")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Profile of epratuzumab and its potential in the treatment of systemic lupus erythematosus
Authors Al Rayes H, Touma Z
Received 9 September 2014
Accepted for publication 22 October 2014
Published 17 November 2014 Volume 2014:8 Pages 2303—2310
DOI https://doi.org/10.2147/DDDT.S49778
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Hanan Al Rayes,1 Zahi Touma2
1Department of Medicine, Division of Rheumatology, Prince Sultan Military Medical City, Riyadh, Saudi Arabia; 2University of Toronto Lupus Clinic, Toronto Western Hospital, Centre for Prognosis Studies in the Rheumatic Diseases, Toronto, ON, Canada
Abstract: Management of systemic lupus erythematosus (SLE) represents a fascinating, emerging field. Research has recently provided us with a better understanding of the immunologic alterations of SLE, leading to the creation of immunomodulatory agents designed to disrupt specific cell targets and pro-inflammatory pathways. Despite the improvement in the prognosis of SLE in the last 50 years with the use of immunosuppressive therapy such as cyclophosphamide and mycophenolate mofetil, cytotoxicity remains a major complication of these medications and the need for more specific targeted immunotherapy is increasing. Early recognition and treatment of SLE with targeted immunotherapy has the potential to improve quality of life and reduce the risk of disease flare-ups and complications. In this review, we will explore the role of B-cells in the pathogenesis of SLE highlighting current insights into SLE development and management. In addition, we will discuss epratuzumab’s role in the treatment of SLE. Epratuzumab is a humanized anti-CD22 monoclonal antibody that targets CD22 on B-cell and its role in B-cell modulation, migration, function, and inhibition of B-cell receptor signaling. Epratuzumab is currently in a Phase III study evaluating its efficacy in the management of moderate to severe SLE. All published trials on epratuzumab have shown great promise with safe profiles.
Keywords: epratuzumab, SLE, lupus, anti-CD22, monoclonal antibody
Introduction
Systemic lupus erythematosus (SLE) is a chronic complex autoimmune disease with variable clinical presentations and disease courses that can be mild, moderate, or life-threatening depending on the severity of the organs involved.1,2 Patients with SLE have hyper-activated B-cells resulting in the production of autoantibodies that contribute to different clinical phenotypes.3–5 These autoantibodies contribute to organ involvement by various mechanisms such as: immune complex-mediated type III hypersensitivity reactions, type II antibody-dependent cytotoxicity, and production of interferon-α, tumor necrosis factor, and interleukin-1.6,7 New insights into SLE pathogenesis have enhanced the development of biological therapies that specifically target key molecules and cells.7,8 Recent therapies have focused on targeting different B-cell compartments.9 These include agents that deplete B-cells like anti-CD20 antibodies (rituximab and ocrelizumab), agents that modulate B-cell activity (anti-CD22, CD40 ligand inhibitors), and agents that affect the development of B-cells via B-lymphocyte stimulator/B-cell-activating factor of the tumor necrosis factor family (BAFF) or proliferation inducing ligand (APRIL) pathways.10–13 In terms of B-cell compartment targeted therapy, clinical experience and reports based on a small series of patients who received anti-CD20 (rituximab) have demonstrated impressive results but unfortunately have failed to achieve the primary outcome in large controlled trials. Several factors could have resulted in the failure of rituximab trials including: the trials’ design, in which enrolled patients received highly efficacious standard of care (SOC) treatment that made the interpretation of the results difficult; underpowered trials with a small sample size; as well as stringent endpoints that are hard to achieve.10,11 Epratuzumab (Anti-CD22 monoclonal antibody) was investigated in moderate to severe SLE with promising results. The results of a Phase III trial, Epratuzumab Versus Placebo in Subjects with Moderate to Severe General Systemic Lupus Erythematosus (EMBODY 1), are still pending. In this review, we will explore the role of B-cell and CD22 in the pathogenesis of SLE and we will summarize the published epratuzumab clinical trials.
Pathogenic role of B-cells and CD22 in SLE
The role of B-cells in the pathogenesis of lupus is very important and involves antibody-dependent and -independent mechanisms. The autoantibody-independent mechanism is characterized by antigen presentation, T-cell activation and polarization, and dendritic cell modulation.6,14 B-cells interact with antigens through B-cell antigen receptors (BCRs).9 BCR of auto reactive B-cells can be activated by unclear nuclear material, leading to B-cell activation and expression of the B-cell survival molecule receptor, BAFF and APRIL.15
There are co-receptors expressed on B-cell surfaces that modulate BCR signaling either positively or negatively.16 CD22, CD72, and Ig (FcRγIIB) are called inhibitory BCR co-receptors which prevent over stimulation of B-cells.7 The inhibitory BCR co-receptors prevent BCR activation signaling cascades through the recruitment of inhibitory intracellular signaling proteins.17–19 Lyn is a novel member of the Src family tyrosine kinase which plays a key role in B-cell activation (and is able to activate some negative regulators of signaling such as CD22).11,18 On the basis of proposed mechanisms outlined above, targeting B-cell membrane antigen receptors such as CD20, CD22, and other receptors was of interest.
