")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Profile of deferasirox for the treatment of patients with non-transfusion-dependent thalassemia syndromes
Authors Ricchi P , Marsella M
Received 26 August 2015
Accepted for publication 25 November 2015
Published 16 December 2015 Volume 2015:9 Pages 6475—6482
DOI https://doi.org/10.2147/DDDT.S40694
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Paolo Ricchi,1 Maria Marsella1,2
1UOSD Malattie Rare del Globulo Rosso, Azienda Ospedaliera di Rilievo Nazionale “Antonio Cardarelli”, Naples, 2UOC Pediatria, Azienda Ospedaliera di Rilievo Nazionale G. Rummo, Benevento, Italy
Abstract: It has been clearly shown that iron overload adds progressively significant morbidity and mortality in patients with non-transfusion-dependent thalassemia (NTDT). The lack of physiological mechanisms to eliminate the excess of iron requires effective iron chelation therapy. The reduced compliance to deferoxamine and the risk of severe hematological adverse events during deferiprone treatment have limited the use of both these drugs to correct iron imbalance in NTDT. According to the principles of evidence-based medicine, following the demonstration of the effectiveness and the safety of deferasirox (Exjade®) in a prospective, randomized, controlled trial, deferasirox was approved by the US Food and Drug Administration in May 2013 for the treatment of iron overload associated with NTDT. This review, assessing the available scientific literature, will focus on the profile of DFX in the treatment of non-transfusional hemosiderosis in patients with NTDT.
Keywords: non-transfusion-dependent thalassemia, deferasirox, profile, iron overload
Introduction
The data presented are based on the retrieval of relevant medical literature by searching PubMed with the terms “deferasirox (DFX)” and “non-transfusion-dependent thalassemia (NTDT)” for studies published between 2000 and 2015.
The Cochrane database for systematic reviews and clinical trial registries was also searched, and pertinent reviews were identified (searches last updated August 1, 2015).
Thalassemia is a complex entity related to a group of inherited diseases, caused by defective or absent hemoglobin chain synthesis leading to anemia. In general, the severity of the disease depends on the genotype inherited without a definite genotype–phenotype correlation due to the presence of several genetic, along with environmental factors, which can alter clinical expression jointly to secondary and tertiary genetic modifiers.1 However, patients with NTDT do not require regular RBC transfusions for survival, but may require occasional transfusions owing to infection or pregnancy or may require more regular transfusions later in life due to splenomegaly or other complications.2 Therefore, NTDT encompasses a great variety of syndromes mixed in terms of their molecular background, clinical course, and severity, with the sole common characteristic of independence from regular transfusions.3 Currently, beta-thalassemia intermedia, alpha-thalassemia (mainly HbH disease), and mild/moderate forms of HbE/beta-thalassemia are the most prevalent forms in the world.4
Despite the lack of a stable transfusional iron overload, the majority of patients with NTDT accumulate iron. Ineffective and expanded erythropoiesis are both responsible for the activation of known and unknown signals of epcidin suppression and of a consequent increased intestinal absorption of iron.5 Thus, patients with NTDT progressively increase their iron stores which may become clinically significant in the second decade of life and is responsible, along with chronic anemia and hemolysis, for most complications observed in older untreated patients.6 However, it is conceivable that, particularly in the more severe forms, some complications could be ascribed to transfusion therapy (either intermittent or regular) as observed for the increased risk of endocrinopathy.7
Patients with NTDT accumulate iron chiefly in the liver and scantly in the heart, which may explain the tendency to not develop myocardial siderosis as compared to patients with thalassemia major (TM).8,9 However, the liver iron overload shown in patients with NTDT has been found to be similar to that of patients with beta TM.10 The elevated iron burden, despite occurring with differences in iron metabolism, pathophysiology and loading rate, is directly involved in the development of several complications or may add in some way to their severity.11 In fact, evaluating a series of unchelated patients by R2 and R2* magnetic resonance imaging (MRI), a liver iron concentration (LIC) of 5 or more mg/g dry weight (dw) was found to be the cut-off able to accurately discriminate between patients with and without morbidities.12 Recently, patients with NTDT were also found to have an increased risk of hepatocellular carcinoma and those affected showed a LIC of 8.5 mg/g dw (median: 8.5; interquartile range: 4.5–17.8).13
Chelation in NTDT
Obviously, chelation practice has been routinely performed in the management of iron overload in patients with NTDT, as their anemia contraindicates phlebotomy, but for a long time only as good clinical practice and without both the use of guidelines and the evidence of clinical benefit as observed in their TM counterparts. The optimal care study was the first retrospective study highlighting a protective effect of iron chelation therapy against several complications of NTDT such as pulmonary hypertension and endocrinopathies and thus reinforced the indication to accurately chelate this category of patients.7 On the other hand, it was demonstrated that an increase in serum ferritin level over time was associated with worsening of hepatic fibrosis in patients with NTDT who are not receiving iron chelation treatment.14 The evidence of these studies suggested to scrupulously estimate iron loading particularly at liver level to control and prevent such iron burden with effective iron chelation therapy.
