")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 11
Profile of dupilumab and its potential in the treatment of inadequately controlled moderate-to-severe atopic dermatitis
Authors Awosika O, Kim L, Mazhar M , Rengifo-Pardo M, Ehrlich A
Received 16 September 2017
Accepted for publication 8 November 2017
Published 24 January 2018 Volume 2018:11 Pages 41—49
DOI https://doi.org/10.2147/CCID.S123329
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Olabola Awosika,1 Lori Kim,2 Momina Mazhar,2 Monica Rengifo-Pardo,1,2 Alison Ehrlich1,2
1Department of Dermatology, The George Washington Medical Faculty Associates, Washington, DC, USA; 2George Washington University School of Medicine & Health Sciences, Washington, DC, USA
Abstract: Atopic dermatitis (AD) is a common inflammatory skin disorder that manifests as eczematous lesions, often associated with allergic rhinitis and asthma. Historically, moderate-to-severe disease has been managed with systemic immunosuppression, such as oral corticosteroids, which result in relapse and limiting side effects. Due to recent advancements in the identification of interleukin (IL)-4 and IL-13 as key mediators in AD, new biological agents have been developed for treatment. Dupilumab is a recently approved monoclonal antibody that targets the alpha subunit of the IL-4 receptor and, thus, downregulates activity of IL-4 and IL-13. This review discusses the profile of dupilumab and its potential for efficacy and safety in treating moderate-to-severe AD by reviewing data from Phase I–III clinical trials. Results suggest that dupilumab shows great therapeutic promise for AD. Further studies investigating extended use of dupilumab and dupilumab in comparison to other agents are needed to establish long-term efficacy and safety.
Keywords: atopic dermatitis, dupilumab, IL-4, IL-13, biologics
Background
Atopic dermatitis (AD) is a chronic inflammatory skin disease characterized by erythema, pruritus, and scaling of skin. AD is clinically diagnosed by these essential features, and the diagnosis is supported by early age of onset, personal or family history of atopy, and xerosis.1 AD has a complex, heterogeneous etiology that includes barrier defects, intrinsic immunological hyperactivity, and extrinsic triggers.2,3 According to a recent study of lifetime worldwide AD prevalence over the period of 1990–2010, there is no overall global trend in the prevalence of AD.4 However, a steady increase in AD prevalence in developing countries has been observed. In general, AD is estimated to affect up to 20% of children in developed countries and 3% of adults worldwide, with up to 50% of pediatric AD cases persisting into adulthood.5,6 In the USA, 10.7% of children and 3.2% of adults are living with AD.7,8
For patients with moderate-to-severe AD, skin lesions encompassing large surface areas are often associated with severe itching. These lesions can cause sleep disturbances and, in turn, symptoms of anxiety, depression, and poor quality of life.9 Topical steroids, topical immunomodulators, and phototherapy are often inadequate in providing sustained improvement in these patients, despite the additive benefit of topicals in decreasing inflammation and restoring epidermal barrier function.2 For patients with poor response to these topicals, the mainstay of treatment is systemic immunosuppression, including oral corticosteroids, cyclosporine, or mycophenolate mofetil. Though these medications may provide moderate relief in AD, the risks often outweigh the benefits of long-term use. In particular, these oral agents are associated with significant side effects such as increased infections, nausea/vomiting, hypertension, and headaches.10,11 In a 10-year chart review of the use of oral immunosuppressive drugs in patients with severe AD, frequent reasons for discontinuation of these therapies included ineffectiveness and adverse events, such as neurological symptoms, gastrointestinal upset, and fatigue/flu-like symptoms.11 Furthermore, for patients with renal impairment and uncontrolled hypertension, oral corticosteroids, mycophenolate mofetil, and cyclosporine are less viable treatment options due to their high risk to exacerbate the underlying comorbidities.
Given the poor therapeutic responses, inconveniences, and therapy-limiting side effects of conventional systemic AD therapeutics, there is a substantial unmet need for more efficacious and promising agents for moderate-to-severe AD with minimal adverse effects. With recent advances in the understanding of the pathological mechanisms of AD, new biological agents have been developed and are being evaluated in clinical trials. In March 2017, dupilumab became the first biologic to be approved by the US Food and Drug Administration for inadequately controlled moderate-to-severe AD.12 The drug received this approval because of its significant demonstration of efficacy and safety in treating AD in three randomized Phase III pivotal trials. The purpose of this article is to review the findings on dupilumab in clinical trials and examine the potential of the drug for inadequately controlled moderate-to-severe AD.
Methods
Relevant articles on disease activity in AD and clinical trials of dupilumab were searched and selected from the databases of PubMed and ClinicalTrials.gov using the following terms: dupilumab, AD, disease activity, interleukin (IL)-4, IL-13, thymus and activation-regulated chemokine (TARC), eotaxin 3 (CCL26), comorbidity, quality of life, efficacy, biologic, and adverse effects. Additional publications were collected from references identified in articles and related citations in PubMed. As of June 2017, Phase I, II, and III clinical trials of dupilumab have been published. In total, 31 relevant papers were reviewed and referenced.