CD22 is a 135 kDa sialo-glycoprotein receptor and a B-lymphocyte-restricted member of the immunoglobulin superfamily. CD22 is involved in BCR inactivation, control of B-cell activation and interaction with T-cells, and produces a costimulatory signal in primary B-cells.20,21 CD22 is expressed in pro-B-cells, pre-B-cells, and mature B-cells while absent in plasma cells.15 CD22 is essential for the development and survival of B-cells.22,23 Elevated expression of CD22 and other BCR associated proteins on B-lymphocytes has been associated with SLE, chronic autoimmune disease, and certain cancers. Current therapies for SLE seek to minimize CD22 and other BCR-protein expression by destroying B-cells.
Targeting B-cells with epratuzumab
Epratuzumab is an anti-CD22 (recombinant) “humanized” IgG1 monoclonal antibody (hLL2), and is 95% of human origin with reduced immunogenicity.24,25 Epratuzumab has a mean serum half-life of 23.9 days, which is comparable to the half-life of human IgG1 and the highest serum levels increased with subsequent doses. Epratuzumab is able to reduce CD22 with a minimized B-cell destruction effect and a minimized impact on the immune system.26 This justifies the partial depletion of B-cell numbers with epratuzumab as compared to total reduction with rituximab.26 Indeed, epratuzumab eliminates up to 45% of circulating B-cells while rituximab eliminates >90% of B-cells.27,28
Rossi et al showed that the mechanism of action of epratuzumab on B-cells is twofold; one via binding to CD22, which also occurs with F(ab)2, and the other via engagement of FcγR-bearing effector cells.28 Epratuzumab also induces a marked decrease of CD22 (>80%), CD19 (>50%), CD21 (>50%), and CD79b (>30%), on B-cells’ surface in peripheral blood mononuclear cells obtained from normal control or SLE patients.28 The other mechanism of action of epratuzumab is trogocytosis which is Fc dependent and causes the transfer of epratuzumab-opsonized B-cells to FcγR-expressing monocytes, natural killer cells, and granulocytes. Epratuzumab also induces moderate antibody-dependent cellular cytotoxicity, without complement-dependent cytotoxicity and this can explain the absence of infusion related reactions in humans.23,24,29,30 The pronounced and persistent loss of CD22 on B-cells by epratuzumab-mediated internalization and trogocytosis is expected to render B-cells less active and less viable, and the accompanied decrease of CD19 could further enhance this effect.28
Unlike rituximab which acts only as cytotoxic, epratuzumab acts as an immunomodulatory and cytotoxic agent.31 In the EMBLEM and other trials related to epratuzumab, no decreases in immunoglobulin levels were observed, thus it speculated that epratuzumab will have a lower risk of infection compared to rituximab.