Guidelines on NTDT chelation treatment have been recently established and recommend initiation of chelation therapy in patients with either ferritin levels higher than 800 ng/L or LIC above 5 mg/g dw.15 However, measurement of ferritin may underestimate the level of iron and sometimes among the main NTDT subtypes an irregular correlation exists between ferritin level and LIC, as measured by R2 MRI or superconducting quantum imaging device.16,17 Therefore, a decision-making algorithm has also been created to aid monitoring of iron burden and initiation of chelation therapy.18
Until 2005, the two drugs were available to treat iron overload in a variety of hematologic disorders: deferoxamine (DFO), administered mainly using continuous slow subcutaneous infusion, and the oral iron chelator deferiprone (DFP).19,20 The DFO, a hexadentate chelator binding iron at a 1:1 molar ratio, was introduced in clinical practice over 40 years ago to decrease iron overload.21 However, the need for frequent and prolonged subcutaneous administration has been associated with undesirable adverse effects and noncompliance to treatment resulting in elevated risk of iron-induced complications.22,23 The oral three times a day agent DFP, a bidentate hydroxypyridone with a small molecular weight, was first used and approved in Europe in 1999 for the treatment of iron overload in TM when DFO was contraindicated or inadequate. US Food and Drug Administration approval for clinical use of DFP was delayed until October 2011 because of the lack of traditional safety and efficacy trials comparing DFP and DFO. While DFO and DFP alone and in combination have been widely used in several trials involving patients with TM and proved to be effective in the reduction of iron burden,24–27 the scientific evidence of their efficacy in the setting of NTDT is very incomplete and limited to specific subpopulations.28 Recently, Calvaruso et al described the first 5-year long-term randomized clinical trial comparing the effectiveness of DFP vs DFO in patients with thalassemia intermedia (TI) showing that long-term iron chelation therapy with DFP is similarly effective as DFO. However, the use of DFP was accompanied by the occurrence of rare but serious adverse events during treatment, such as neutropenia and agranulocytosis.29
The once daily oral iron chelator, DFX (Exjade®, Novartis Pharmaceuticals Corporation, Basel, Switzerland), was introduced later, but has rapidly shown to be effective for reduction of body iron in iron-overloaded patients with transfusion-dependent anemias. It is a tridentate chelator that mobilizes iron stores by binding selectively to the ferric (Fe3+) form of iron.30,31 The efficacy of DFX has also been demonstrated in sickle-cell disease and in bone marrow failure syndromes, such as myelodysplastic and aplastic syndromes, Diamond–Blackfan and Fanconi anemia.32–34 In a Phase I/II trial with 49 patients with HF-related hereditary hemochromatosis, DFX also proved to be effective in reducing iron overload.35 In the past few years, several clinical trials have shown that accurate monitoring and dose adjustment of DFX was safe and effective in the long-term management of iron-overloaded patients with TM. Briefly, it has been assessed that DFX at a dose of 20 mg/kg per day can stabilize serum ferritin levels and LIC, while at doses of 30–40 mg/kg per day is able to reduce these parameters and achieve negative iron balance in patients with transfusional iron overload.36
The DFX profile in NTDT
Pharmacokinetic and pharmacodynamic properties of DFX have been mainly assessed among patients with TM and have been extensively reviewed elsewhere.37,38 However, studies from the myelodysplasia population seem to indicate that DFX has a constant pharmacological profile, independently from the underlying disease or race.39
The DFX is currently the only chelator to have gained approval for the treatment of iron overload in patients with NTDT in the USA (patients ≥10 years of age with an LIC ≥5 mg Fe/g dw and SF >300 ng/mL) and in Europe (LIC ≥5 mg Fe/g dw and SF >800 ng/mL with inadequate response or contraindication to DFO).40 To date three small studies and one large randomized trial have evaluated the safety and efficacy of DFX in patients with NTDT with iron overload (Table 1).