Results
Pathogenesis and disease activity in AD
Ongoing debates regarding the pathogenesis of AD have led to the following description of two forms of the disease: 1) the intrinsic form resulting from abnormal epidermal structure and function, and 2) the extrinsic form due to cutaneous inflammation caused by immune responses to extrinsic antigens.13,14 Moreover, there has been identification of increased activity of T lymphocytes secreting effector cytokines and dysregulation of both cell-mediated immune responses and inflammatory cascades in AD skin.13 Yet, over the past several years, attention has been turned specifically toward type 2 inflammatory mediators as targets for AD therapy.
In particular, IL-4 and IL-13, cytokines from T helper type 2 (Th2) cells, are key mediators of the inflammation in AD. These cytokines upregulate the expression of chemokines such as TARC and CCL26.10,15–18 This increased expression of TARC and CCL26 relates to increased disease activity, including the selective migration of Th2 lymphocytes and eosinophils into AD lesions, which cause inflammation.15–18 Additionally, in synergy with Toll-like receptor 2 ligands, IL-4 potentiates the chronicity of AD through IL-4-mediated suppression of IL-10.19 IL-4 and IL-13 also decrease the keratinocyte expression of barrier proteins, such as filaggrin, involucrin, and loricrin, triggering epidermal hyperplasia.20,21 Additional effects of these cytokines on keratinocytes include the suppression of keratinocyte differentiation and the synthesis of antimicrobial peptides and lipids.13,22,23 This activity further disrupts the skin barrier through aberrant expression of human β-defensins (hBD-2 and hBD-3) and cathelicidin (LL-37) leading to an increase in the susceptibility of lesional skin to infection by microorganisms, such as Staphylococcus aureus.13,24,25 Moreover, IL-4 and IL-13 induce B-cell differentiation leading to immunoglobulin E (IgE) class switching.26 This phenomenon explains the elevation of IgE levels in AD, which is often associated with the extrinsic form of the disease.10,27,28 Given that both IL-4 and IL-13 act through a common receptor (IL-4 receptor alpha; IL-4Rα), IL-4Rα has become a target of interest in treating AD. Inhibition of IL-4Rα has resulted in normalization of skin gene expression from lesional toward nonlesional skin, reduction of TARC expression, and efficacy in the suppression of itch.10,28–30
Dupilumab: mechanism of action
Dupilumab is a fully human monoclonal antibody that binds to the IL-4Rα, resulting in inhibition of both IL-4 and IL-13 signaling.10,12 This blockade by dupilumab reduces the type 2 helper T-cell-mediated inflammation cascade in AD. Specifically, competitive inhibition at the IL-4Rα inhibits activation of the signal transducer and activator of transcription 6 (STAT6)/Janus kinase 1 (JAK1) signaling cascade (Figure 1).31,32 Overexpression of STAT6 has been demonstrated to decrease epidermal differentiation complex genes, such as the genes for loricrin and involucrin, and enhance penetration of pathogens across the skin barrier leading to AD-like skin disease in mice models.20,33
