")
Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 4
Profile of elotuzumab and its potential in the treatment of multiple myeloma
Authors Liu Y, Szmania S, van Rhee F
Received 7 April 2014
Accepted for publication 3 May 2014
Published 3 June 2014 Volume 2014:4 Pages 15—27
DOI https://doi.org/10.2147/BLCTT.S49780
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Yi-Chang Liu,1,2 Susann Szmania,1 Frits van Rhee1
1Myeloma Institute for Research and Therapy, University of Arkansas for Medical Sciences, Little Rock, AR, USA; 2Faculty of Medicine, College of Medicine, Kaohsiung Medical University and Department of Hematology-Oncology, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan
Abstract: Although the introduction of novel drugs has improved outcome significantly in multiple myeloma (MM), many patients still eventually relapse. Monoclonal antibodies (mAbs) targeting MM-related antigens can complement currently available therapies. CS1 (also known as CD2 subunit 1, SLAMF7, CD319, and CRACC), a cell surface glycoprotein receptor that is a member of the signaling lymphocytic activation molecule (SLAM) family, is highly and nearly uniformly expressed in myeloma cells at the gene and protein level, but not expressed in other tissues, including hematopoietic stem cells, making CS1 a compelling target for the design of immunotherapies directed at MM. Elotuzumab (formerly HuLuc63), which is a humanized IgG1 mAb recognizing the extracellular region of human CS1, has been shown to be effective in preclinical and early stage clinical investigations, and its efficacy and safety will be further validated in ongoing Phase III trials. Integration of elotuzumab into multidrug therapeutic paradigms seems logical, as elotuzumab is more effective when combined with other agents, such as immunomodulatory drugs or proteasome inhibitors. The functional role of CS1 in MM pathogenesis and the consequences of elotuzumab on normal immune cells should be further investigated. Identification of potential biomarkers and exploration of resistance mechanisms are important issues for elotuzumab-based therapies, as is determining the best clinical placement of elotuzumab, not only in the relapsed/refractory setting but also in upfront therapy for high-risk frank MM, smoldering MM at high-risk of progression, and in maintenance regimens. This review will cover the biological characteristics of CS1 in normal immune cells and MM cells, the efficacy profile and mechanisms of action of elotuzumab from preclinical and clinical investigations, and its potential impact on the treatment of MM.
Keywords: CS1, monoclonal antibody, immunotherapy
Introduction
The plasma cell malignancy multiple myeloma (MM) has undergone a marked evolution over the past decade, both in terms of an increased understanding of the pathobiology of the disease as well as in the development of a number of novel agents. The introduction of several new drugs, especially proteasome inhibitors such as bortezomib and carfilzomib and immunomodulatory drugs including thalidomide, lenalidomide, and pomalidomide, has improved outcomes in patients afflicted with MM when applied in the relapsed/refractory setting or incorporated into the treatment paradigms of pre- and post-autologous stem cell transplantation in newly diagnosed MM.1–5 Although it has been postulated that MM can be a curable disease, many patients still eventually relapse and become refractory to all presently available therapies6 or experience treatment-related toxicities. High-risk myeloma as defined by gene expression profiling is characterized by similar remission rates as standard risk myeloma, but as early relapse often occurs, this entity remains a therapeutic challenge.7 Taken together, these observations suggest that there is an unmet need and the development of further novel therapies is required.
Recently, a number of novel agents with different mechanisms of action have been developed based upon an increasing knowledge of the pathogenesis of MM. These include newer generations of proteasome inhibitors and immunomodulatory drugs, monoclonal antibodies (mAbs) and other immunotherapies, histone deacetylase inhibitors, signal transduction pathway inhibitors, kinase inhibitors, and heat shock protein inhibitors, to name but a few.8–10
Therapeutic mAbs may have one or more of the following mechanisms of action. MAbs could directly induce growth inhibition by interfering with receptor-ligand interactions, stimulate apoptosis signaling cascades, or act as a carrier of cytotoxic agents such as chemotherapeutic drugs or radioisotopes. MAbs could also induce tumor cell killing via indirect mechanisms mediated by the immune system. These include antibody-dependent cellular cytotoxicity (ADCC), macrophage-mediated phagocytosis, and complement-dependent cytotoxicity (CDC).11–14 ADCC is triggered by interactions between the Fc region of mAbs bound to the tumor cell and Fc receptors on immune effector cells such as natural killer (NK) cells, macrophages, and neutrophils, inducing subsequent cytotoxicity by immune cells. CDC is triggered by the interactions between the Fc region of mAb with C1q, resulting in the accumulation of C3b and subsequent formation of membrane attack complex.
MAb-based therapies have been widely used in several human malignancies, including B-cell malignancies. One of the most actively utilized mAbs is rituximab, a chimeric anti-CD20 mAb which is indicated in the treatment of B-cell non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, and Waldenström’s macroglobulinemia, both in combination with cytotoxic chemotherapy and as a single agent in maintenance therapy. Rituximab is the first mAb that was evaluated in the treatment of MM. CD20 is expressed by myeloma cells of the CD2 subgroup comprising 15%–20% of MM patients.15 However, rituximab has limited efficacy in CD20-positive MM. Potential explanations are heterogeneous CD20 expression in some patients, the inhibition of CDC by complement regulator molecules such as CD55 or CD59 on MM cells, or Fc receptor polymorphism, which may limit the efficacy of ADCC.11–14,16,17 Theoretically, the ideal target for mAb therapy should be exclusively expressed or have a significantly higher expression level on tumor cells than nontarget cells. Since myeloma cells could interact with various cellular or noncellular components in the bone marrow (BM) microenvironment, the search of the potential targets has led to the development of mAbs not only targeting the antigens expressing on the myeloma cell, but also targeting adhesion of myeloma cells to BM stromal cells (BMSC), neutralizing growth factors in the BM microenvironment and inhibiting the function of their receptors, targeting mediators of bone disease, and improving anti-MM immune responses.11–14 Currently, a number of novel mAbs have been developed and investigated in preclinical studies, and some of them have entered early stage clinical trials.11–14 MAbs which have been investigated in various stages of clinical trials in the treatment of MM are listed in Table 1.