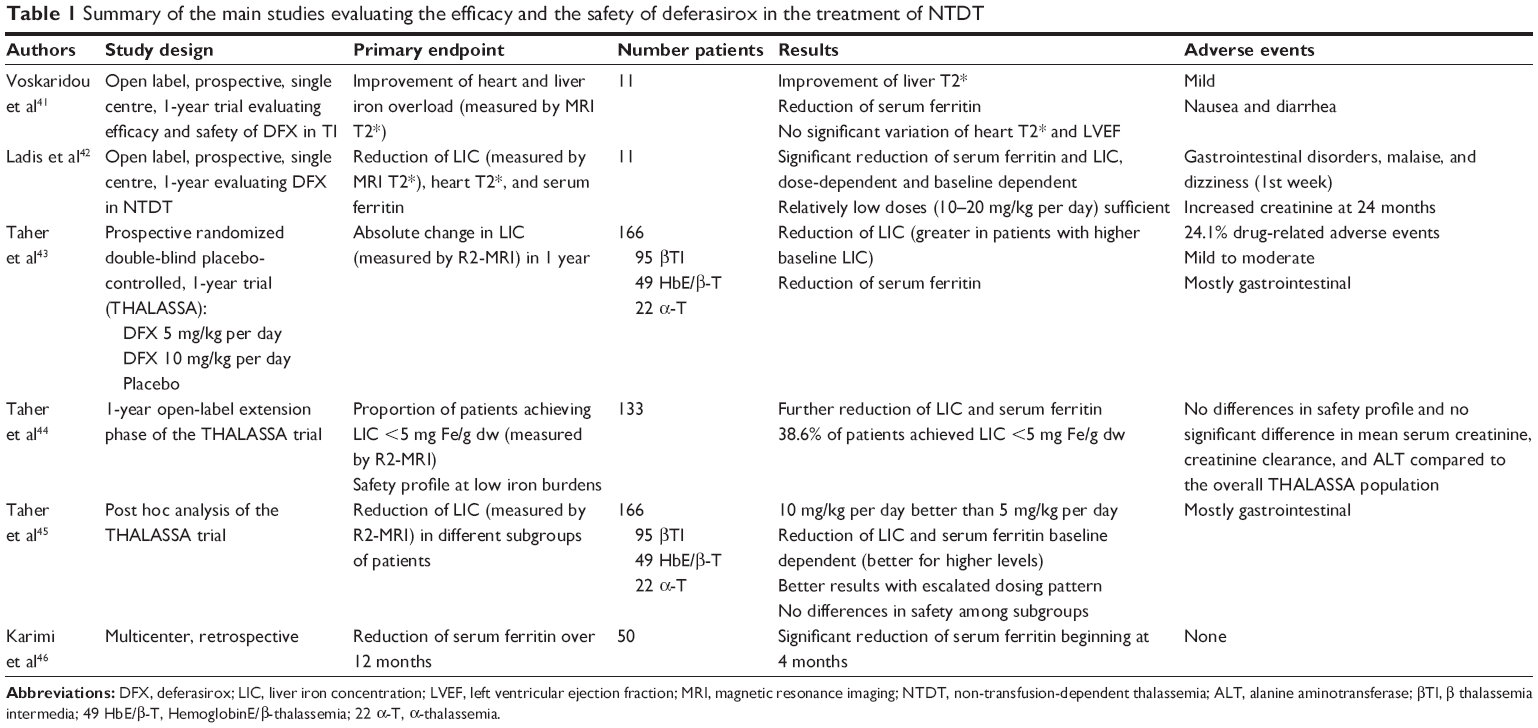 | Table 1 Summary of the main studies evaluating the efficacy and the safety of deferasirox in the treatment of NTDT |
In the prospective open label study by Voskaridou et al 11 patients received DFX at a starting dose of 10–20 mg/kg per day, according to the baseline iron burden, with subsequent dose adjustments according to efficacy and adverse events. After 1 year of treatment, significant improvement of mean liver T2* (P=0.02) was observed in the eight patients who completed the study.41
In the study by Ladis et al,42 eleven patients were enrolled to receive DFX at 10 or 20 mg/kg per day for 24 months. Mean LIC, measured by MRI-T2*, and mean serum ferritin were significantly reduced (P=0.005 and P<0.05, respectively) in the nine patients completing the study both at 12 and 24 months and five patients achieved LIC <3 mg Fe/g dw.