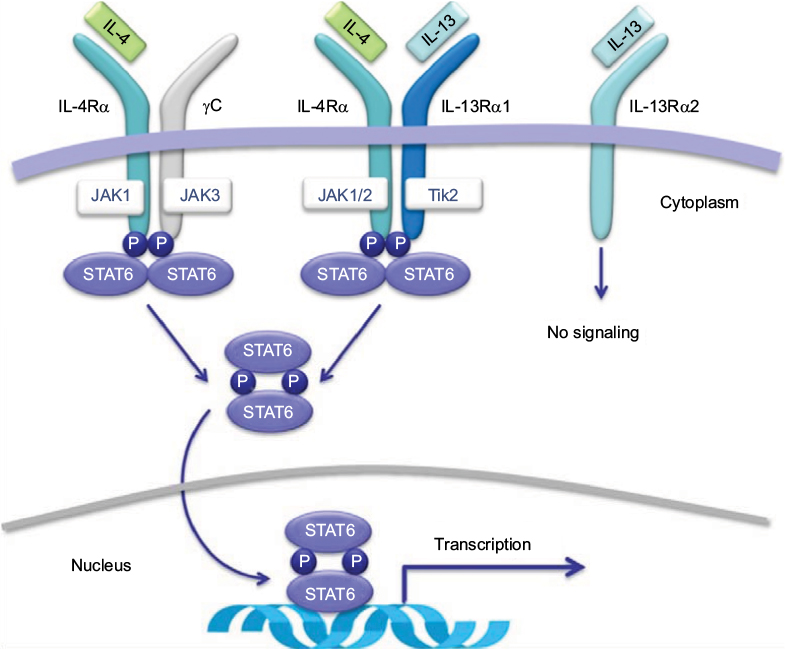 | Figure 1 Receptor signaling for IL-4 and IL-13. Notes: In hematopoietic cells, binding of IL-4 to type I IL-4Rα induces heterodimerization with γC, which activates JAK kinases and leads to the phosphorylation of STAT6. Similarly, in nonhematopoietic cells, such as keratinocytes, hair follicles, and epithelial/smooth muscle cells, STAT6 is phosphorylated by the induction of the heterodimerization of type II IL-4Rα and IL-13Rα1 after binding of the IL-4 or IL-13 to their respective receptors. Of note, IL-13 may bind to IL-13Rα2; however, this receptor lacks a signaling motif. Dupilumab binds the IL-4R subunit of both type I and type II IL-4 receptors leading to inhibition of the JAK/STAT signaling cascade. Source: Copyright 2014. Dove Medical Press. Reproduced from Vatrella A, Fabozzi I, Calabrese C, Maselli R, Pelaia G. Dupilumab: a novel treatment for asthma. J Asthma Allergy. 2014;7:123–130.32 Abbreviations: IL, interleukin; IL-4Rα, IL-4 receptor alpha subunit; IL-13α1, IL-13 receptor alpha 1 subunit; IL-13α2, IL-13 receptor alpha 2 subunit; JAK, Janus kinase; STAT, signal transducer and activator of transcription. |
Dupilumab: Phase I trials
In two 4-week randomized, double-blind, placebo-controlled, dose-increasing Phase I trials, dupilumab was evaluated as a monotherapy for moderate-to-severe AD in adults.28,34 In the M4A trial, 30 subjects received 75, 150, or 300 mg of subcutaneous dupilumab or placebo weekly for 4 weeks. The subjects were randomly assigned to receive placebo or dupilumab in a 1:4 ratio. In the M4B trial, 37 subjects were studied with 150 or 300 mg of subcutaneous dupilumab or placebo weekly for 4 weeks. These subjects were also randomized to receive placebo or dupilumab, but in a 1:3 ratio. Both the M4A and M4B trials were designed to assess safety as the primary end point. From composite analysis of both studies, by day 29, 59% of patients receiving dupilumab showed 50% reduction in the Eczema Area and Severity Index (EASI) score (EASI-50) compared to 19% of the placebo group (Table 1). In addition, significant improvements in Investigator Global Assessment (IGA) scores and pruritus scores for all dupilumab doses combined were observed in both studies. Concerning safety data, nasopharyngitis and headache were the most common side effects with no evidence of serious adverse events in either trial.28,34
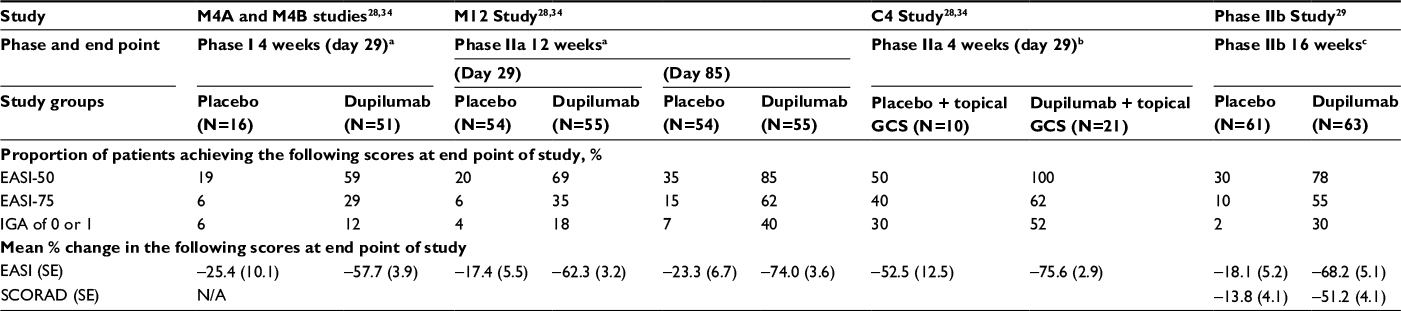 | Table 1 Clinical efficacy and safety in Phase I–II trials Notes: aSubjects were adults older than 18 years with moderate-to-severe AD as defined by an IGA score of ≥3, a baseline EASI score of ≥12, and duration of disease of ≥3 years. Dupilumab refers to subjects receiving all doses. bSubjects were adults older than 18 years with moderate-to-severe AD as defined by an IGA score of ≥3, a baseline EASI score of ≥16, and duration of disease of ≥3 years. Dupilumab refers to subjects receiving all doses. cSubjects were adults older than 18 years with moderate-to-severe AD as defined by duration ≥3 years, screening EASI ≥12, baseline EASI ≥16, baseline IGA ≥3. Dupilumab refers to patients receiving the 300 mg dose every other week. All mean percent changes were calculated as percent change in least squares mean. Abbreviations: AD, atopic dermatitis; EASI, Eczema Area and Severity Index; EASI-50, proportion of patients at 50% reduction in EASI; EASI-75, proportion of patients at 75% reduction in EASI; GCS, glucocorticoids; IGA, Investigator’s Global Assessment; N/A, not assessed; SCORAD, Scoring Atopic Dermatitis; SE, standard error. |
In addition to clinical improvement and safety, gene expression profiles of lesional sites after 150 and 300 mg dupilumab shifted to a more nonlesional molecular phenotype within 4 weeks.34,35 Lesional skin showed overall improvement in transcriptome by 24% in the 150 mg dose group and 49% in 300 mg dose group compared to 21% in the placebo group.28,34 Notably, markers of epidermal proliferation (K16 and K6B, which reduced epidermal hyperplasia) were downregulated by dupilumab.34,35 T-cell markers and other inflammatory mediators, such as chemokines, were also downregulated by dupilumab. Dupilumab also upregulated genes related with skin barrier functions such as structure-related gene, MATN4, lipid-metabolism-related genes, PLIN4, ADIPOQ, and PLIN1, and barrier-related function genes, CLDN8, ELN, and CLDN11.35