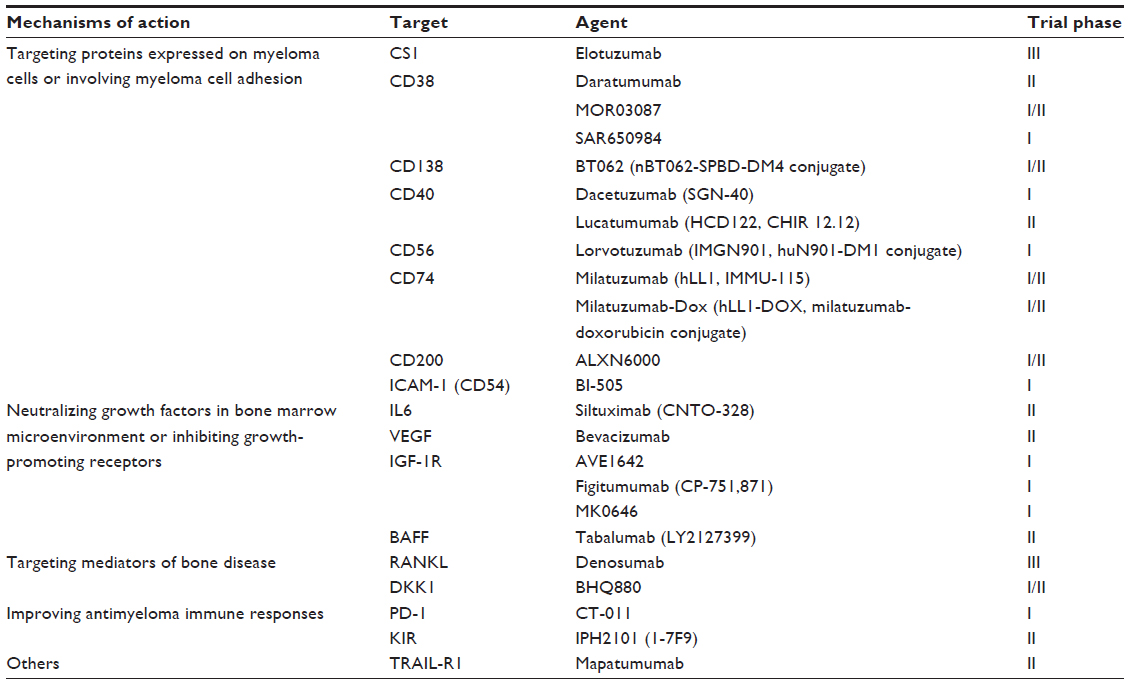 | Table 1 Monoclonal antibodies in various stages of clinical investigation for the treatment of multiple myeloma |
CS1, a cell surface glycoprotein receptor that is a member of the signaling lymphocytic activation molecule (SLAM) family, is highly and nearly uniformly expressed in patient MM cells at the gene and protein levels while expression in healthy cells is limited to certain immune cell subsets, making CS1 a compelling target for the design of immunotherapies for MM.18–20 Elotuzumab (formerly HuLuc63) is a fully humanized IgG1 monoclonal antibody recognizing the extracellular region of human CS1 that has been shown to be effective in preclinical investigations both in vitro and in vivo,18–20 and has prompted several early stage clinical trials in patients with relapsed and refractory MM.21–25 Currently, elotuzumab is the most extensively evaluated mAb for the treatment of MM, and thus is further along in its clinical development.26
In this review, we will first discuss the biological characteristics of CS1, its expression and function in normal cells, as well as the possible role of CS1 in the pathogenesis of MM. Next, we will review the preclinical data and the mechanisms of action for elotuzumab, which support its use to treat MM. Lastly, we will review the results of the completed early stage clinical trials, mention ongoing clinical trials with elotuzumab, and discuss the potential applications and future directions of elotuzumab in the treatment of MM.
Biological characteristics of CS1 and the SLAM family
CS1 (also known as CD2 subunit 1, SLAMF7, CD319, CRACC) is a cell surface glycoprotein receptor that belongs to the SLAM family.27,28 The SLAM family receptors are a subset of the CD2 superfamily of immunoglobulin domain-containing molecules that are expressed on the surface of various hematopoietic cells and are recognized as important immunomodulatory receptors.29–31 The SLAM family receptors in humans consist of SLAM (CD150, SLAMF1), CD48 (SLAMF2), LY9 (CD229, SLAMF3), 2B4 (CD244, SLAMF4), CD84 (SLAMF5), NK-T-B antigen (NTB-A, CD352, SF2000, SLAMF6), CS1, BLAME (CD353, SLAMF8), and SF2001 (CD84H1, SLAMF9). The genes encoding the SLAM family receptors, including CS1, are located on human chromosome 1q23. Interestingly, amplification of chromosome 1q is frequently found in aggressive MM and is associated with poor prognosis.7 The SLAM family receptors have a similar structure characterized by an extracellular domain containing an Ig variable-like domain and a C2-like domain, a transmembrane domain, and a cytoplasmic domain containing one or more immunoreceptor tyrosine-based switch motifs (ITSMs), except LY9 that has duplicated V-C2-like sequences, and BLAME and SF2001 which have short intracellular tails that lack tyrosine motifs.29–31 The six core SLAM family receptors, SLAMF1 and SLAMF3 to SLAMF7, are self-ligands except 2B4, whose ligand is CD48. When the receptor is engaged, the ITSM undergoes phosphorylation and enables the recruitment of SLAM-associated protein (SAP) family adaptors, including SAP and EWS-Fli1-activated transcript-2 (EAT-2) in humans. The gene encoding EAT-2 is also located on chromosome 1q23. Through recruiting downstream effectors in the presence or absence of the SAP family adaptors, the SLAM family receptors may mediate activating or inhibitory effects on immune cells.29–31
CS1 has some distinctive features compared to other SLAM family receptors.29–31 CS1 was found to be highly expressed in normal and malignant plasma cells.18–20 CS1 only possesses one ITSM in its cytoplasmic domain, compared with at least two ITSMs in other SLAM family members, with the exception of LY9. CS1 only recruits EAT-2 to mediate the activating effects,32,33 unlike other SLAM family members that interact with both SAP and EAT-2. CS1 has two isoforms, CS1-long (CS1-L) and CS1-short (CS1-S), which differ in cytoplasmic domain and exhibit differential expression patterns among immune cells.34
CS1 expression and its functional role in normal cells
CS1 expressed in hematopoietic cells but not body tissue cells
In the initial report identifying CS1, it was found to be expressed on virtually all NK cells, most CD8+ T cells, activated B cells, mature dendritic cells (DCs), and a small percentage of CD4+ T cells, but was not expressed on monocytes and immature DCs using flow cytometric analysis of human PBMC with anti-CS1 mAb 162.27 Many studies have shown that CS1 is expressed in mature NK cells and plays an important role in NK cell activation.27,28,32,33,35 CS1 was also found to be highly expressed in normal plasma cells from healthy donors, but was not expressed in 332 normal tissue samples from 86 different organs using a DNA microarray technique.18 At the protein level CS1 was detected in normal plasma cells, but not in a variety of normal tissues using immunohistochemical (IHC) staining with anti-CS1 mAb MuLuc63 and 1G9.18
In other hematopoietic cells, low CS1 expression was found on resting B cells by anti-CS1 mAb 1G10 and the expression was upregulated upon B cell activation. Only the CS1-L isoform was found on human B cells compared with both CS1-L and CS1-S on human NK cells.34 Another study showed a bimodal pattern of CS1 expression in B cell lineage. CS1 was highly expressed on pro-B cells, dropped during B cell maturation, and again became abundantly expressed in terminally differentiated plasmablasts and plasma cells.36
Functional role of CS1 in normal NK and other immune cells
Most studies have shown that CS1 plays an important role in NK cell activation,27,28,32,33,35 while a study exploring NK cell function in CS1-deficient mice showed that CS1 does not appear to be critical for NK cell development in vitro and in vivo.32 Initial findings suggested that engagement of CS1 by mAb 162 promotes NK cell-mediated cytotoxicity in a SAP-independent manner.27 This engagement has no cytotoxic effect on neighboring NK cells due to the binding of inhibitory receptors with major histocompatibility complex class I on neighboring NK cells.35 In addition to CS1–CS1 interaction between NK cells and target cells, the CS1–CS1 interaction between NK cells may further enhance NK cell function.32 The positive regulation of NK cell function by CS1 is determined by the expression of EAT-2 but not SAP.32 CS1 recruits EAT-2 and activates PI3K and phospholipase C-γ signaling pathways in human NK cells.33 Further experiments showed that in the presence of EAT-2, CS1 has a positive effect on NK cell activation, while in the absence of EAT-2 CS1 has an inhibitory effect.32
Some studies have explored the functional role of CS1 in B cells and T cells. Ligation of surface CS1 with anti-CS1 mAb 1G10 enhanced proliferation of activated human B lymphocytes, and the CS1 activation enhanced mRNA expression of several growth-promoting cytokines, suggesting CS1 may play a role in the regulation of B lymphocyte proliferation during immune response.34 In T cells, CS1 may have an inhibitory effect. Engagement of CS1 on mouse CD4+ T cells, which naturally lacked EAT-2, resulted in an inhibition of antigen-induced T cell proliferation and cytokine production.32 A low level of CS1 expression was also reported in other immune cells like NKT cells and DCs; however, the functional role of CS1 in these cells remains unclear.