Beum et al found a substantial loss of CD20 on B-cells of chronic lymphocytic leukemia patients when rituximab plasma concentrations were high, which is related to a phenomenon called antigenic modulation.32 In this phenomenon, the removal of rituximab-CD20 complexes is mediated by trogocytosis to monocytes, enabling the malignant cells to escape the effects of the antibody.32 Williams et al showed that reducing the dose of rituximab decreases CD20 loss, by limiting trogocytosis, and this resulted in an improvement of the therapeutic effects of rituximab.33 Rossi et al reported that a similar process of antigen modulation via trogocytosis induced by anti-CD22 or anti-CD20 antibodies can be encountered and affect their therapeutic efficacy.28 This could also explain the findings in SLE clinical trials where higher doses of epratuzumab did not show an improvement compared to the lower doses.28,34
Evidence of clinical efficacy of epratuzumab in SLE
Epratuzumab was initially developed to treat non-Hodgkin’s lymphoma and leukemia. It was also tried for the treatment of Sjögren’s disease and SLE.35–38 Nearly all of the published studies on epratuzumab as an additive to the SOC treatment in moderate to severe SLE patients showed improvement in the disease activity after the first cycle of therapy. The benefits were persistent in those who were maintained on regular epratuzumab every 12 weeks as in SL0006 trial.24
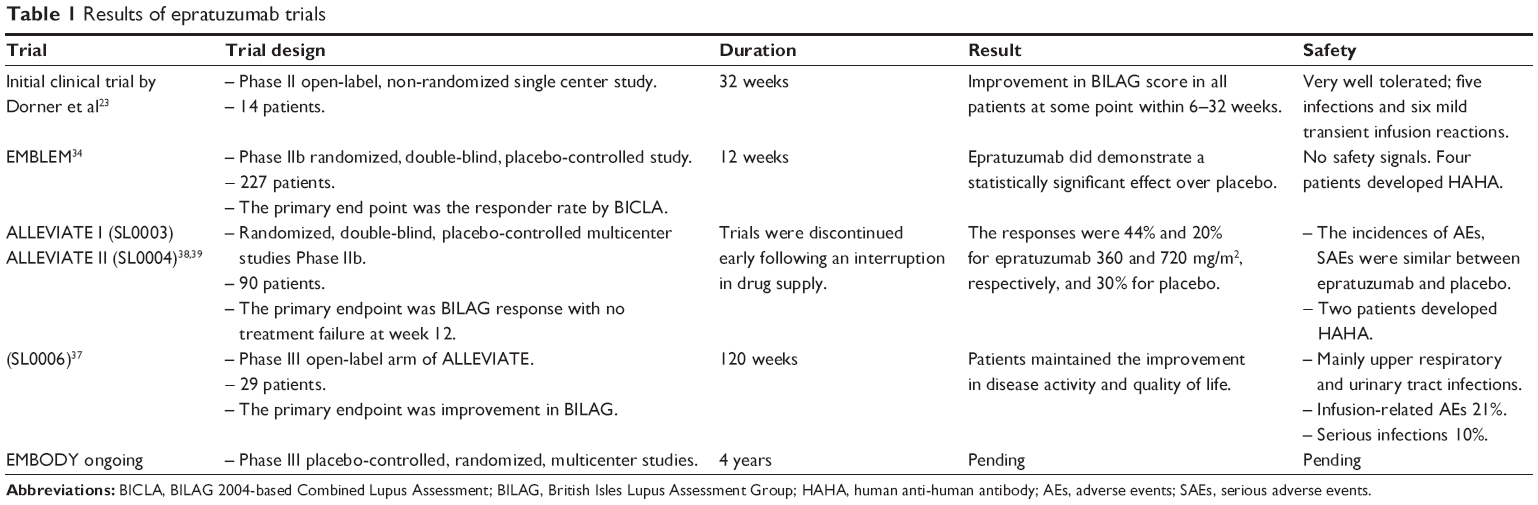
The first trial in SLE was by Dorner et al in 2006.23 It was a Phase II open labeled single center study. All patients received 360 mg/m2 epratuzumab intravenously every 2 weeks for a total of four doses. A total of 14 patients with moderate disease activity were enrolled in this study. Disease activity and the total British Isles Lupus Assessment Group (BILAG) index 2004 scores were determined at 6, 10, 18, and 32 weeks. In all patients, a clinically important improvement was achieved with a decrease in BILAG scores by ≥50% at some point during the study. At 6 months, 77% of the patients had a ≥50% decrease in BILAG scores. In three patients with multiple BILAG B organ involvement at baseline, a complete resolution in all B-level disease activities by 18 weeks was noticed. This clinical improvement was associated with a decrease in B-cell levels by 35% at 18 weeks and remained low at 6 months post-treatment. There were no safety signals and no evidence of immunogenicity or significant changes in T-cells, immunoglobulins, or autoantibody levels.23
EMBLEM (NCT00624351)
This is a Phase IIb, 12 week, multicenter, randomized, controlled study that was published in 2013 by Wallace et al. The main objective of this trial was to identify appropriate safe and effective epratuzumab dosing regimens in patients with moderate to severe SLE disease activity.34 The primary outcome measure in this study was the BILAG 2004-based Combined Lupus Assessment (BICLA). This is a composite index with five components: 1) BILAG-2004 improvement (all A scores at baseline improved to B/C/D, and all B scores improved to C or D); 2) no worsening in disease activity (no new BILAG-2004 A scores and ≤1 new B score); 3) no worsening of total SLEDAI-2000 (Systemic Lupus Erythematosus Disease Activity Index-2K) score from baseline; 4) no significant deterioration (<10% worsening) in 100 mm visual analog physician global assessment; and 5) no treatment failure (defined as non-protocol treatment, ie, new or increased immunosuppressives or antimalarials; or increased or parenteral corticosteroids; or premature discontinuation from study treatment).34 The BICLA response required the achievement of all five components.
