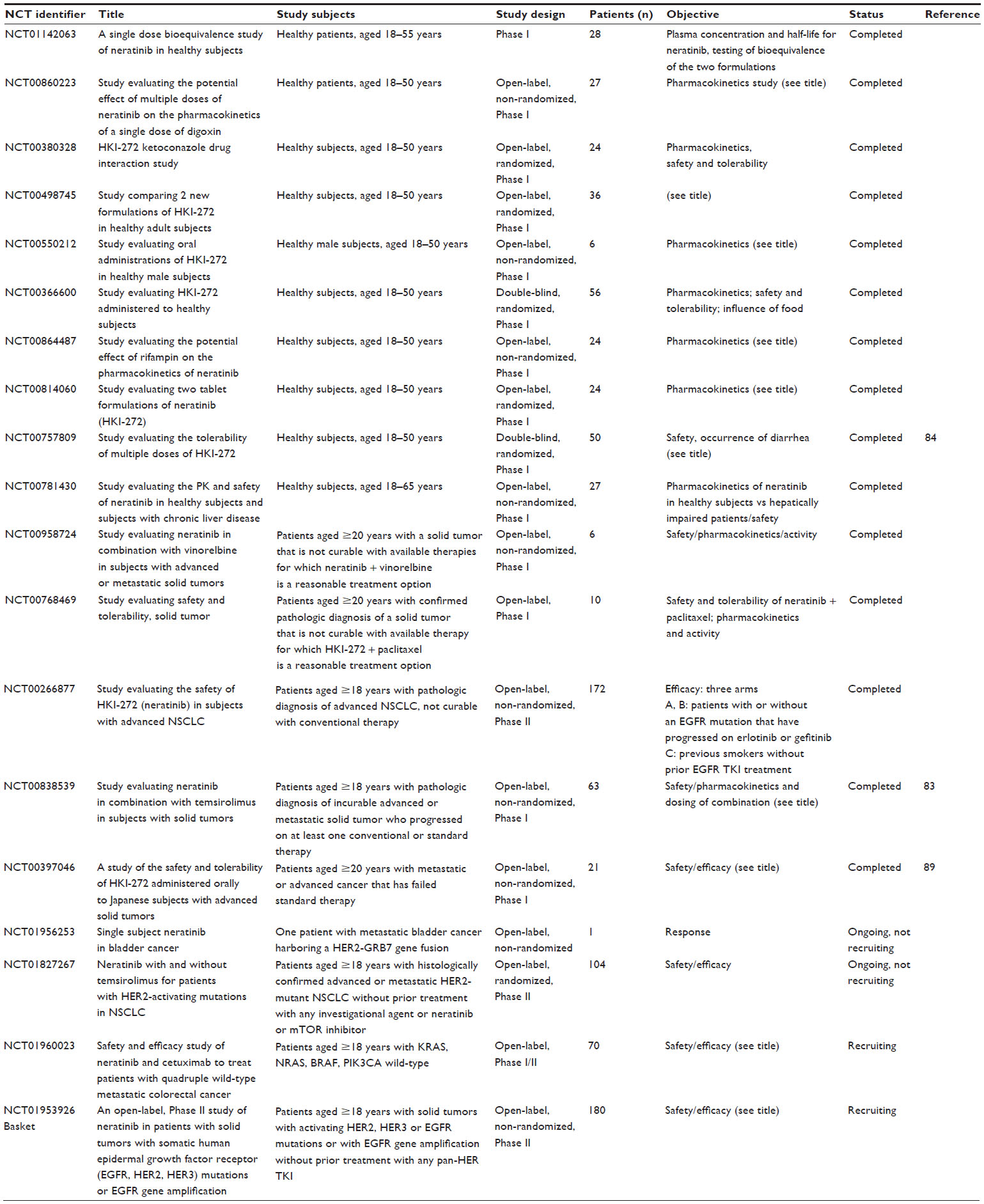 | Table 3 Clinical trials involving neratinib in other cancers |
Clinical trials in breast cancer
A Phase I dose-escalation study examined the safety and efficacy of neratinib at doses of 40–400 mg daily in patients with solid tumors. The absorption of neratinib was measured as median time to peak plasma concentration over 3–6.5 hours. The steady-state peak plasma concentration and area under the concentration–time curve for neratinib increased in a dose-dependent manner from 40 to 320 mg, but plateaued at higher doses.78 Patients who received 40, 80, or 120 mg experienced no dose-limiting toxicities. This study determined the maximum tolerated dose at 320 mg, with 240 mg being the therapeutic dose.