CS1 expression in malignant plasma cells and the possible role in MM pathogenesis
CS1 is highly expressed in MM cells from most MM cell lines and from more than 95% of MM patients both at the mRNA and protein levels.18,19,37 CS1 is also highly expressed in CD138 purified plasma cells from patients with monoclonal gammopathy of undetermined significance and smoldering multiple myeloma (SMM) (Figure 1A).18 CS1 expression in myeloma cells was seen across all MM subpopulations and was not restricted to certain cytogenetic abnormalities, high-risk or low-risk molecular profiles, or molecular subtypes.18 CS1 expression persisted at the time of disease relapse and after bortezomib treatment.18,20 Higher CS1 expression in malignant plasma cells compared to normal plasma cells has not been consistently reported.18,37
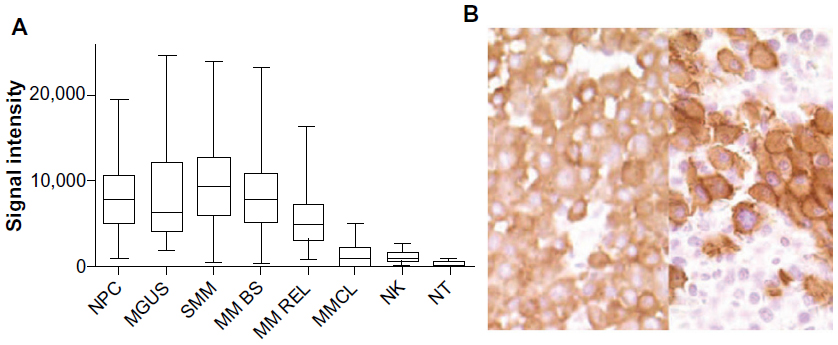 | Figure 1 CS1 expression quantified by gene expression (A), and immunohistochemistry (B). |
CS1 protein is present on many MM cell lines and on primary MM cells by flow analysis with anti-CS1 mAb,19 and in tissue samples from patients with MM and extramedullary plasmacytoma by IHC staining with anti-CS1 mAb 1G9 (Figure 1B).18 CS1 protein was not found in a variety of solid tumors, acute leukemias, mature B cell neoplasms without plasmacytic differentiation, and Hodgkin’s lymphomas, but could be found in 25% of peripheral T cell lymphomas at a modest level.18 Low levels of CS1 could be detected in sera of MM patients, but was not found in healthy donors. The correlation of circulating soluble CS1 level and the International Staging System (ISS) grade is moderate, with a higher level in ISS II/III patients.19 CS1 surface protein expression did not change significantly in patient-derived myeloma cells before and after bortezomib treatment.20
The functional role of CS1 in myeloma cells is not well characterized. CS1 may play an important role in the interaction between myeloma cells and its adhesion to BMSCs, as CS1 was shown to be concentrated and colocalized with CD138 at uropod membranes of polarized myeloma cells, which promote cell–cell adhesion.19 Myeloma cells from cell lines showed a decreased binding to BMSCs when transduced with CS1 short interfering RNA; similarly, an inhibited adhesion to BMSCs was observed when these cells were treated with elotuzumab.19 It is well known that the survival and growth of MM cells are dependent on the interactions between MM cells and surrounding microenvironment, including BMSCs. One report showed that CS1 activates ERK1/2, STAT3, and AKT signaling pathways, thereby promoting myeloma growth and survival; however, further confirmation of these results is required.38
Targeting CS1 on myeloma cells with elotuzumab-preclinical studies
Development of elotuzumab
The characteristics of CS1 expression suggested CS1 is a potential antibody target and led to the generation of anti-CS1 mAb. Initially the murine anti-CS1 mAbs MuLuc63 (IgG2a) and MuLuc90 (IgG2b), recognizing the extracellular portion of CS1, were developed by generating hybridoma cell lines from the spleen cells of mice immunized with purified CS1 protein.18 Both MuLuc63 and MuLuc90 showed significant antitumor activity in vivo in a human myeloma cell line/SCID mouse xenograft model. MuLuc63 was more potent and showed rapid tumor eradication and was therefore selected for humanization.39 HuLuc63, the humanized IgG1 version of MuLuc63, later named elotuzumab, exhibited significant antitumor activity in several xenograft models. It is a fully humanized IgG1 mAb and recognizes the extracellular region of human CS1.