Dupilumab: Phase IIa trials
In a 12-week-long monotherapy study, M12, dupilumab demonstrated significant improvement in reduction of involvement and severity of AD in adults with moderate-to-severe disease. In the M12 study, subjects were randomized to receive subcutaneous 300 mg dupilumab (n=55) or placebo (n=54) weekly for 12 weeks. Dupilumab resulted in substantial improvement in multiple clinical measures in EASI-50, EASI-75, and IGA scores (Table 1), and pruritus numerical rating scale (NRS) score by day 85. Of note, 85% of patients on dupilumab achieved EASI 50 compared to 35% in the placebo group. Overall, pruritus NRS scores decreased by 56% in the dupilumab group compared to 15% in the placebo group. Serum TARC and serum IgE levels also decreased significantly more in dupilumab patients.28,34
In a 4-week-long randomized, double-blind, parallel-group, placebo-controlled clinical trial (C4), dupilumab was also evaluated in combination with topical corticosteroids (TCSs). Subjects were randomized in a 2:1 ratio to receive subcutaneous 300 mg dupilumab (n=21) or placebo (n=10) weekly for 4 weeks in combination with a regimen of TCSs. The primary end points were the occurrence and severity of adverse events. All subjects in the dupilumab plus TCS group achieved EASI-50 by 4 weeks, while only 50% in the placebo plus TCS group achieved the same (Table 1).28 Again, the dupilumab group showed significant improvement in pruritus NRS and IGA scores compared to the placebo group. Moreover, the dupilumab group used 50% less TCSs in comparison to the placebo group. There was also a greater decline of TARC and IgE levels in the dupilumab group.28,34 In the combined safety data for studies of M4A, M4B, M12, and C4, adverse events occurred at similar rates in the dupilumab and placebo groups.34
Dupilumab: Phase IIb trial
In an international, multicenter, randomized, double-blind, dose-ranging, placebo-controlled clinical trial, the efficacy and safety of five different dosings of dupilumab were evaluated over a period of 16 weeks. Subjects were randomly assigned to receive subcutaneous placebo (n=61) or dupilumab 100 mg (n=65) monthly, 300 mg (n=65) monthly, 200 mg (n=61) every 2 weeks, 300 mg (n=64) biweekly, and 300 mg (n=63) weekly. By week 16, dupilumab showed improved EASI scores and resulted in significant improvement in SCORAD (Scoring Atopic Dermatitis) scores in a dose-dependent manner (Table 1). All of the 300 mg dupilumab dose regimens resulted in more than 3 points decrease in pruritus NRS scores in 37%–54% of subjects versus 8% of subjects in the placebo group. Furthermore, dupilumab resulted in early and sustained improvement in depression, anxiety, and quality-of-life scores. Mean percentage changes in TARC at week 16 correlated with clinical outcomes such as EASI, SCORAD, and IGA scores.29
Dupilumab: Phase III trials