In both studies, cardiac T2* and left ventricular ejection fraction remained normal in all patients.41,42
THALASSA, a randomized, double-blind, placebo controlled phase II trial, is the largest study to investigate the safety and efficacy of DFX in reducing iron overload in patients with NTDT.43 The trial included 166 patients with β-thalassemia intermedia (n=95), α-thalassemia (n=22) and HbE/β-thalassemia (n=49), and was conducted over 1 year. Iron-overloaded patients (≥10 years of age with LIC ≥5 mg Fe/g dw and serum ferritin >300 ng/mL) were randomized to DFX starting doses of 5 or 10 mg/kg per day or matching placebo. After 1 year of treatment with DFX mean LIC decreased significantly from baseline in both starting dose groups (DFX 5 mg/kg per day: −2.33±0.70 mg Fe/g dw; P=0.001 and 10 mg/kg per day: −4.18±0.69 mg Fe/g dw; P<0.001) compared to placebo. The decrease in LIC, measured by R2-MRI, was more significant (P=0.009) for the DFX 10 mg/kg per day group compared with the lower dose group (−1.85±0.70 mg Fe/g dw). For both groups LIC reductions were observed from week 24. Similarly, serum ferritin decreased significantly with DFX from baseline to week 52 compared to placebo (least squares mean 121 ng/mL in 5 mg/kg per day group, −222 ng/mL in 10 mg/kg per day group, +115 ng/mL for placebo; P<0.001).
Patients who completed the THALASSA core study were eligible to enter the 1-year extension phase, where patients were continued on DFX or were switched from placebo.44 Overall, 133 patients entered the extension phase, including 48 who crossed over from placebo. The LIC and serum ferritin continued to decrease throughout the extension phase. Patients randomized to receive DFX in both phases (n=110) achieved a mean absolute reduction of LIC of 7.14 mg Fe/g dw from baseline and a mean decrease of serum ferritin of 450 ng/mL.
Patients who switched over from placebo achieved a mean absolute reduction of −6.66 mg Fe/g dw from baseline. At the end of the extension phase the proportion of patients achieving an LIC of <5 mg Fe/g was 38.6% overall (39.1% randomized to DFX in the core study; 37.5% originally randomized to placebo). Serum ferritin also continued to decrease over the extension study.
A subanalysis was performed evaluating LIC reduction in various subgroups, based on baseline LIC, baseline serum ferritin, age, sex, race, splenectomy status, and underlying NTDT syndrome.45 Across all subgroups, patients receiving DFX showed a greater reduction in LIC compared with patients who received placebo with better results in patients in the 10 mg/kg per day starting dose group.
DFX was in general well tolerated in all studies. The most frequently reported adverse events were mild to moderate in severity. In the THALASSA study adverse events assessed to be drug related by the investigators were reported in 24.1% of the patients and were mostly gastrointestinal disorders (mostly nausea and diarrhea), which resolved spontaneously or with dose adjustment or drug interruption. Eight patients discontinued the study because of adverse events. The safety profile in the extension phase and in the smaller studies was comparable.
As for renal function, no progressive increases in serum creatinine were observed. In patients treated for 2 years, five experienced two consecutive increases >33% above baseline and above upper limit of normal.43,44 No similar reports by Voskaridou et al41 and Ladis et al,42 although in the latter at the end of the 2nd year of treatment serum creatinine increased and creatinine clearance decreased compared to baseline, but neither reached abnormal values.