Two hundred and twenty-seven patients were randomly assigned to one of six treatment groups: placebo or epratuzumab 200 mg cumulative dose (cd) (100 mg every other week [EOW]), 800 mg cd (400 mg EOW), 2,400 mg cd (600 mg weekly), 2,400 mg cd (1,200 mg EOW), or 3,600 mg cd (1,800 mg EOW). Although the percentage of responders was greater in all epratuzumab groups compared to placebo, this was not statistically significant. In the exploratory analysis, the patients who received 600 mg weekly (2,400 mg cd) have the higher percentage of responders and this was statistically significant with an odds ratio of 3.2. In the groups of patients with <2,400 mg cd or 3,600 mg cd, the percentage of responders was lower than placebo. Starting from week 8, differences in BICLA responders were noticed. In this trial, epratuzumab was safe and well tolerated with similar rates of adverse events.
Human anti-human antibody was found in four patients. Both ALLEVIATE and EMBLEM Phase IIb showed a low response rate in patients receiving a higher dose of epratuzumab (3,600 mg cd). Wallace et al explained that the high doses of epratuzumab may affect a specific function of B-cells or it can induce alternative signaling events not seen at lower doses.34 The results from EMBODY 1 are very crucial to clarify this phenomenon and are necessary before one can draw solid conclusions.
ALLEVIATE-1 (SL0003) and ALLEVIATE-2 (SL0004) – Phase IIb
ALLEVIATE-1 and ALLEVIATE-2 were terminated early in September 2006 due to the interruption of drug supply. Both studies were randomized, double-blinded, placebo-controlled, and multicenter studies. SL0006 was an open-label extension study of patients enrolled in ALLEVIATE.37,38
Ninety patients were randomized to 36 patients (severe BILAG A) in ALLEVIATE I and 54 patients (moderate BILAG B) in ALLEVIATE II.38,39 In ALLEVIATE I, patients were given either SOC treatment plus repeated administrations of epratuzumab 360 mg/m2 (14 patients) or 720 mg/m2 (eleven patients) or individualized SOC treatment plus placebo (eleven patients). Twenty-eight patients in ALLEVIATE II were given SOC treatment plus repeated administrations of epratuzumab 360 mg/m2, and 26 patients were given SOC treatment plus placebo. In ALLEVIATE, patients had severe lupus with 43% having at least one BILAG A. The tapering goals in ALLEVIATE-1 and ALLEVIATE-2 were 7.5–10 mg/day and 5–7.5 mg/day prednisone (or equivalent) by weeks 20 and 24.
The primary endpoint was BILAG response at week 24 where all BILAG A scores were reduced to B/C/D and B scores to C/D, and no new A and <2 new B scores. The primary endpoint response was subsequently redefined for 12 weeks due to the premature discontinuation of drug supply and termination of ALLEVIATE. The exploratory pooled analyses of both studies found that responses at week 12 were 15/34 (44%) and 2/10 (20%) for epratuzumab 360 and 720 mg/m2, respectively, versus 9/30 (30%) for placebo. Total BILAG scores were lower in both epratuzumab groups versus placebo (Table 1).
 | Table 1 Results of epratuzumab trials |
The incidence of adverse events was similar between groups with no major side effects. Responders had an improvement in the health related quality of life as determined by SF-36.37,38 In these studies, epratuzumab was well tolerated with no safety signals. The frequency of adverse and severe adverse events was comparable between epratuzumab and placebo arms.
An open-label extension study (SL0006)
Twenty-nine patients from the ALLEVIATE trials continued participating in the SL0006 trial. It is important to note that there was a delay between completion of the ALLEVIATE studies and entry into SL0006 study of a median of 165 days (range 1–400 days) which was secondary to the interruption of the drug supply. Patients received 12 week cycles of 360 mg/m2 epratuzumab over 100 weeks. Patients in SL0006 maintained their improvement in disease activity as determined by SLEDAI/BILAG as well as their improvement in SF-36 scores.37,38,40
Although the ALLEVIATE studies were discontinued early due to the interruption in drug supply, the analysis from the available data showed a potential role for epratuzumab in treating SLE. This in turn has led to a Phase III study, EMBODY 1, which is currently ongoing and the results are expected to be released by early 2015. In ALLEVIATE trials and the extension study SL0006, epratuzumab treatment has led to a clinically important and sustained improvement in physicians’ and patients’ global assessment. There was also a sustained improvement in health related quality of life as determined by SF-36 scores as well as a reduction in corticosteroid doses.37
Phase III study (EMBODY 1) (NCT01261793; NCT01262365)
These are Phase III placebo-controlled, randomized, multicenter studies to assess the efficacy and safety of epratuzumab in patients with moderate to severe SLE over four treatment cycles, each 12 weeks in duration (48 weeks total). The results of these studies are still pending.