In 2016, two identically designed Phase III trials of dupilumab were carried out for subjects with moderate-to-severe AD. Subjects were randomly assigned in a 1:1:1 ratio to receive, subcutaneous 300 mg dupilumab or placebo weekly or the same dose of dupilumab every other week alternating with placebo for 16 weeks. The primary outcome was the proportion of subjects who had both a score of 0 or 1 (clear or almost clear) on IGA and a reduction of 2 points or more in that score from baseline at week 16. Over 600 patients participated in each trial, with 671 subjects for SOLO 1 and 708 subjects for SOLO 2 randomized to receive dupilumab or placebo. In SOLO 1, the primary outcome point was achieved by 38% of patients receiving dupilumab every other week, 37% of those receiving dupilumab weekly, and 10% of subjects who received placebo (Table 2). SOLO 2 demonstrated comparable results, with 36% of patients in both dupilumab groups and 8% of the placebo group reaching the primary outcome point. Additionally, those in the placebo group received more rescue treatment than those in the dupilumab groups.30
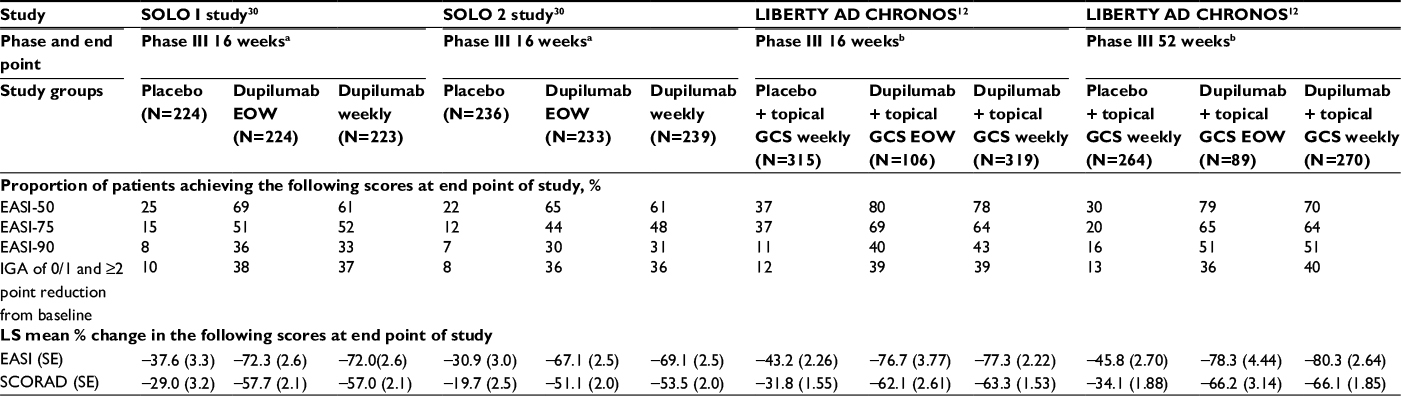 | Table 2 Clinical efficacy and safety in Phase III trials Notes: aSubjects were adults older than 18 years with moderate-to-severe AD as defined by an IGA score of ≥3, a baseline EASI score of ≥16, baseline BSA of ≥10%, and duration of disease of ≥3 years. Dupilumab refers to the 300 mg dose. bSubjects were adults older than 18 years with moderate-to-severe AD as defined by duration ≥3 years, screening EASI ≥12, baseline EASI ≥16, baseline IGA ≥3. Dupilumab refers to 300 mg dose. Abbreviations: AD, atopic dermatitis; EASI, Eczema Area and Severity Index; EASI-50, 50% reduction in EASI; EASI-75, 75% reduction in EASI; EASI-90, 90% reduction in EASI; EOW, every other week; GCS, glucocorticoids; IGA, Investigator’s Global Assessment; LS mean, least squares mean; SCORAD, Scoring Atopic Dermatitis; SE, standard error. |
In both trials, dupilumab significantly decreased patient-reported symptoms of AD, with improvement in sleep, anxiety, depression, and, therefore, quality of life of subjects. In Dermatology Life Quality Index (DLQI) and Patient-Oriented Eczema Measure (POEM) scores, dupilumab groups demonstrated twice as much improvement compared to placebo groups. At week 16, among the subjects who had Hospital Anxiety and Depression Scale (HADS)-Anxiety or HADS-Depression scores ≥8 at baseline, significantly more dupilumab-treated subjects had HADS scores of <8 compared to the placebo group.30
In a 1-year-long randomized, double-blinded, placebo-controlled clinical trial (LIBERTY AD CHRONOS), dupilumab was evaluated as concomitant therapy with TCSs in adults with moderate-to-severe AD and inadequate response to TCSs alone.12 Subjects were randomized in a 3:1:3 ratio to receive subcutaneous 300 mg dupilumab weekly (n=319), 300 mg dupilumab every 2 weeks (n=106), or placebo (n=315), with all three groups receiving concomitant TCSs with or without topical calcineurin inhibitors (TCI) tapered, discontinued, or restarted on the basis of disease activity. By week 16, 39% of patients in each dupilumab group achieved the coprimary endpoint of IGA 0/1 compared to 12% in the placebo group (Table 2). The other coprimary endpoint of 75% reduction in EASI score (EASI-75) was achieved by 64% of the weekly dupilumab group, 69% of the dupilumab every other week group, and 23% of the placebo group. Overall, patients receiving dupilumab had more days free of TCSs/TCI and/or systemic rescue medication use than those in the placebo group at 16 weeks and 52 weeks. Additionally, corresponding SCORAD, POEM, HADS, and DLQI scores were significantly reduced in the dupilumab groups compared to the placebo group.12 Improvement of NRS and DLQI scores in all three Phase III trials for dupilumab patients is demonstrated in Figures 2 and 3.12,30