In vitro binding of elotuzumab to leukocyte subsets in human blood and BM samples was primarily to NK cells, NK-like T cells, and CD3+CD8+ T cells, while significant binding to monocytes, B cells, and granulocytes was not observed. No binding was found to CD34+ hematopoietic stem cells, and elotuzumab is not stem cell toxic.18 A modest 20% average decrease in NK cell count and no significant depletion of other major lymphocyte subsets were observed when blood samples from healthy individuals were cultured with different concentrations of HuLuc63, compared with a 70% decrease of B cells following rituximab and 65%–100% decrease on all subsets following alemtuzumab in the same experiment.18
Mechanisms of actions of elotuzumab
Most work suggests that the predominant mechanism of action of elotuzumab is NK cell-mediated ADCC (Figure 2) and that elotuzumab does not induce CDC. After binding to CS1 on the MM cell surface, the Fc portion of elotuzumab binds to the Fc receptor (CD16) on NK cells, triggering NK cell activation, release of cytotoxic granules, and MM cell killing. In vitro evidence shows that elotuzumab is capable of triggering ADCC in the presence of human peripheral blood mononuclear cells (PBMC), inducing myeloma cell killing of CS1-positive MM cell lines in a dose-dependent manner.19 Elotuzumab also induces lysis of primary myeloma cells by purified autologous NK cells and NK cells from healthy allogeneic donors.18 Blocking the Fc receptor on NK cells by anti-CD16 Ab or depletion of NK cells from PBMC significantly decreased the lytic effect, supporting the notion that NK cells are the dominant effectors.18,20 Further, elotuzumab could trigger autologous effector cell mediated ADCC of primary MM cells resistant or refractory to conventional or novel therapies, including bortezomib or a heat shock protein-90 inhibitor.19
 | Figure 2 Mechanisms of action of elotuzumab. The primary mechanism of action of elotuzumab against myeloma cells is NK cell-mediated ADCC. Elotuzumab can also interfere with the adhesion of myeloma cells to BMSC, or can induce NK cell activation directly through binding CS1 expressed on NK cells. |
The studies of elotuzumab in combination with other agents in vitro provided a rationale for combination therapy. Pretreatment of PBMC effector cells with lenalidomide could enhance elotuzumab-mediated ADCC against MM cell lines and primary MM cells.19 When cells from the MM1R cell line were pretreated with dexamethasone, bortezomib, lenalidomide, perifosine, or the MEK inhibitor U2106, an increase of myeloma cell killing induced by elotuzumab-mediated ADCC could be observed using PBMC from healthy donors.19 Bortezomib could significantly enhance elotuzumab-mediated ADCC in CS1-positive myeloma cell lines using NK cells or PBMCs from healthy donors and in primary myeloma cells using autologous NK effector cells.20
Antitumor activity of elotuzumab could also be observed in vivo. Significant tumor volume decrease following treatment with elotuzumab compared with control Ab was observed in L363-, OPM2-, and MMS1-inoculated SCID mice, and an elotuzumab dose-dependent effect was found in OPM2 xenograft mice.19 Elotuzumab also inhibited tumor growth in OPM2-engrafted SCID mice in another study, with higher activity observed when the affinity of the Ab to the Fc receptor was increased, and lower activity when NK cells were depleted, further suggesting that antitumor activity depends on the Fc–Fc receptor interaction and the presence of functional NK cells.18 Experiments targeting patient-derived primary myeloma cells in a SCID-hu mouse model demonstrated significant reduction of tumor volume and circulating IgG level in MuLuc63-treated mice, suggesting that the antitumor activity occurs in the setting of the human bone microenvironment.20 Furthermore, elotuzumab in combination with bortezomib exhibited enhanced antitumor activity in OPM2-engrafted SCID mice, compared with elotuzumab or bortezomib monotherapy.20
In addition to ADCC, several alternative mechanisms of action have been proposed for elotuzumab, including interfering with MM cell adhesion to BMSC and enhancing NK cell cytotoxicity directly (Figure 2). As mentioned previously, CS1 mediates MM cell adhesion to BMSC and elotuzumab appeared to induce NK cell activation through binding to CS1 on NK cells, which enhances cytotoxicity against CS1-positive MM cells but not against autologous NK cells.19 Elotuzumab may also promote CS1–CS1 interaction between NK cells and CS1-positive target cells to enhance cytotoxicity.40
The presence of functional effector cells is important for ADCC, and the functional immune response of NK cells toward MM cells is impaired due to immunoevasive strategies by MM cells.41 We have reported that NK cell function can be augmented by coculture with K562 transfectants expressing membrane bound IL15 and the costimulatory molecule 4-1BBL.42 Ex vivo expanded NK (ENK) cells derived from healthy donors or MM patients were capable of killing both allogeneic and autologous primary MM cells in vitro, whereas resting NK cells were not, and adoptive transfer of ENK cells inhibited growth of OPM2 and high-risk primary MM tumors grown in mice and protected bone from osteolysis.42 In a subsequent clinical trial on patients with high-risk relapsed/refractory MM, the infusion of high-dose ENK cells was safe and in vivo expansion was observed.43 Pretreatment of autologous MM cells with elotuzumab could enhance ENK cell-mediated killing by ADCC in vitro,44 providing the rationale for a combination therapy of elotuzumab and ENK cells. Elotuzumab was observed to be able to enhance both ENK cell-mediated, and to a much lesser extent, resting NK cell-mediated killing of autologous MM cells in vitro, implying that the activation status of NK effector cells contributes to elotuzumab activity (Figure 3).
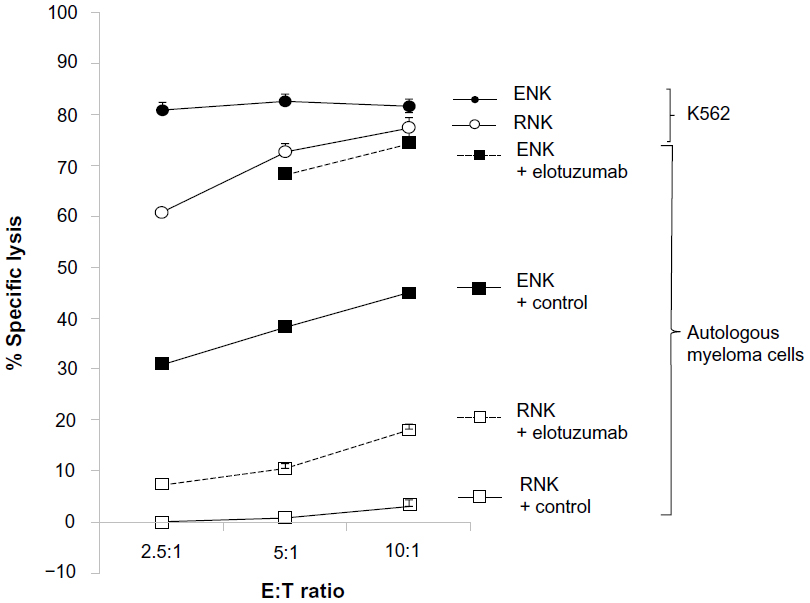 | Figure 3 Elotuzumab enhanced both ex vivo expanded NK cell-mediated, and to a lesser extent, resting NK cell-mediated killing of autologous myeloma cells in a standard 4-hour 51Cr release assay. |
Clinical trials with elotuzumab
Phase I clinical trials
The above preclinical findings of elotuzumab prompted several Phase I clinical trials in patients with relapsed/refractory MM. In a monotherapy Phase I study using dose-escalated (0.5 to 20 mg/kg) intravenous elotuzumab every 2 weeks in 35 patients with relapsed/refractory MM (≥2 prior therapies, median 4.5), no maximum tolerated dose (MTD) was identified up to the maximum planned dose of 20 mg/kg.21 Toxicities were acceptable; the frequent treatment-related adverse events (AEs) are mostly due to infusion reactions, and mostly grade 1 to 2, including chills, pyrexia, and flushing, which prompted the protocol amendment to include premedication with antihistamine and acetaminophen before each infusion and corticosteroid before the first dose of elotuzumab in 20 mg/kg cohort. CS1 on BM-derived MM cells was saturated (>95%) at the dose level of 10 and 20 mg/kg without dose-limiting toxicity. A transient decrease of absolute lymphocyte count, including both CS1-positive and CS1-negative cells subsets, was found after the first dose but subsequently recovered to near baseline in the following dosing without chronic depletion of T cells. In terms of efficacy, however, no objective responses were observed; the best response was stable disease, which was found in 9 (26.5%) patients.