In this dose-escalation study, 25 patients with breast cancer were evaluated for treatment efficacy; a partial response was observed in eight patients (32%), and stable disease for ≥24 weeks was found in one patient. The majority of responders had HER2-positive disease (IHC score 3+) and all had previously received trastuzumab, anthracyclines, and taxanes.78
Neratinib monotherapy in patients with HER2-positive advanced breast cancer was associated with a 16-week progression-free survival rate of 59% with a median progression-free survival of 22.3 weeks in patients who had received prior trastuzumab treatment; and a rate of 78% with a median progression-free survival of 39.6 weeks in patients who were trastuzumab-naïve. The objective response rates were 24% in patients with prior trastuzumab treatment and 56% in the trastuzumab-naïve cohort.79 Similarly, a Phase I/II study exploring the combination of neratinib with vinorelbine in pretreated HER2-positive metastatic breast cancer showed an objective response of 8% in patients with prior lapatinib and 41% in patients who had no prior lapatinib, which was also mirrored in the respective progression-free survival rates.80
On the other hand, neratinib in combination with paclitaxel showed an overall response rate of 71% in patients who had not previously been treated with lapatinib and 77% in those with prior lapatinib.81 However, the number of patients in this trial was small. Overall, the combination was found to have increased toxicity but it achieved a better response rate relative to neratinib monotherapy. The overall response rate was 73% (95% confidence interval 62.9–81.2); seven patients (7%) had a complete response and another nine patients (9%) had stable disease for ≥24 weeks. The median progression-free survival was 57.0 weeks (95% confidence interval 47.7–81.6).81
Another study compared the combination of neratinib with weekly paclitaxel and trastuzumab in heavily pretreated patients with HER2-positive metastatic breast cancer and showed an overall clinical benefit (complete response + partial response + stable disease ≥24 weeks) in eleven patients (52%) with a median time to disease progression of 3.7 months.82 The addition of neratinib and/or trastuzumab to neoadjuvant paclitaxel followed by doxorubicin and cyclophosphamide chemotherapy is currently being tested in a Phase II randomized trial (ClinicalTrials.gov identifier NCT 01008150).82 Neratinib in combination with the mammalian target of rapamycin inhibitor temsirolimus was found to be tolerable and to be potentially beneficial in HER2-positive, trastuzumab-resistant breast cancer and HER2-mutant NSCLC in a Phase I study; however, numbers were small.83
Throughout the studies, the most common adverse event was diarrhea of any grade (up to 93%), especially during the first week of treatment; other adverse events included nausea, fatigue, vomiting, neutropenia, and anorexia.79,81 In the initial dose-escalation study, diarrhea (32%) was also the most common grade 3 or higher adverse event. In addition, it was the cause of dose reduction in 19 of 22 patients and was the adverse event that most frequently led to treatment discontinuation (14%).26 This dose-escalation study suggested a potential relationship between neratinib dose/exposure and diarrhea,78 but a follow-up study by Abbas et al revealed that there was no difference in the onset or severity of diarrhea in healthy subjects who were given neratinib either 240 mg once daily or 120 mg twice daily.84 However, several studies have reported that diarrhea could be controlled with concurrent use of antidiarrheal agents.79,81 Thus, primary prophylactic use of antidiarrheal medication is now mandatory for all patients taking neratinib in recent clinical trials, which has helped to reduce the incidence of diarrhea.
A common side effect of anti-HER2 treatments, including trastuzumab, is cardiac in nature, ie, a decrease in left ventricular ejection fraction. Neratinib was investigated in healthy subjects with regard to its effect on cardiac repolarization and was shown not to prolong the QTc interval in comparison with controls.85 However, the possible long-term cardiac toxicity from addition of neratinib to trastuzumab and/or chemotherapy will not be known until the ongoing clinical trials yield mature safety data.