In all three studies mean ALT levels decreased over time, suggesting a corresponding improvement in liver function. Only one patient in the THALASSA extension study experienced hepatitis, which was suspected to be drug related.42
In general efficacy was achieved at relatively lower doses compared to patients with transfusional iron overload. However, monitoring for efficacy and adverse events with adequate dose adjustments remains pivotal in all patients.
The most recent study was a multicenter trial performed in Iran by Karimi et al who treated 50 β-thalassemia intermedia patients with serum ferritin >1,000 ng/mL with DFX for 12 months, observing a significant decrease of ferritin starting at 4 months of treatment.46
Conclusion
The recent definition of serum ferritin thresholds to predict clinically relevant LICs together with more and more wide access to MRI technology have led to the frequent diagnosis of iron overload in patients with NTDT, where initiation of chelation therapy is indicated. As a consequence, to control iron imbalance and to prevent iron toxicity, the long term use of a safe and effective chelator is required. An extensive experience gained from studies in patients with TM with over 150,000 patient years of drug exposure has definitively shown that DFX has a favorable side-effect profile in patients with transfusion-dependent thalassemia (TDT), with treatment-related adverse events comprising gastrointestinal, renal and dermatologic effects that were generally mild and reversible on cessation of treatment.47 In this review the restricted experience from the results on 238 patients with NTDT under continuous DFX treatment were evaluated. These studies involve small numbers of patients followed for 1–3 years who generally had never been transfused and encompasses two pilot studies, a prospective, placebo-randomized trial with its 2-year extension study and a retrospective study.41–46 Overall, DFX, at doses ranging from 10 to 20 mg and in a dose-dependent manner, was effective in reducing LIC and/or ferritin in patients with different subgroups of NTDT. Similarly to TDT, drug-related adverse events reported during these studies, were manageable, self-resolving, and typically did not necessitate discontinuing therapy, even as patients achieved low LIC levels toward the end of the study.48 Following monthly monitoring, incidences of liver and renal abnormalities were low and nonprogressive. Mild, nonprogressive increases in serum creatinine levels were also observed in a few patients; these increases were generally within the normal range and rarely exceeded twice the upper limit of normal.
Future perspectives and open questions
Taken together, these data suggest that the use of DFX can successfully manage iron overload in patients with NTDT. However, further data and larger study populations are needed to explore the safety profile of this drug in this group of patients (Table 2). Post-marketing surveillance studies and information from the ongoing THETIS trial will provide additional assessment of the long-term efficacy and safety of DFX.49 Given the need for lower doses as compared to TM, less toxicity and less morbidity are expected in NTDT than that observed when DFX was initially administered in patients with TM. Obviously, in most NTDT forms, lifelong treatment with iron chelation may not be necessary and several attempts at improving drug efficacy and reducing drug toxicity have been given particular attention, as the need to interrupt chelation if treatment goals are reached. In fact, a prudent and conservative approach has been adopted in the THALASSA trial in the sense that LIC <3 mg Fe/g dw was used as an indicator to interrupt iron chelation therapy in the hypothesis that it could represent a low, acceptable iron burden.43 In the described post hoc analysis, a consistent safety profile was nevertheless demonstrated as patients approached this target, indicating that such low iron burdens may be obtained with minimal risk of overchelation.48 This approach, now included in current guidelines for chelation therapy in NTDT, obviously will guarantee from the observed risk in TDT that overchelation could further reduce glomerular filtration rate also in NTDT.50 However, it should be taken into account that cases of glomerular hyperfiltration may be observed in patients with NTDT and that the administration of DFX could bring the glomerular filtration rate levels under control.51 Thus, it could be argued that occasionally a potential adverse event may become a useful strategy against the potential damage of persistent glomerular hyperfiltration.