Despite the limited activity in monotherapy, the combination with other agents has shown encouraging results. In another Phase I study, dose-escalated (2.5 to 20 mg/kg) elotuzumab in combination of a fixed, usually scheduled dose of bortezomib was used in 28 patients with relapsed/refractory MM (one to three prior therapies [median 2] bortezomib pretreated were eligible). Treatment comprised intravenous elotuzumab on Days 1 and 11 and intravenous bortezomib 1.3 mg/m2 on Days 1, 4, 8, and 11 cycled on a 21-day basis.22 Infusion reactions were observed in 71% of patients, mostly grade 1 or 2 and one with grade 3 toxicity. The protocol was amended to require a premedication with methylprednisolone, diphenhydramine, and acetaminophen before each elotuzumab infusion to mitigate infusion reactions. MTD was not reached up to the maximum planned dose of 20 mg/kg. The most frequent grade 3–4 AEs were lymphopenia (25%), fatigue (14%), thrombocytopenia (11%), hyperglycemia (11%), neutropenia (11%), peripheral neuropathy (11%), and pneumonia (11%). An objective response rate (ORR) was 48% (13 of 27 evaluable patients), including two patients who achieved a complete response (CR) and two bortezomib-refractory patients achieving a partial response (PR). A median time to progression (TTP) was 9.5 months. This compared favorably to the Phase III study (APEX trial) in patients with relapsed/refractory MM, patients receiving bortezomib only, and had an ORR of 38% with a median TTP of 6.2 months.45
In another Phase I study, dose-escalated elotuzumab (5 to 20 mg/kg) in combination with lenalidomide and low-dose dexamethasone was used to treat 29 patients with relapsed/refractory MM (at least one prior therapy [median 3], lenalidomide pretreatment eligible). Protocol therapy consisted of a 28-day cycle of intravenous elotuzumab on Days 1, 8, 15, and 22 for the first two cycles and on Day 1 and 15 for the remaining cycles, lenalidomide 25 mg daily on Days 1 through 21, and dexamethasone 40 mg weekly.23 MTD was not reached up to the maximum planned dose of 20 mg/kg. Infusion reactions were again common, mostly mild to moderate, with 89% of patients experiencing at least one; two patients experienced grade 3 or 4 infusion reactions leading to treatment discontinuation. Again, the protocol was amended for patients to receive premedication. The most frequent grade 3 to 4 AEs (>10%) were neutropenia (36%), thrombocytopenia (21%), fatigue (11%), and diarrhea (11%). The ORR was 82% (23 of 28 evaluable patients), including one CR (4%) and eight very good PRs (29%). Objective responses were observed in two of six patients with prior exposure to lenalidomide, in 15 (94%) of 16 patients with prior therapy with thalidomide, and in 15 (75%) of 20 patients who had been treated with bortezomib. The median TTP was not reached for 22 patients receiving 20 mg/kg after a median follow-up of 16.4 months, suggesting that these responses were durable.
Preliminary data of Phase II trials and ongoing Phase III trials
The results of Phase I studies prompted subsequent Phase II trials. In a Phase II study of 73 patients with relapsed/refractory MM (one to three prior therapies, lenalidomide naïve), patients were randomized to receive elotuzumab 10 mg/kg (n=36) or 20 mg/kg (n=37), on Days 1, 8, 15, 22 for the first two cycles and on Day 1 and 15 for the remaining cycles, in combination with lenalidomide 25 mg daily on Days 1 through 21 and dexamethasone 40 mg orally weekly or 28 mg orally plus 8 mg intravenously on elotuzumab dosing days, in a 28-day cycle.24 The primary objective was efficacy (ORR≥PR by International Myeloma Working Group criteria). In an updated analysis,25 the ORR was 84% in the overall population, and 92% and 76% in the 10 mg/kg and 20 mg/kg cohorts, respectively. The median progression-free survival (PFS) was not reached in the 10 mg/kg cohort and was 18.6 months in the 20 mg/kg cohort, with a median follow-up of 20.8 months. When combining both Phase I and Phase II studies to evaluate the long-term safety profile, the most common grade 3 to 4 AEs were lymphopenia (19%), neutropenia (18%), thrombocytopenia (16%), and anemia (14%). Fifteen patients discontinued due to AEs and four patients developed secondary malignancies; however, none were found after 18 months of treatment. AEs emergent after 18 months of treatment were consistent with AEs during the initial 18 months, suggesting that there were no cumulative drug toxicities.25 These trials demonstrated the efficacy of combining lenalidomide, dexamethasone, and elotuzumab, and favorable toxicity profiles were observed. In comparison of two Phase III studies (MM009 and MM010 trials) in patients with relapsed/refractory MM treated with lenalidomide and dexamethasone, an ORR of 61% with a median TTP of 11.1 months and an ORR of 60.2% with a median TTP of 11.3 months were noted, respectively.46,47 A dose of 10 mg/kg elotuzumab was chosen for subsequent Phase III trials.
These findings led to the initiation of two currently ongoing Phase III trials. The first Phase III study (ELOQUENT-2, NCT01239797) has currently recruited approximately 640 patients with relapsed/refractory MM. Eligible patients had one to three prior lines of therapy, and prior lenalidomide treatment was permitted provided the best response was ≥PR. Patients refractory to lenalidomide, having drug discontinuation due to grade ≥3 AEs, or having received ≥9 cycles were ineligible. Finally, not more than 9 months should have elapsed between the last dose of lenalidomide and progression; hence, patients enrolled in this trial were likely to be lenalidomide sensitive. Patients were randomized to 28-day cycles of lenalidomide and dexamethasone with or without elotuzumab (Table 2). In the other ongoing Phase III study of newly diagnosed, previously untreated MM patients (n=750) (ELOQUENT-1, NCT01335399), a regimen of lenalidomide and dexamethasone with or without elotuzumab is being utilized (Table 2). In both ELOQUENT-1 and ELOQUENT-2, the primary end point is PFS and results are eagerly anticipated. The efficacy and safety of elotuzumab in combination with lenalidomide and dexamethasone will be further validated in these two Phase III trials.