Ongoing trials
Since neratinib was often more potent than lapatinib in the preclinical setting, there is interest in comparing the clinical efficacy of these two TKIs. Neratinib monotherapy has previously been compared with the combination of lapatinib and capecitabine; however, this study could determine neither inferiority nor noninferiority of neratinib.86 Overall survival was slightly better in the lapatinib + capecitabine arm, but this difference was not statistically significant.86 In addition, neratinib monotherapy may not be directly comparable with a combination of lapatinib and chemotherapy since the anti-HER2 treatment response could be enhanced by addition of concurrent chemotherapy. A subsequent study comparing neratinib + capecitabine with lapatinib + capecitabine in patients with HER2-positive metastatic breast cancer who have received two or more prior HER2-directed regimens in the metastatic setting (ClinicalTrials.gov identifier NCT01808573, NALA) is currently ongoing.
The ExteNET trial is comparing extended adjuvant neratinib vs placebo in HER2-positive patients with prior adjuvant trastuzumab. The primary endpoint is disease-free survival, which was increased by 33% in the neratinib arm (hazard ratio 0.67, P=0.0046) compared with the placebo arm. With regard to the secondary endpoint, ie, disease-free survival including for ductal carcinoma in situ, there was a 37% improvement in the neratinib arm compared with the placebo arm (hazard ratio 0.63, P=0.0009). Full results are expected soon. However, based on these results, Puma Biotechnology is planning to apply for approval of neratinib in the extended adjuvant setting in the first half of 2015.87 In view of the effect of neratinib on HER receptor somatic mutations (see above), Puma Biotechnology has started an open-label Phase II study of neratinib in patients with solid tumors carrying somatic mutations of EGFR, HER2 or HER3, or EGFR gene amplification (BASKET study).88
Conclusion
Neratinib is a potent, irreversible, pan-HER inhibitor that has shown promising activity in preclinical as well as clinical studies with an acceptable safety profile in humans. However, further elucidation is needed to determine when and in which combination this compound should be used. Although most studies showed that neratinib offers the greatest benefit to patients with HER2-positive breast cancer, some data indicate that some HER2-negative patients may also benefit. Moreover, neratinib is now being tested in many solid tumors carrying EGFR, HER2, or HER3 mutations, or EGFR amplification. It will be interesting to see where neratinib will be positioned in relation to other available HER-targeting antibodies as well as TKIs. Ongoing studies will help to better define patients likely to benefit most from neratinib-containing treatments and the associated predictive biomarkers.
Disclosure
The authors report no conflicts of interest in this work.
References
Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9(7):463–475. | |
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182. | |
Linggi B, Carpenter G. ErbB receptors: new insights on mechanisms and biology. Trends Cell Biol. 2006;16(12):649–656. | |
Klapper LN, Glathe S, Vaisman N, et al. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc Natl Acad Sci U S A. 1999;96(9):4995–5000. | |
Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125(6):1137–1149. | |
Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. | |
Dankort D, Maslikowski B, Warner N, et al. Grb2 and Shc adapter proteins play distinct roles in Neu (ErbB-2)-induced mammary tumorigenesis: implications for human breast cancer. Mol Cell Biol. 2001;21(5):1540–1551. | |
Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26(45):6469–6487. | |
Cho HS, Mason K, Ramyar KX, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421(6924):756–760. | |
Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997;16(7):1647–1655. | |
Wissner A, Mansour TS. The development of HKI-272 and related compounds for the treatment of cancer. Arch Pharm (Weinheim). 2008;341(8):465–477. | |
Britten CD. Targeting ErbB receptor signaling: a pan-ErbB approach to cancer. Mol Cancer Ther. 2004;3(10):1335–1342. | |