Discussion
We have witnessed an advance in the management of SLE in the last 5 decades which has led to an improvement in patients’ survival.41 A better understanding of lupus pathogenesis has facilitated the development of new drugs for lupus but the conduction and the results of clinical trials have been challenging.8 Following the successful story of belimumab, there is hope for the future.42 The lupus pipeline has several promising drug candidates currently in development by different companies.14 The current SOC therapy for lupus patients with the use of corticosteroids and immunosuppressive drugs is associated with an increased risk of infections, hepatic and bone marrow toxicity, and other complications. In addition SOC therapy has proven ineffective for certain patients, thus, there is an unmet need for new drugs in lupus.
Several biological drugs have been studied for the management of active SLE aiming to have safer immunosuppression especially with regards to cytotoxicity and serious infections. The results of open-label studies of rituximab which is a chimeric anti-CD20 monoclonal antibody have been promising but their effect was not proven in randomized controlled trials.10,11,43,44 Rituximab failed to achieve the primary end points in large randomized clinical trials (The Exploratory Phase II/III SLE Evaluation of Rituximab [EXPLORER] trial and the Lupus Nephritis Assessment with Rituximab study [LUNAR] trials) on the efficacy in treatment of moderate to severe SLE.11 Nevertheless, the results from observational studies are encouraging and rituximab has been efficient in the treatment of proliferative and membranous lupus nephritis.45–47 The discrepancy between the results of clinical trials and observational studies regarding the efficacy of rituximab suggests that it should not be disregarded in the management of lupus.48
Several lessons have been learned from previous trials and it is well accepted that the success and failure of a clinical trial depends on several factors in addition to the efficacy of the studied drug. The study design and the choice of outcomes and endpoints are crucial and affect the results of the trials.49,50 The measurement of disease activity along with other domains, as recommended by the Outcome Measures in Rheumatology, continues to be challenging.50 Several lupus disease activity measures have been developed and validated and of these, two are commonly adopted in clinical trials, SLEDAI-2K – a global disease activity index, and the BILAG 2004 – an organ-specific index, along with the physician global assessment.49,51 SLEDAI was developed in 1985 and published in 1992, and BILAG was published in 1988.52,53 Both indices have their advantages and disadvantages. BILAG is a more comprehensive index, contains 97 items, and captures disease activity over the last 4 weeks. It measures improvement, worsening, resolution, persistence, and new occurrences of manifestations (not present, improving, same, worse, and new). However, BILAG is difficult to use because of its complicated glossary and scoring systems.54 SLEDAI-2K contains 24 descriptors and captures disease activity over 4 weeks and records the clinical manifestations as present or absent. SLEDAI-2K use is easy to administer and the scoring is intuitive which makes it a more viable candidate index for use in everyday practice.55,56 Nevertheless, in SLEDAI-2K, to demonstrate improvement, a manifestation has to resolve completely. Thus, to be able to measure a partial improvement in SLEDAI-2K descriptors, it is recommended to use SLEDAI-2K Responder Index-50.57,58 SLEDAI-2K Responder Index-50 is able to capture ≥50% improvement in each descriptor and it is currently being used in clinical trials for new drugs in lupus.59
Currently, in clinical trials there is a trend to use composite indices. Both composite indices, SLE Responder Index (SRI) and BICLA incorporate the SELENA-SLEDAI (derivation of SLEDAI), BILAG, and physician global assessment. However, in SRI, SELENA-SLEDAI is used as the key component and in the BICLA, the key component is the BILAG.34,60 SRI and BICLA performances were compared to physician-rated improvement (derived based on charts’ review) in the Oklahoma cohort study retrospectively. This study showed that the BICLA may be less sensitive than SRI in capturing improvement. This resulted from the fact that “BICLA improvement requires that all A scores at baseline improved to B/C/D, and all B scores improved to C or D, which might be more difficult to achieve in patients with multiple organ involvement at baseline”, as interpreted by the authors.61 In a post hoc analysis, the EMBLEM data were used to compare BICLA versus SRI. The SRI response rate was higher than the BICLA response rate in the placebo arm and in the epratuzumab arm. Authors found that the disagreement in BICLA and SRI response rates was attributed to the discrepancies between the individual scoring of SLEDAI and BILAG items, thus it is very difficult to draw conclusions from this post hoc analysis.62 A similar analysis was conducted on the preliminary data of the Biomarkers of Lupus Disease (BOLD) study, a study of 100 patients with SLE on immunosuppressive therapy. The performance of different outcome measures in detecting improvement was determined. The analysis at 4 weeks showed that the BICLA-like end point was superior to SRI in detecting improvement (SRI-4: 48%, SRI-5: 26%, BICLA-similar: 68%) and at 8 weeks (SRI-4: 67%, SRI-5: 39% and BICLA-similar: 43%).63 Although the analyses from the above three studies did not agree on the performance of BICLA and SRI response rates, one can assume that BICLA response rates are superior to SRI response rates. This can be attributed to the fact that it is easier to capture partial improvement with BILAG while it is not possible with SRI where an improvement is based on a complete resolution of the manifestation. In conclusion, the potential variability in the application of SLEDAI, BILAG, and other indices by physicians in multicenter trials, requires the preparedness of investigators on the use of specific measures, and highlights the role of the centralized adjudication committees.8,64
Epratuzumab improved disease activity measures in the trials included in this review and was found to have a low safety profile. The EMBLEM trial provided us with the most effective safe dose of epratuzumab which is a 2,400 mg cd/cycle (600 mg weekly or 1,200 mg EOW). The ALLEVIATE trial analysis showed clinical meaning improvement in BILAG scores as well as improvement in the health related quality of life and physician global assessments of disease activity with statistically insignificant reduction in corticosteroid doses over 12–47 weeks. Nevertheless, some of these results were based on pooled analyses of interrupted randomized controlled trials and a small number of patients. Thus, further results of the ongoing EMBODY 1 study might give us better answers on the efficacy of epratuzumab in the near future.