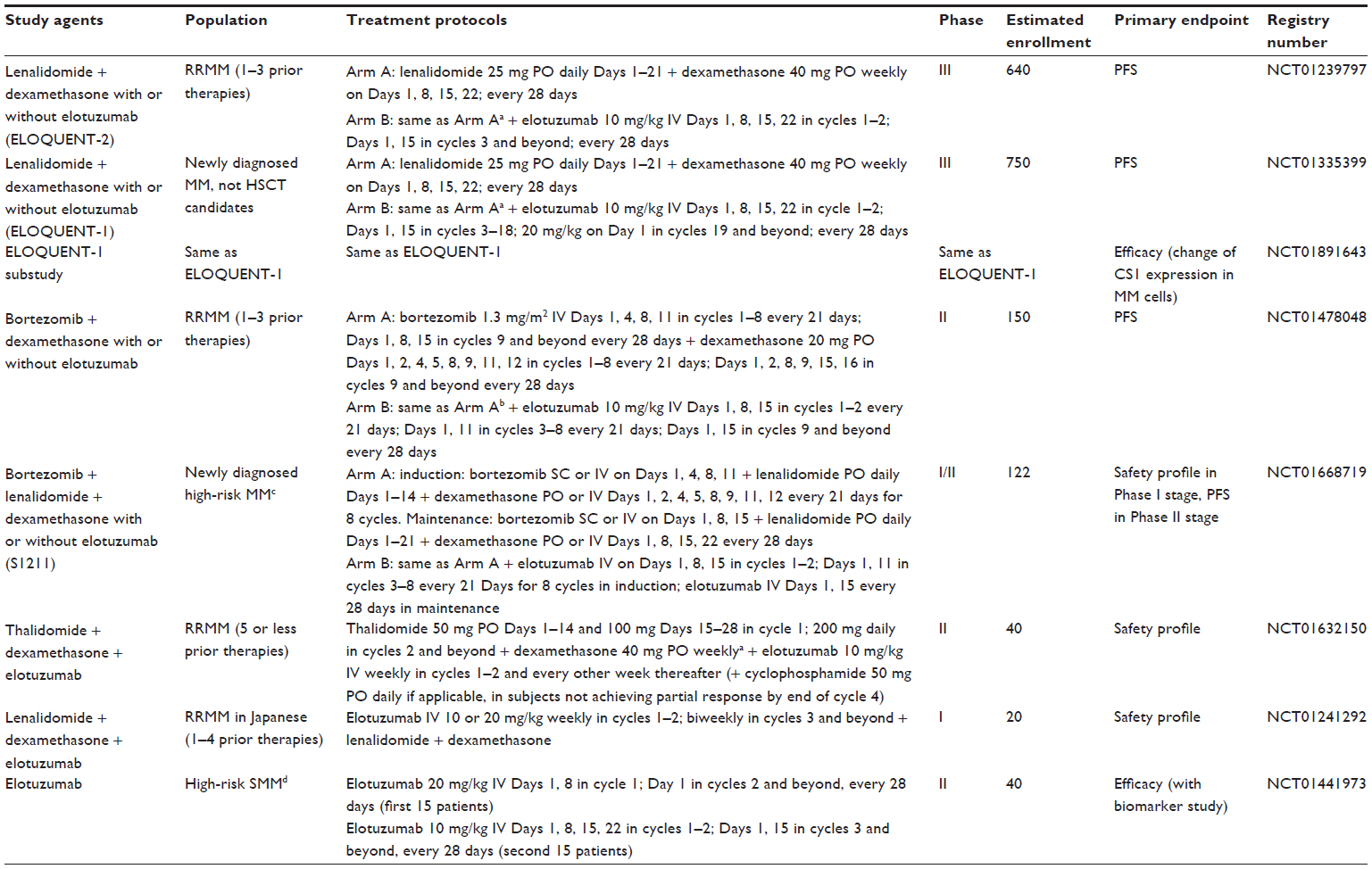 | Table 2 Ongoing clinical trials using elotuzumab for the treatment of multiple myeloma |
Other registered clinical trials
Other currently ongoing clinical trials of elotuzumab are listed in Table 2. An ELOQUENT-1 substudy (NCT01891643) is being conducted to see the changes of CS1 expression in MM cells from baseline to progression in both treatment arms. A randomized Phase II study (NCT01478048) is being conducted to compare the efficacy of bortezomib and dexamethasone with or without elotuzumab in patients with relapsed/refractory MM, with a primary endpoint of PFS. A randomized SWOG Phase I/II study focusing on newly diagnosed MM patients with high-risk profiles (NCT01668719) is being conducted to determine the optimal dose of elotuzumab to use in combination of bortezomib, dexamethasone, and lenalidomide in the Phase I stage, and to assess whether incorporation of elotuzumab into the treatment algorithm will improve the PFS in the subsequent Phase II stage. A Phase IIa single arm study (NCT01632150) has been designed to see the safety profile when elotuzumab is used in combination with thalidomide and dexamethasone in patients with relapsed/refractory MM. Another Phase I study in a Japanese population (NCT01241292) is underway to access the safety and tolerability of elotuzumab in combination with lenalidomide and dexamethasone in patients with relapsed/refractory MM. In the premalignancy setting, a Phase II biomarker study of elotuzumab monotherapy (NCT01441973) is in progress to see the association between elotuzumab-induced changes in monoclonal protein and the baseline percentage of NK cells in BM as well as the efficacy of elotuzumab in patients with high-risk SMM.
In the early studies, the principal side effects were infusion reactions, which have been mitigated by premedications. One concern was that elotuzumab therapy would lead to depletion of immune cells, given the fact that certain normal immune cells express CS1. In an analysis reviewing all the three Phase I studies, a transient decrease of total lymphocyte count, both CS1-positive and CS1-negative lymphocyte subsets, was observed following the first dose of elotuzumab, and was resolved as dosing was continued without evidence of elotuzumab-mediated CS1-positive lymphocyte depletion.48 The transient decrease is likely to be associated with lymphocyte trafficking mediated by chemokines or cytokines such as interferon-inducible protein 10.21,48
Future directions
Other immunotherapeutic approaches targeting CS1
In addition to elotuzumab, several other immune-based therapies are developing around the CS1 target. Significant progress has been achieved in the development of adoptive immunotherapy using the genetic modification of T cells or NK cells with chimeric-antigen receptor (CAR) that directly target tumor associated antigens. Successful experiences using genetically engineered CAR-T cells were reported in patients with CD19+ acute lymphoblastic leukemia and chronic lymphocytic leukemia.49,50 Recently a CS1-specific CAR incorporating a CD28-CD3ζ costimulatory signaling domain was designed and was expressed on human NK cells using lentiviral vector transduction to generate CS1-CAR-NK cells.51 The CS1-CAR-NK cells showed an enhanced cytolysis of CS1-positive myeloma cells and more interferon (IFN)-γ production in vitro, and also showed similarly enhanced activities to primary MM cells ex vivo. Further, inhibited myeloma tumor growth and prolonged survival of tumor bearing mice in an orthotopic xenograft MM model could be observed.51
Another immunotherapy approach is to trigger cytotoxic T lymphocytes (CTL) with immunogenic peptides, which have specific activity against MM-related antigens expressed on myeloma cells.52 Recently, a novel immunogenic HLA-A2-specific CS1 239-247 (SLFVLGLFL) peptide was identified that was capable of inducing CS1-specific CTL to MM cells.37 The CTL showed antigen-specific cytotoxicity, proliferation, degranulation, and IFN-γ production against HLA-A2-positive/CS1-positive primary MM cells and MM cell lines.