Solca F, Dahl G, Zoephel A, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343(2):342–350. | |
Rabindran SK, Discafani CM, Rosfjord EC, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64(11):3958–3965. | |
Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67(24):11924–11932. | |
National Center for Biotechnology Information. PubChem Compound Database; CID=208908. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/208908#section=2D-Structure. Accessed March 28, 2015. | |
National Center for Biotechnology Information. PubChem Compound Database; CID=123631. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/123631#section=2D-Structure. Accessed March 28, 2015. | |
National Center for Biotechnology Information. PubChem Compound Database; CID=176870. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/176870#section=2D-Structure. Accessed March 28, 2015. | |
National Center for Biotechnology Information. PubChem Compound Database; CID=9915743. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/9915743#section=2D-Structure. Accessed March 28, 2015. | |
National Center for Biotechnology Information. PubChem Compound Database; CID=6445562. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/6445562#section=2D-Structure. Accessed March 28, 2015. | |
National Center for Biotechnology Information. PubChem Compound Database; CID=10184653. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/10184653#section=2D-Structure. Accessed March 28, 2015. | |
Schwartz PA, Kuzmic P, Solowiej J, et al. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc Natl Acad Sci U S A. 2014;111(1):173–178. | |
Bose P, Ozer H. Neratinib: an oral, irreversible dual EGFR/HER2 inhibitor for breast and non-small cell lung cancer. Expert Opin Investig Drugs. 2009;18(11):1735–1751. | |
Tsou HR, Overbeek-Klumpers EG, Hallett WA, et al. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J Med Chem. 2005;48(4):1107–1131. | |
Wissner A, Overbeek E, Reich MF, et al. Synthesis and structure-activity relationships of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles. The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER-2). J Med Chem. 2003;46(1):49–63. | |
Canonici A, Gijsen M, Mullooly M, et al. Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget. 2013;4(10):1592–1605. | |
Nafi S, Generali D, Kramer-Marek G, et al. Nuclear HER4 mediates acquired resistance to trastuzumab and is associated with poor outcome in HER2 positive breast cancer. Oncotarget. 2014;5(15):5934–5949. | |
Seyhan AA, Varadarajan U, Choe S, et al. A genome-wide RNAi screen identifies novel targets of neratinib sensitivity leading to neratinib and paclitaxel combination drug treatments. Mol Biosyst. 2011;7(6):1974–1989. | |
O’Neill F, Madden SF, Clynes M, et al. A gene expression profile indicative of early stage HER2 targeted therapy response. Mol Cancer. 2013;12:69. | |
Cobleigh MA, Vogel CL, Tripathy D, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17(9):2639–2648. | |
Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. | |
Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353(16):1673–1684. | |
Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353(16):1659–1672. | |
Pandolfi PP. Breast cancer – loss of PTEN predicts resistance to treatment. N Engl J Med. 2004;351(22):2337–2338. | |
Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–127. | |
Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65(7):2554–2559. | |
Nahta R, Takahashi T, Ueno NT, Hung MC, Esteva FJ. P27(kip1) down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res. 2004;64(11):3981–3986. | |
Scaltriti M, Rojo F, Ocana A, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007;99(8):628–638. | |
Nagy P, Friedlander E, Tanner M, et al. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a Herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005;65(2):473–482. | |
Palyi-Krekk Z, Barok M, Isola J, Tammi M, Szollosi J, Nagy P. Hyaluronan-induced masking of ErbB2 and CD44-enhanced trastuzumab internalisation in trastuzumab resistant breast cancer. Eur J Cancer. 2007;43(16):2423–2433. | |
Shattuck DL, Miller JK, Carraway KL 3rd, Sweeney C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008;68(5):1471–1477. | |