Acknowledgments
Dr Zahi Touma is supported by the Great-West Life, London Life, and Canada Life Fellowship. The University of Toronto Lupus Clinic is supported by the University Health Network.
Disclosure
Dr Z Touma has consulted for GSK, Serono, and Janssen. The authors report no other conflicts of interest.
References
Bootsma H, Spronk P, Derksen R, et al. Prevention of relapses in systemic lupus erythematosus. Lancet. 1995;345(8965):1595–1599. | ||
Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–2121. | ||
Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358(9):929–939. | ||
Gatto M, Zen M, Ghirardello A, et al. Emerging and critical issues in the pathogenesis of lupus. Autoimmun Rev. 2013;12(4):523–536. | ||
Touma Z, Gladman DD, Tulloch-Reid D, et al. Burden of autoantibodies and association with disease activity and damage in systemic lupus erythematosus. Clin Exp Rheumatol. 2010;28(4):525–531. | ||
Chan O, Shlomchik MJ. A new role for B-cells in systemic autoimmunity: B-cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J Immunol. 1998;160(1):51–59. | ||
Harris DP, Haynes L, Sayles PC, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol. 2000;1(6):475–482. | ||
Touma Z, Urowitz MB, Gladman DD. Systemic lupus erythematosus: an update on current pharmacotherapy and future directions. Expert Opin Biol Ther. 2013;13(5):723–737. | ||
Ding HJ, Gordon C. New biologic therapy for systemic lupus erythematosus. Curr Opin Pharmacol. 2013;13(3):405–512. | ||
Rovin BH, Furie R, Latinis K, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012;64(4):1215–1226. | ||
Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62(1):222–233. | ||
Mysler EF, Spindler AJ, Guzman R, et al. Efficacy and safety of ocrelizumab in active proliferative lupus nephritis: results from a randomized, double-blind, phase III study. Arthritis Rheum. 2013;65(9):2368–2379. | ||
Mok MY. The immunological basis of B-cell therapy in systemic lupus erythematosus. Int J Rheum Dis. 2010;13(1):3–11. | ||
Traczewski P, Rudnicka L. Treatment of systemic lupus erythematosus with epratuzumab. Br J Clin Pharmacol. 2011;71(2):175–182. | ||
Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B-cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416(6881):603–607. | ||
Fattah Z, Isenberg DA. Recent developments in the treatment of patients with systemic lupus erythematosus: focusing on biologic therapies. Expert Opin Biol Ther. 2014;14(3):311–326. | ||
Nitschke L. The role of CD22 and other inhibitory co-receptors in B-cell activation. Curr Opin Immunol. 2005;17(3):290–297. | ||
Looney RJ, Anolik J, Sanz I. A perspective on B-cell-targeting therapy for SLE. Mod Rheumatol. 2010;20(1):1–10. | ||
Dorner T, Jacobi AM, Lipsky PE. B-cells in autoimmunity. Arthritis Res Ther. 2009;11(5):247. | ||
Ding C, Foote S, Jones G. B-cell-targeted therapy for systemic lupus erythematosus: an update. BioDrugs. 2008;22(4):239–249. | ||
Tedder TF, Tuscano J, Sato S, Kehrl JH. CD22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu Rev Immunol. 1997;15:481–504. | ||
Otipoby KL, Andersson KB, Draves KE, et al. CD22 regulates thymus-independent responses and the lifespan of B-cells. Nature. 1996;384(6610):634–637. | ||
Dorner T, Kaufmann J, Wegener WA, Teoh N, Goldenberg DM, Burmester GR. Initial clinical trial of epratuzumab (humanized anti-CD22 antibody) for immunotherapy of systemic lupus erythematosus. Arthritis Res Ther. 2006;8(3):R74. | ||
Wallace DJ, Goldenberg DM. Epratuzumab for systemic lupus erythematosus. Lupus. 2013;22(4):400–405. | ||
Goldenberg DM. Epratuzumab in the therapy of oncological and immunological diseases. Expert Rev Anticancer Ther. 2006;6(10):1341–1353. | ||
Dorner T, Goldenberg DM. Targeting CD22 as a strategy for treating systemic autoimmune diseases. Ther Clin Risk Manag. 2007;3(5):953–959. | ||