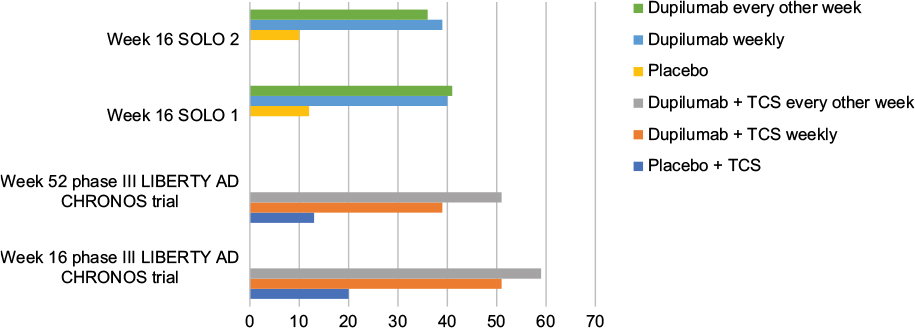 | Figure 2 Percentage of patients who achieved peak pruritus numerical rating scale score improvement ≥4 points from baseline in Phase III trials. Abbreviations: AD, atopic dermatitis; TCS, topical corticosteroid. |
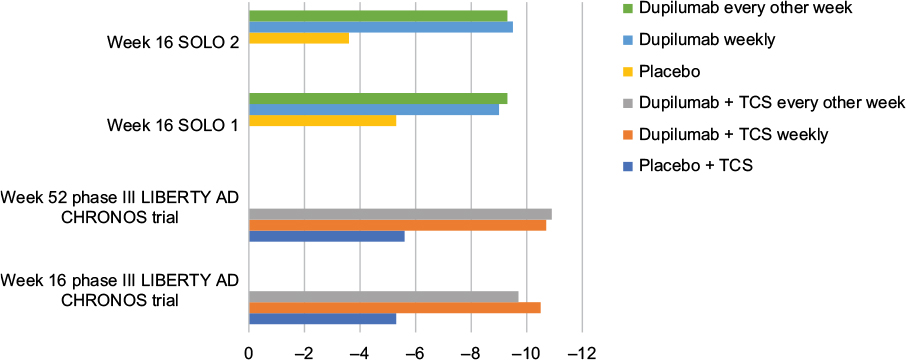 | Figure 3 Least squares mean change in Dermatology Life Quality Index score from baseline in Phase III trials. Abbreviations: AD, atopic dermatitis; TCS, topical corticosteroid. |
Discussion
Literature concerning the impact of dupilumab in the pathogenesis of AD includes Phase I, II, and III clinical trials. Results from these trials show that dupilumab improves clinical symptoms of moderate-to-severe AD and decreases T-cell markers, markers of epidermal proliferation, and inflammatory mediators and chemokines.28 In particular, compared to placebo and lower doses, the 300 mg dose of dupilumab demonstrated the greatest improvement in EASI and NRS scores, and transcriptome of lesional skin in Phase I and II studies.28,34 Moreover, the 300 mg dose every other week resulted in similar efficacy to the 300 mg weekly dose in achieving primary and secondary outcome measures in Phase III trials.12,30 Notably, the greatest percentage of patients achieved improvement in EASI or IGA when dupilumab was administered at 300 mg every other week with concomitant TCS use.12
Additionally, the frequency of adverse events was demonstrated to be similar between placebo and dupilumab groups, with the most commonly reported adverse events including headaches and nasopharyngitis in Phase I and II trials.28 In comparison, the most common adverse events reported in Phase III trials were exacerbations of AD (10%–18%), injection-site reactions (15%–19%), and nasopharyngitis (10%–23%), with conjunctivitis also occurring in 14% or more of patients on dupilumab in the 1-year-long Phase III trial.12,30 Of note, in Phase I and II trials dupilumab also demonstrated decreased total number of skin infections compared to placebo (4%–5% versus 10%–24%).28 Moreover, across the four Phase I and II trials, the rate of skin infections in the placebo groups was 0.2 per patient compared to 0.05 infections per patient in the dupilumab groups.28 This particular finding supports the concept that dupilumab improves epidermal barrier function.
In addition to its clinical efficacy, dupilumab also demonstrated improved quality of life as well, with significant reduction of DLQI and POEM scores.30 Overall, these results suggest that IL-4 and IL-13 are important mediators in the pathogenesis and morbidity of AD. However, additional trials over an extended period of time are necessary to establish a long-term safety and efficacy profile of dupilumab.
The recent recognition of AD as a predominantly Th2-mediated disease has led the way for the investigation of a variety of therapeutics that target specific inflammatory mediators involved in innate immunity. Multiple biologics are currently being investigated in clinical trials, including antibodies that specifically target IL-13, IL-17, IL-22, IL-31, and IL-12/IL-23p40.36 Topical and oral phosphodiesterase-4 inhibitors are also being investigated in Phase II and Phase III clinical trials, along with a JAK inhibitor and therapeutics targeting thymic stromal lymphopoietin and chemoattractant receptor-homologous molecule expressed on Th2 cells.36 These novel therapies have shown promising results. Notably, IL-31 inhibition has shown significant reduction of pruritus in patients with AD.37 Yet, despite these ongoing investigations into the use of multiple biologics for treatment of AD, dupilumab remains the first and only biologic to be approved for moderate-to-severe AD.