Potential applications and future directions of elotuzumab
Presently, no mAbs are approved for the treatment of MM. Elotuzumab is the most extensively evaluated mAb and ongoing Phase III trials, if positive, may lead to treatment indications in both the newly diagnosed and relapsed/refractory setting. Another important consideration is where elotuzumab is best positioned. Clearly there is an unmet need for novel agents in the relapsed/refractory setting, and elotuzumab is one of the agents that is furthest in its clinical development. It will be important to analyze the efficacy in lenalidomide-pretreated patients in the ELOQUENT-2 study.
Based on the preclinical and clinical data thus far, it is clear that elotuzumab will be more effective when used in combination with other agents. Therefore, it will be important to search for novel agents which when partnered with elotuzumab enhance anti-MM activity without adding toxicity. Since the immunomodulatory drugs enhance ADCC, combining elotuzumab with lenalidomide as maintenance therapy seems an attractive and rational option to explore. High-risk myeloma, especially when defined by gene expression profiling, still has a poor outcome; it will be of interest to evaluate whether addition of elotuzumab can improve outcome, which is being studied in a SWOG study. A groundbreaking study in patients with high-risk SMM has reported a longer time to progression and survival in patients treated with lenalidomide and dexamethasone compared to observation only.53 These findings have raised the speculation that nontoxic mAbs such as elotuzumab could be applied in this setting too.
More than 20 novel mAbs will be clinically studied in MM. Daratumumab, an anti-CD38 mAb which has demonstrated single agent anti-MM activity, seems a particularly interesting agent.54,55 Future attempts to directly compare the efficacy of different mAbs will likely be difficult. It will be important to look for biomarkers which predict response to specific mAbs and study mechanisms of tumor escape.
Conclusion
MAb therapy targeting specific MM-related antigens is an exciting therapeutic strategy currently being vigorously investigated. The unique expression pattern of CS1 makes it an attractive target for designing mAbs and other immunotherapies. Currently, elotuzumab has shown to be active in preclinical and early stage clinical trials, and its efficacy and safety will be further validated in ongoing Phase III trials. It will be important to define the best way to further integrate elotuzumab into currently available therapeutic paradigms, as well as to determine the best clinical placement of elotuzumab, not only in the relapsed/refractory setting but also as upfront therapy in high-risk MM, maintenance therapy, or even high-risk SMM, to achieve better disease control and quality of life, maintain longer remission duration, and decrease treatment-related toxicities. Elotuzumab is more effective when used in combination with other agents; therefore, it will be important in the future to design rational combinations, such as newer generation of immunomodulatory drugs or proteasome inhibitors, small molecules or other mAbs, and cellular therapies such as ENK cells, with an enhanced anti-MM effect and a favorable AE profile. Future basic research will likely explore the biology of CS1, including the action of CS1 in immune cells other than NK cells, the functional consequences of elotuzumab on normal immune cells, and the functional role and signaling pathways of CS1 during the malignant transformation of plasma cells, which need to be clarified.
Acknowledgments
This work was supported in part by National Institutes of Health grants CA55819 and CA134522.
Disclosure
F van Rhee has received research funding from PDL Biopharma. The authors declare no further conflicts of interest in this work.
References
Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;17; 364(11):1046–1060. | |
Mahindra A, Laubach J, Raje N, Munshi N, Richardson PG, Anderson K. Latest advances and current challenges in the treatment of multiple myeloma. Nat Rev Clin Oncol. 2012;9(3):135–143. | |
Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516–2520. | |
Mateos MV, Ocio EM, San Miguel JF. Novel generation of agents with proven clinical activity in multiple myeloma. Semin Oncol. 2013;40(5):618–633. | |
Moreau P, Richardson PG, Cavo M, et al. Proteasome inhibitors in multiple myeloma: 10 years later. Blood. 2012;120(5):947–959. | |
Kumar SK, Lee JH, Lahuerta JJ, et al; International Myeloma Working Group. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia. 2012;26(1):149–157. | |
Shaughnessy JD, Zhan F, Burington BE, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109(6):2276–2284. | |
Ocio EM, Richardson PG, Rajkumar SV, et al. New drugs and novel mechanisms of action in multiple myeloma in 2013: a report from the International Myeloma Working Group (IMWG). Leukemia. 2014;28(3):525–542. | |
Munshi NC, Anderson KC. New strategies in the treatment of multiple myeloma. Clin Cancer Res. 2013;19(13):3337–3344. | |
Anderson KC. The 39th David A. Karnofsky Lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. J Clin Oncol. 2012;30(4):445–452. | |
Allegra A, Penna G, Alonci A, et al. Monoclonal antibodies: potential new therapeutic treatment against multiple myeloma. Eur J Haematol. 2013;90(6):441–468. | |
van de Donk NW, Kamps S, Mutis T, Lokhorst HM. Monoclonal antibody-based therapy as a new treatment strategy in multiple myeloma. Leukemia. 2012;26(2):199–213. | |
Tai YT, Anderson KC. Antibody-based therapies in multiple myeloma. Bone Marrow Res. 2011;2011:924058. | |
Richardson PG, Lonial S, Jakubowiak AJ, Harousseau JL, Anderson KC. Monoclonal antibodies in the treatment of multiple myeloma. Br J Haematol. 2011;21. doi: 10.1111/j.1365-2141.2011.08790.x | |
Zhan F, Huang Y, Colla S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–2028. | |
Kapoor P, Greipp PT, Morice WG, Rajkumar SV, Witzig TE, Greipp PR. Anti-CD20 monoclonal antibody therapy in multiple myeloma. Br J Haematol. 2008;141(2):135–148. | |
Moreau P, Voillat L, Benboukher L, et al; IFM group. Rituximab in CD20 positive multiple myeloma. Leukemia. 2007;21(4):835–836. | |
Hsi ED, Steinle R, Balasa B, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res. 2008;14(9):2775–2784. | |