Edwards JC, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol. 2006;6(5):394–403. | ||
Rossi EA, Goldenberg DM, Michel R, Rossi DL, Wallace DJ, Chang CH. Trogocytosis of multiple B-cell surface markers by CD22 targeting with epratuzumab. Blood. 2013;122(17):3020–3029. | ||
Jacobi AM, Goldenberg DM, Hiepe F, Radbruch A, Burmester GR, Dorner T. Differential effects of epratuzumab on peripheral blood B-cells of patients with systemic lupus erythematosus versus normal controls. Ann Rheum Dis. 2008;67(4):450–457. | ||
Carnahan J, Wang P, Kendall R, et al. Epratuzumab, a humanized monoclonal antibody targeting CD22: characterization of in vitro properties. Clin Cancer Res. 2003;9(10 Pt 2):3982S–3990S. | ||
Carnahan J, Stein R, Qu Z, et al. Epratuzumab, a CD22-targeting recombinant humanized antibody with a different mode of action from rituximab. Mol Immunol. 2007;44(6):1331–1341. | ||
Beum PV, Kennedy AD, Williams ME, Lindorfer MA, Taylor RP. The shaving reaction: rituximab/CD20 complexes are removed from mantle cell lymphoma and chronic lymphocytic leukemia cells by THP-1 monocytes. J Immunol. 2006;176(4):2600–2609. | ||
Williams ME, Densmore JJ, Pawluczkowycz AW, et al. Thrice-weekly low-dose rituximab decreases CD20 loss via shaving and promotes enhanced targeting in chronic lymphocytic leukemia. J Immunol. 2006;177(10):7435–7443. | ||
Wallace DJ, Kalunian K, Petri MA, et al. Efficacy and safety of epratuzumab in patients with moderate/severe active systemic lupus erythematosus: results from EMBLEM, a phase IIb, randomised, double-blind, placebo-controlled, multicentre study. Ann Rheum Dis. 2014;73(1):183–190. | ||
Leonard JP, Coleman M, Ketas JC, et al. Phase I/II trial of epratuzumab (humanized anti-CD22 antibody) in indolent non-Hodgkin’s lymphoma. J Clin Oncol. 2003;21(16):3051–3059. | ||
Steinfeld SD, Tant L, Burmester GR, et al. Epratuzumab (humanised anti-CD22 antibody) in primary Sjogren’s syndrome: an open-label phase I/II study. Arthritis Res Ther. 2006;8(4):R129. | ||
Strand V, Petri M, Kalunian K, et al. Epratuzumab for patients with moderate to severe flaring SLE: health-related quality of life outcomes and corticosteroid use in the randomized controlled ALLEVIATE trials and extension study SL0006. Rheumatology (Oxford). 2014;53(3):502–511. | ||
Wallace DJ, Gordon C, Strand V, et al. Efficacy and safety of epratuzumab in patients with moderate/severe flaring systemic lupus erythematosus: results from two randomized, double-blind, placebo-controlled, multicentre studies (ALLEVIATE) and follow-up. Rheumatology (Oxford). 2013;52(7):1313–1322. | ||
Petri M, Hobbs K, Gordon C. Randomized controlled trials (RCTS) of epratuzumab (anti-CD22 MAB targeting B-cells) reveal clinically meaningful improvements in patients (pts) with moderate/severe SLE flares. Ann Rheum Dis. 2008;67(Suppl II):53. | ||
Wallace D, Hobbs K, Houssiau F, et al. Randomized controlled trials of epratuzumab (anti-CD22 MAB targeting B-cells) reveal clinically meaningful reductions in corticosteroid use with favorable safety profile in moderate and severe flaring SLE patients. Ann Rheum Dis. 2008;67(Suppl II):212. | ||
Urowitz MB, Gladman DD, Tom BD, Ibanez D, Farewell VT. Changing patterns in mortality and disease outcomes for patients with systemic lupus erythematosus. J Rheumatol. 2008;35(11):2152–2158. | ||
Navarra SV, Guzman RM, Gallacher AE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9767):721–731. | ||
Davies RJ, Sangle SR, Jordan NP, et al. Rituximab in the treatment of resistant lupus nephritis: therapy failure in rapidly progressive crescentic lupus nephritis. Lupus. 2013;22(6):574–582. | ||
Lightstone L. Minimising steroids in lupus nephritis – will B-cell depletion pave the way? Lupus. 2013;22(4):390–399. | ||