Gijsen M, King P, Perera T, et al. HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 Herceptin in breast cancer. PloS Biol. 2010;8(12):e1000563. | |
Feldinger K, Generali D, Kramer-Marek G, et al. ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget. 2014;5(16):6633–6646. | |
Xia W, Petricoin EF 3rd, Zhao S, et al. An heregulin-EGFR-HER3 autocrine signaling axis can mediate acquired lapatinib resistance in HER2+ breast cancer models. Breast Cancer Res. 2013;15(5):R85. | |
Leung WY, Roxanis I, Sheldon H, et al. Combining lapatinib and pertuzumab to overcome lapatinib resistance due to NRG1-mediated signaling in HER2 amplified breast cancer. Oncotarget. January 21, 2015. [Epub ahead of print.] | |
Sanchez-Martin M, Pandiella A. Differential action of small molecule HER kinase inhibitors on receptor heterodimerization: therapeutic implications. Int J Cancer. 2012;131(1):244–252. | |
Mallon R, Feldberg LR, Lucas J, et al. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res. 2011;17(10):3193–3203. | |
Pierce A, McGowan PM, Cotter M, et al. Comparative antiproliferative effects of iniparib and olaparib on a panel of triple-negative and non-triple-negative breast cancer cell lines. Cancer Biol Ther. 2013;14(6):537–545. | |
Modi S, Stopeck A, Linden H, et al. HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res. 2011;17(15):5132–5139. | |
Mehta AI, Brufsky AM, Sampson JH. Therapeutic approaches for HER2-positive brain metastases: circumventing the blood-brain barrier. Cancer Treat Rev. 2013;39(3):261–269. | |
Polli JW, Humphreys JE, Harmon KA, et al. The role of efflux and uptake transporters in [N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino }methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos. 2008;36(4):695–701. | |
Zhao XQ, Xie JD, Chen XG, et al. Neratinib reverses ATP-binding cassette B1-mediated chemotherapeutic drug resistance in vitro, in vivo, and ex vivo. Mol Pharmacol. 2012;82(1):47–58. | |
Hegedus C, Truta-Feles K, Antalffy G, et al. Interaction of the EGFR inhibitors gefitinib, vandetanib, pelitinib and neratinib with the ABCG2 multidrug transporter: implications for the emergence and reversal of cancer drug resistance. Biochem Pharmacol. 2012;84(3):260–267. | |
Freedman RA Gelman RS, Wefel JS, et al. TBCRC 022: Phase II trial of neratinib for patients (Pts) with human epidermal growth factor receptor 2 (HER2+) breast cancer and brain metastases (BCBM). J Clin Oncol. 2014;32(5 Suppl):Abstr 528. | |
Birner P, Oberhuber G, Stani J, et al. Evaluation of the United States Food and Drug Administration-approved scoring and test system of HER-2 protein expression in breast cancer. Clin Cancer Res. 2001;7(6):1669–1675. | |
Wolff AC, Hammond ME, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med. 2007;131(1):18–43. | |
Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31(31):3997–4013. | |
Allison M. The HER2 testing conundrum. Nat Biotechnol. 2010;28(2):117–119. | |
Paik S, Kim C, Wolmark N. HER2 status and benefit from adjuvant trastuzumab in breast cancer. N Engl J Med. 2008;358(13):1409–1411. | |
Robidoux A, Tang G, Rastogi P, et al. Evaluation of lapatinib as a component of neoadjuvant therapy for HER2+operable breast cancer: NSABP protocol B-41. J Clin Oncol. 2012;30 Suppl 18:Abstr LBA506. | |
Puma Biotechnology Inc. Puma Biotechnology announces presentation of positive PB272 Phase II data from I-SPY 2 trial: neratinib graduates from I-SPY 2 trial. 2014. Available from: http://www.pumabiotechnology.com/pr20140407.html. Accessed March 28, 2015. | |
Corkery B, Crown J, Clynes M, O’Donovan N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann Oncol. 2009;20(5):862–867. | |
McGowan PM, Mullooly M, Caiazza F, et al. ADAM-17: a novel therapeutic target for triple negative breast cancer. Ann Oncol. 2013;24(2):362–369. | |
Carlson RH. I-SPY 2 trial: neoadjuvant neratinib improves pathologic complete response in HR-/HER2+ breast cancer. Oncology Times. 2014;36(10):25–26. | |
Carey LA, Rugo HS, Marcom PK, et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. 2012;30(21):2615–2623. | |
Baselga J, Gomez P, Greil R, et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2013;31(20):2586–2592. | |
Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–13311. | |
Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105(6):2070–2075. | |
Katakami N, Atagi S, Goto K, et al. LUX-Lung 4: a phase II trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J Clin Oncol. 2013;31(27):3335–3341. | |
Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13(5):528–538. | |
Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334. | |
Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3(2):224–237. | |
Jaiswal BS, Kljavin NM, Stawiski EW, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013;23(5):603–617. | |
Prickett TD, Agrawal NS, Wei X, et al. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet. 2009;41(10):1127–1132. | |
Seyhan AA, Varadarajan U, Choe S, Liu W, Ryan TE. A genome-wide RNAi screen identifies novel targets of neratinib resistance leading to identification of potential drug resistant genetic markers. Mol Biosyst. 2012;8(5):1553–1570. | |
Corcoran C, Rani S, Breslin S, et al. miR-630 targets IGF1R to regulate response to HER-targeting drugs and overall cancer cell progression in HER2 over-expressing breast cancer. Mol Cancer. 2014;13:71. | |
Rani S, Corcoran C, Shiels L, et al. Neuromedin U: a candidate biomarker and therapeutic target to predict and overcome resistance to HER-tyrosine kinase inhibitors. Cancer Res. 2014;74(14):3821–3833. | |
Wong KK, Fracasso PM, Bukowski RM, et al. A phase I study with neratinib (HKI-272), an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin Cancer Res. 2009;15(7):2552–2558. | |
Burstein HJ, Sun Y, Dirix LY, et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol. 2010;28(8):1301–1307. | |
Awada A, Dirix L, Manso Sanchez L, et al. Safety and efficacy of neratinib (HKI-272) plus vinorelbine in the treatment of patients with ErbB2-positive metastatic breast cancer pretreated with anti-HER2 therapy. Ann Oncol. 2013;24(1):109–116. | |
Chow LW, Xu B, Gupta S, et al. Combination neratinib (HKI-272) and paclitaxel therapy in patients with HER2-positive metastatic breast cancer. Br J Cancer. 2013;108(10):1985–1993. | |
Jankowitz RC, Abraham J, Tan AR, et al. Safety and efficacy of neratinib in combination with weekly paclitaxel and trastuzumab in women with metastatic HER2-positive breast cancer: an NSABP Foundation Research Program phase I study. Cancer Chemother Pharmacol. 2013;72(6):1205–1212. | |
Gandhi L, Bahleda R, Tolaney SM, et al. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J Clin Oncol. 2014;32(2):68–75. | |
Abbas R, Hug BA, Leister C, Sonnichsen D. A double-blind, randomized, multiple-dose, parallel-group study to characterize the occurrence of diarrhea following two different dosing regimens of neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor. Cancer Chemother Pharmacol. 2012;70(1):191–199. | |
Hug B, Abbas R, Leister C, Burns J, Sonnichsen D. A single-dose, crossover, placebo- and moxifloxacin-controlled study to assess the effects of neratinib (HKI-272) on cardiac repolarization in healthy adult subjects. Clin Cancer Res. 2010;16(15):4016–4023. | |
Martin M, Bonneterre J, Geyer CE Jr, et al. A phase two randomised trial of neratinib monotherapy versus lapatinib plus capecitabine combination therapy in patients with HER2+ advanced breast cancer. Eur J Cancer. 2013;49(18):3763–3772. | |
Puma Biotechnology Inc. Puma Biotechnology announces positive top line results from phase III PB272 trial in adjuvant breast cancer (ExteNET Trial). 2014. Available from: http://www.pumabiotechnology.com/pr2014072202.html. Accessed March 28, 2015. | |
Puma Biotechnology Inc. Puma Biotechnology expands cohort in phase II trial of PB272 (neratinib) in HER2 mutation-positive solid tumor patients. 2014. Available from: http://investor.pumabiotechnology.com/press-release/puma-biotechnology-expands-cohort-phase-ii-trial-pb272-neratinib-her2-mutation-positiv. Accessed March 28, 2015. | |
Ito Y, Suenaga M, Hatake K, et al. Safety, efficacy and pharmacokinetics of neratinib (HKI-272) in Japanese patients with advanced solid tumors: a phase 1 dose-escalation study. Jpn J Clin Oncol. 2012;42(4):278–286. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.