Tai YT, Dillon M, Song W, et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood. 2008;112(4):1329–1337. | |
van Rhee F, Szmania SM, Dillon M, et al. Combinatorial efficacy of anti-CS1 monoclonal antibody elotuzumab (HuLuc63) and bortezomib against multiple myeloma. Mol Cancer Ther. 2009;8(9):2616–2624. | |
Zonder JA, Mohrbacher AF, Singhal S, et al. A Phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood. 2012;120(3):552–559. | |
Jakubowiak AJ, Benson DM, Bensinger W, et al. Phase I trial of anti-CS1 monoclonal antibody elotuzumab in combination with bortezomib in the treatment of relapsed/refractory multiple myeloma. J Clin Oncol. 2012;30(16):1960–1965. | |
Lonial S, Vij R, Harousseau JL, et al. Elotuzumab in combination with lenalidomide and low-dose dexamethasone in relapsed or refractory multiple myeloma. J Clin Oncol. 2012;30(16):1953–1959. | |
Richardson PG, Jagannath S, Moreau P, et al. A Phase 2 study of elotuzumab (elo) in combination with lenalidomide and low-dose dexamethasone (ld) in patients (pts) with relapsed/refractory multiple myeloma (r/r mm): updated results. Blood (ASH Annual Meeting Abstracts). 2012;120: Abstract 202. | |
Lonial S, Jagannath S, Moreau P, et al. Phase (Ph) I/II study of elotuzumab (Elo) plus lenalidomide/dexamethasone (Len/dex) in relapsed/refractory multiple myeloma (RR MM): Updated Ph II results and Ph I/II long-term safety. J Clin Oncol. 2013;31:15(Suppl): Abstract 8542. | |
Lonial S, Kaufman J, Laubach J, Richardson P. Elotuzumab: a novel anti-CS1 monoclonal antibody for the treatment of multiple myeloma. Expert Opin Biol Ther. 2013;13(12):1731–1740. | |
Bouchon A, Cella M, Grierson HL, Cohen JI, Colonna M. Activation of NK cell-mediated cytotoxicity by a SAP-independent receptor of the CD2 family. J Immunol. 2001;167(10):5517–5521. | |
Kumaresan PR, Lai WC, Chuang SS, Bennett M, Mathew PA. CS1, a novel member of the CD2 family, is homophilic and regulates NK cell function. Mol Immunol. 2002;39(1–2):1–8. | |
Cannons JL, Tangye SG, Schwartzberg PL. SLAM family receptors and SAP adaptors in immunity. Annu Rev Immunol. 2011;29:665–705. | |
Schwartzberg PL, Mueller KL, Qi H, Cannons JL. SLAM receptors and SAP influence lymphocyte interactions, development and function. Nat Rev Immunol. 2009;9(1):39–46. | |
Veillette A, Guo H. CS1, a SLAM family receptor involved in immune regulation, is a therapeutic target in multiple myeloma. Crit Rev Oncol Hematol. 2013;88(1):168–177. | |
Cruz-Munoz ME, Dong Z, Shi X, Zhang S, Veillette A. Influence of CRACC, a SLAM family receptor coupled to the adaptor EAT-2, on natural killer cell function. Nat Immunol. 2009;10(3):297–305. | |
Tassi I, Colonna M. The cytotoxicity receptor CRACC (CS-1) recruits EAT-2 and activates the PI3K and phospholipase Cgamma signaling pathways in human NK cells. J Immunol. 2005;175(12):7996–8002. | |
Lee JK, Mathew SO, Vaidya SV, Kumaresan PR, Mathew PA. CS1 (CRACC, CD319) induces proliferation and autocrine cytokine expression on human B lymphocytes. J Immunol. 2007;179(7):4672–4678. | |
Stark S, Watzl C. 2B4 (CD244), NTB-A and CRACC (CS1) stimulate cytotoxicity but no proliferation in human NK cells. Int Immunol. 2006;18(2):241–247. | |
De Salort J, Sintes J, Llinàs L, Matesanz-Isabel J, Engel P. Expression of SLAM (CD150) cell-surface receptors on human B-cell subsets: from pro-B to plasma cells. Immunol Lett. 2011;134(2):129–136. | |
Bae J, Song W, Smith R, et al. A novel immunogenic CS1-specific peptide inducing antigen-specific cytotoxic T lymphocytes targeting multiple myeloma. Br J Haematol. 2012;157(6):687–701. | |
Tai YT, Soydan E, Song W, et al. CS1 promotes multiple myeloma cell adhesion, clonogenic growth, and tumorigenicity via c-maf-mediated interactions with bone marrow stromal cells. Blood. 2009;113(18):4309–4318. | |
Rice A, Dillon M, van Abbema A, et al. Eradication of tumors in pre-clinical models of multiple myeloma by anti-cs1 monoclonal antibody HuLuc63: mechanism of action studies. Blood (ASH Annual Meeting Abstracts). 2006;108: Abstract 3503. | |
Collins SM, Bakan CE, Swartzel GD, et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother. 2013;62(12):1841–1849. | |
Godfrey J, Benson DM. The role of natural killer cells in immunity against multiple myeloma. Leuk Lymphoma. 2012;53(9):1666–1676. | |
Garg TK, Szmania SM, Khan JA, et al. Highly activated and expanded natural killer cells for multiple myeloma immunotherapy. Haematologica. 2012;97(9):1348–1356. | |
Szmania S, Garg TK, Lapteva N, et al. Fresh ex vivo expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma (mm) patients. Blood (ASH Annual Meeting Abstracts). 2012;120: Abstract 579. | |
Garg TK, Szmania S, Shi J, et al. Ex vivo activated natural killer (NK) cells from myeloma patients kill autologous myeloma and killing is enhanced by elotuzumab. Blood (ASH Annual Meeting Abstracts). 2008;112: Abstract 3666. | |
Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352(24):2487–2498. | |
Weber DM, Chen C, Niesvizky R, et al; Multiple Myeloma (009) Study Investigators. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357(21):2133–2142. | |
Dimopoulos M, Spencer A, Attal M, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;22;357(21):2123–2132. | |
Neyer L, Ding H, Chen D, et al. Effect of elotuzumab on circulating lymphocytes, chemokines, and cytokines in multiple myeloma patients. Blood (ASH Annual Meeting Abstracts). 2010;116: Abstract 4070. | |
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. | |
Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. | |
Chu J, Deng Y, Benson DM, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917–927. | |
Bae J, Smith R, Daley J, et al. Myeloma-specific multiple peptides able to generate cytotoxic T lymphocytes: a potential therapeutic application in multiple myeloma and other plasma cell disorders. Clin Cancer Res. 2012;18(17):4850–4860. | |
Mateos MV, Hernández MT, Giraldo P, et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N Engl J Med. 2013;369(5):438–447. | |
de Weers M, Tai YT, van der Veer MS, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186(3):1840–1848. | |
Plesner T, Lokhorst H, Gimsing P, Nahi H, Lisby S, Richardson PG. Daratumumab, a CD38 monoclonal antibody in patients with multiple myeloma – data from a dose-escalation Phase I/II study. Blood (ASH Annual Meeting Abstracts). 2012;120: Abstract 73. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.