Condon MB, Ashby D, Pepper RJ, et al. Prospective observational single-centre cohort study to evaluate the effectiveness of treating lupus nephritis with rituximab and mycophenolate mofetil but no oral steroids. Ann Rheum Dis. 2013;72(8):1280–1286. | ||
Gunnarsson I, Jonsdottir T. Rituximab treatment in lupus nephritis – where do we stand? Lupus. 2013;22(4):381–389. | ||
Moroni G, Raffiotta F, Trezzi B, et al. Rituximab vs mycophenolate and vs cyclophosphamide pulses for induction therapy of active lupus nephritis: a clinical observational study. Rheumatology (Oxford). 2014;53(9):1570–1577. | ||
Rovin BH. Targeting B-cells in lupus nephritis: should cautions optimism remains Nephrol Dial Transplant. 2014;28(1):7–9. | ||
Touma Z, Gladman DD, Urowitz MB. Clinical measures, metrics and indices. In: Wallace DJ, Hahn BH, editors. Dubois’ Lupus Erythematosus; 8th ed. Lippincott Williams and Wilkins, Saunders; 2012. | ||
Strand V, Gladman D, Isenberg D, Petri M, Smolen J, Tugwell P. Outcome measures to be used in clinical trials in systemic lupus erythematosus. J Rheumatol. 1999;26(2):490–497. | ||
Touma Z, Urowitz MB, Gladman DD. Outcome measures in systemic lupus erythematosus. Indian Journal of Rheumatology. 2013;8(Supp 1):S46–S53. | ||
Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum. 1992;35(6):630–640. | ||
Symmons DP, Coppock JS, Bacon PA, et al. Development and assessment of a computerized index of clinical disease activity in systemic lupus erythematosus. Members of the British Isles Lupus Assessment Group (BILAG). Q J Med. 1988;69(259):927–937. | ||
Isenberg DA, Rahman A, Allen E, et al. BILAG 2004. Development and initial validation of an updated version of the British Isles Lupus Assessment Group’s disease activity index for patients with systemic lupus erythematosus. Rheumatology (Oxford). 2005;44(7):902–906. | ||
Gladman DD, Ibanez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. 2002;29(2):288–291. | ||
Touma Z, Urowitz MB, Ibanez D, Gladman DD. SLEDAI-2K 10 days versus SLEDAI-2K 30 days in a longitudinal evaluation. Lupus. 2011;20(1):67–70. | ||
Touma Z, Gladman DD, Ibanez D, Urowitz MB. Development and initial validation of the systemic lupus erythematosus disease activity index 2000 responder index 50. J Rheumatol. 2011;38(2):275–284. | ||
Touma Z, Urowitz MB, Taghavi-Zadeh S, Ibanez D, Gladman DD. Systemic lupus erythematosus disease activity Index 2000 Responder Index 50: sensitivity to response at 6 and 12 months. Rheumatology (Oxford). 2012;51(10):1814–1819. | ||
Touma Z, Gladman DD, Ibanez D, Urowitz MB. SLEDAI-2K Responder Index 50 captures 50% improvement in disease activity over 10 years. Lupus. 2012;21(12):1305–1311. | ||
Furie RA, Petri MA, Wallace DJ, et al. Novel evidence-based systemic lupus erythematosus responder index. Arthritis Rheum. 2009;61(9):1143–1151. | ||
Thanou A, Chakravarty E, James JA, Merrill J. Which outcome measures in SLE clinical trials best reflect medical judgment? Lupus Science and Medicine. 2014;1(1):e000005. | ||
Petri M, Pike MC, Kelley L, Kilgallen B, Gordon C. Systemic Lupus Erythematosus Responder Index Assessment of Responders in EMBLEM, a Phase IIb Study in Patients with Moderate to Severe Systemic Lupus Erythematosus. Arthritis and Rheumatism. 2011;63(Suppl 10):1378. | ||
Sridharan ST, Zhou T, Immermann F, Lehmann M, Masferrer JL, Honczarenko M, et al. Low placebo responses and clinical components of the biomarkers of lupus disease (BOLD) study may provide useful insights for SLE clinical trial design. Arthritis Rheumatol. 2011;63(Suppl):S543. | ||
Pike MC, Kelley L. Data quality challenges in systemic lupus erythematosus trials: how can this be optimized? Curr Rheumatol Rep. 2012;14(4):324–333. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.