Prior to the development of dupilumab, treatments available for AD included topical and oral glucocorticoids, calcineurin inhibitors, cyclosporine, methotrexate, azathioprine, and mycophenolic acid precursors. Currently, there are no studies comparing dupilumab to other systemic treatments approved for AD. However, prior to the approval of dupilumab, a systematic review of 34 randomized control trials involving 1,653 patients compared the efficacy and safety of 12 systemic treatments using the Grading of Recommendations Assessment, Development, and Evaluation approach.38 Azathioprine and methotrexate were recommended as second- and third-line treatments, respectively, according to moderate-quality evidence.38 Cyclosporine A (CsA) received the strongest recommendation as a first-line treatment for short-term use in AD. Additionally, evidence based on four trials demonstrated that long-term treatment with CsA can be recommended for up to 1 year.38–42 However, comparison of associated risks of long-term use of CsA, such as nephrotoxicity and hypertension, to that of year-long use of dupilumab, such as nasopharyngitis and conjunctivitis, suggests that dupilumab is likely a safer long-term option.12,39–43 Yet, due to variations in trial designs, it is problematic to compare safety and efficacy data between studies of systemic treatments and dupilumab.29,38
Future evaluations should focus on comparing the efficacy and safety of dupilumab against available systemic treatments in head-to-head trials. In addition, further studies observing the long-term efficacy and safety of dupilumab are also needed. In particular, extended observation to assess immunogenicity is warranted. Specifically, an assessment of the development of neutralizing antibodies over long-term use may provide further insight into how immunogenicity may affect long-term efficacy of dupilumab. Overall, the administration of dupilumab every other week and its relative safety and efficacy offer a convenient and lower-risk alternative to currently available systemic treatments for moderate-to-severe AD.
Disclosure
Dr Alison Ehrlich is an investigator for Sun Pharmaceuticals, Abbvie, Pfizer, UCB Biopharma, Merck, Leo Pharma, Eli Lilly, and is a speaker for Eli Lilly, Celgene, and Abbvie. Dr Alison Ehrlich was also a former principal investigator for the SOLO 2 trial. Dr Olabola Awosika’s fellowship is funded by Janssen Biotech, Inc. The authors report no other conflicts of interest in this work.
References
Eichenfeld LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70(2):338–351. | ||
Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387(10023):1109–1122. | ||
Skabytska Y, Kaesler S, Volz T, Biedermann T. The role of innate immune signaling in the pathogenesis of atopic dermatitis and consequences for treatments. Semin Immunopathol. 2016;38(1):29–43. | ||
Deckers IA, McLean S, Linssen S, Mommers M, van Schayck CP, Sheikh A. Investigating international time trends in the incidence and prevalence of atopic eczema 1990–2010: a systematic review of epidemiological studies. PLoS One. 2012;7(7):e39803. | ||
Mei-Yen Yong A, Tay YK. Atopic dermatitis: racial and ethnic differences. Dermatol Clin. 2017;35(3):395–402. | ||
Margolis JS, Abuabara K, Bilker W, Hoffstad O, Margolis DJ. Persistence of mild to moderate atopic dermatitis. JAMA Dermatol. 2014;150(6):593–600. | ||
Silverberg JI, Hanifin JM. Adult eczema prevalence and associations with asthma and other health and demographic factors: A US population– based study. J Allergy Clin Immunol. 2013;132(5):1132–1138. | ||
Shaw TE, Currie GP, Koudelka CW, Simpson EL. Eczema prevalence in the United States: data from the 2003 National Survey of Children’s Health. J Invest Dermatol. 2011;131(1):67–73. | ||
Simpson EL, Beiber T, Eckert L, et al. Patient burden of moderate to severe atopic dermatitis (AD): Insights from a phase 2b clinical trial of dupilumab in adults. J Am Acad Dermatol. 2016;74(3):491–498. | ||
Gandhi NA, Pirozzi G, Graham NM. Commonality of the IL-4/IL-13 pathway in atopic disease. Exp Rev Clin Immunol. 2017;13(5):425–437. | ||
Garritsen FM, Roekevisch E, van der Schaft J, Deinum J, Spuls PI, de Bruin-Weller MS. Ten years’ experience with oral immunosuppressive treatment in adult patients with atopic dermatitis in two academic centres. J Eur Acad Dermatol Venereol. 2015;29(10):1905–1912. | ||
Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomized, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389(10086):2287–2303. | ||
Wang AX, Xu Landén N. New insights into T cells and their signature cytokines in atopic dermatitis. IUBMB Life. 2015;67(8):601–610. | ||
Dharmage SC, Lowe AJ, Matheson MC, Burgess JA, Allen KJ, Abramson MJ. Atopic dermatitis and the atopic march revisited. Allergy. 2014;69(1):17–27. | ||
Kakinuma T, Nakamura K, Wakugawa M, et al. Thymus and activation-regulated chemokine in atopic dermatitis: Serum thymus and activation-regulated chemokine level is closely related with disease activity. J Allergy Clin Immunol. 2001;107(3):535–541. | ||
Hijnen D, De Bruin-Weller M, Oosting B, et al. Serum thymus and activation-regulated chemokine (TARC) and cutaneous T cell– attracting chemokine (CTACK) levels in allergic diseases: TARC and CTACK are disease-specific markers for atopic dermatitis. J Allergy Clin Immunol. 2004;113(2):334–340. | ||
Kagami S, Kakinuma T, Saeki H, et al. Significant elevation of serum levels of eotaxin-3/CCL26, but not of eotaxin-2/CCL24, in patients with atopic dermatitis: serum eotaxin-3/CCL26 levels reflect the disease activity of atopic dermatitis. Clin Exp Immunol. 2003;134(2):309–313. | ||
Kagami S, Saeki H, Komine M, et al. Interleukin-4 and interleukin-13 enhance CCL26 production in a human keratinocyte cell line, HaCaT cells. Clin Exp Immunol. 2005;141(3):459–466. | ||
Kaesler S, Volz T, Skabytska Y, et al. Toll-like receptor 2 ligands promote chronic atopic dermatitis through IL-4-mediated suppression of IL-10. J Allergy Clin Immunol. 2014;134(1):92–99. | ||
Kim BE, Leung DY, Boguniewicz M, Howell MD. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol. 2008;126(3):332–337. | ||
Howell MD, Kim BE, Gao P, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2009;124(3 Suppl 2):R7–R12. | ||
Kamsteeg M, Bergers M, De Boer R, et al. Type 2 helper T-cell cytokines induce morphologic and molecular characteristics of atopic dermatitis in human skin equivalent. Am J Pathol. 2011;178(5):2091–2099. | ||
Sawada E, Yoshida N, Sugiura A, Imokawa G. Th1 cytokines accentuate but Th2 cytokines attenuate ceramide production in the stratum corneum of human epidermal equivalents: an implication for the disrupted barrier mechanism in atopic dermatitis. J Dermatol Sci. 2012;68(1):25–35. | ||
Park KY, Kim DH, Jeong MS, Li K, Seo SJ. Changes of antimicrobial peptides and transepidermal water loss after topical application of tacrolimus and ceramide-dominant emollient in patients with atopic dermatitis. J Korean Med Sci. 2010;25(5):766–771. | ||
Yamasaki K, Gallo RL. Antimicrobial peptides in human skin disease. Eur J Dermatol. 2008;18(1):11–21. | ||
Lebman DA, Coffman RL. Interleukin 4 causes isotype switching to IgE in T cell- stimulated clonal B cell cultures. J Exp Med. 1988; 168(3):853–862. | ||
Bieber T. Atopic dermatitis. N Engl J Med. 2008;358(14):1483–1494. | ||
Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371(2):130–139. | ||
Thaçi D, Simpson EL, Beck LA, et al. Efficacy and safety of dupilumab in adults with moderate- to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet. 2016;387(10013):40–52. | ||
Simpson EL, Bieber T1, Guttman-Yassky E, et al; SOLO 1 and SOLO 2 Investigators. Two Phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375(24):2335–2348. | ||
Bao L, Zhang H, Chan LS. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAKSTAT. 2013;2(3):e24137. | ||
Vatrella A, Fabozzi I, Calabrese C, Maselli R, Pelaia G. Dupilumab: a novel treatment for asthma. J Asthma Allergy. 2014;7:123–130. | ||
Sehra S, Yao Y, Howell MD, et al. IL-4 regulates skin homeostasis and the predisposition toward allergic skin inflammation. J Immunol. 2010;184(6):3186–3190. | ||
Hamilton JD, Ungar B, Guttman-Yassky E. Drug evaluation review: dupilumab in atopic dermatitis. Immunotherapy. 2015;7(10):1043–1058. | ||
Hamilton JD, Suárez-Fariñas M, Dhingra N, et al. Dupilumab improves the molecular signature in skin of patients with moderate-to-severe atopic dermatitis. J Allergy Clin Immunol. 2014;134(6):1293–1300. | ||
Heratizadeh A, Werfel T. Anti-inflammatory therapies in atopic dermatitis. Allergy. 2016;71(12):1666–1675. | ||
Ruzicka T, Hanifin JM, Furue M, et al. Anti–Interleukin-31 receptor a antibody for atopic dermatitis. N Engl J Med. 2017;376(9):826–835. | ||
Roekevisch E, Spuls PI, Kuester D, Limpens J, Schmitt J. Efficacy and safety of systemic treatments for moderate-to-severe atopic dermatitis: a systematic review. J Allergy Clin Immunol. 2014;133(2):429–438. | ||
Harper JI, Ahmed I, Barclay G, et al. Cyclosporine for severe childhood atopic dermatitis: short course versus continuous therapy. Br J Dermatol. 2000;142(1):52–58. | ||
Zonneveld IM, De Rie MA, Beljaards RC, et al. The long-term safety and efficacy of cyclosporin in severe refractory atopic dermatitis: a comparison of two dosage regimens. Br J Dermatol. 1996;135(Suppl 48):15–20. | ||
Granlund H, Erkko P, Remitz A, et al. Comparison of cyclosporin and UVAB phototherapy for intermittent one-year treatment of atopic dermatitis. Acta Derm Venereol. 2001;81(1):22–27. | ||
Haeck IM, Knol MJ, Ten Berge.O, van Velsen SG, de Bruin-Weller MS, Bruijnzeel-Koomen CA. Enteric-coated mycophenolate sodium versus cyclosporin A as long-term treatment in adult patients with severe atopic dermatitis: a randomized controlled trial. J Am Acad Dermatol. 2011;64(6):1074–1084. | ||
Schmitt J, Schakel K, Folster-Holst R, et al. Prednisolone vs. cyclosporine for severe adult eczema. An investigator-initiated double-blind placebo- controlled multicenter trial. Br J Dermatol. 2010;162(3):661–668. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.