 | Table 2 Essential points in the future evaluation of deferasirox in NTDT |
While a severe or >5 mg Fe/g dw LIC was clearly associated with complications, the level of “acceptable” iron burden and iron associated toxicity in NTDT may be different and transiently modifiable by occasional transfusional requirement. Currently, in line with the Thalassemia International Federation guidelines for DFX use in patients with NTDT, iron chelation should be stopped in patients reaching LIC of 3 mg Fe/g dw or SF of 300 ng/mL, when MRI is unavailable, as safety data do not exist to support continued chelation with DFX below this level.15 On the other hand, due to the permanent progressive iron accumulation, therapy should be restarted when patients re-achieve a LIC of 5 mg Fe/g dw thus realizing a gray zone (from 3 to 5 mg Fe/g dw), where patients are either interrupting treatment or waiting to restart it. It has also been observed that each genetic entity of NTDT has different erythroid activity, hepcidin levels and occasional transfusional iron loading, which may generate in some cases and in the presence of nearly normal LIC level, high levels of saturation of transferrin which in turn produces labile iron species and potential organ damage.52,53 Given that DFX has been shown not only to control iron burden but also labile plasma iron, the chelatable form of non-transferrin-bound iron, there could be the space to test DFX for a maintenance and or a transient therapy in such circumstances.54 As DFX remains within the therapeutic range over a 24-hour period, it offers a complete chelation coverage at standard doses and can therefore better control labile plasma iron.55 Further studies are needed to fully evaluate the efficacy and the safety of alternative administration schedules (ie, alternate days, three times/week) and/or reduced doses of DFX to control iron stores in this gray zone, where a maintenance chelation therapy could be acceptable. Further studies are also needed to better delineate the appropriate schedule of treatment of DFX to induce a neutral iron balance according to the increased risk of renal injury when a considered safe or normal iron burden has been reached. On the other hand, consideration should be given in the future also to better tailor initial DFX therapy to the basal iron depot, to different subtypes of NTDT, to splenectomy status and to the rate of occasional transfusional iron intake, all conditions which may affect iron balance in NTDT. Of particular interest, over the long-term use of DFX, will be also the prospective determination of the impact of iron chelation on most common complications and survival in patients with NTDT. The THETIS trial will focus on the effects of 5-year treatment with DFX on the endocrine profile. The favorable outcome of DFX on osteopenia and osteoporosis observed in patients with TM could be expected also in NTDT.56 Similarly, the reported long-term effects of DFX treatment on liver fibrosis should be also readdressed in NTDT.57
Currently, to improve anemia and reduce the occasional transfusional requirements, patients with NTDT are frequently treated with “older” drugs such hydroxycarbamide58 and new agents such as those interfering with the activity of several transforming growth factor-β family cytokines involved in late stages of erythropoiesis59,60 and JAK2 inhibitors.61 Thus, the efficacy, safety, and pharmacokinetics of DFX should also be evaluated in patients concomitantly receiving some of these drugs, taking into account that the potential interference of such drugs with mechanisms regulating the iron absorption, could further address and limit iron overload.
In conclusion, following the results of the THALASSA and extension studies, DFX has become the current gold-standard for iron chelation in patients with NTDT. However, prospective data from a randomized comparison with other chelating agents and from a magnitude of drug-exposure comparable to that obtained for patients with TM should be performed to obtain a more accurate and complete evaluation of its profile in patients with NTDT.
Disclosure
The authors report no conflicts of interest in this work.
References
Galanello R. Recent advances in the molecular understanding of non-transfusion-dependent thalassemia. Blood Rev. 2012;26S:S7–S1. | ||
Weatherall DJ. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl 1):S3–S6. | ||
Ricchi P, Filosa A, Maggio A, Fucharoen S. Non-transfusion-dependent thalassemia: a complex mix of genetic entities yet to be fully discovered. Biomed Res Int. 2015;2015:161434. | ||
Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusion-dependent thalassemias. Haematologica. 2013;98(6):833–844. | ||
Origa R, Galanello R, Ganz T, et al. Liver iron concentrations and urinary hepcidin in b-thalassemia. Haematologica. 2007;92:583–588. | ||
Taher AT, Musallam KM, El-Beshlawy A, et al. Age-related complications in treatment-naive patients with thalassaemia intermedia. Br J Haematol. 2010;150(4):486–489. | ||
Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the optimal care study. Blood. 2010;115(10):1886–1892. | ||
Origa R, Barella S, Argiolas GM, et al. No evidence of cardiac iron in 20 never-or minimally-transfused patients with thalassemia intermedia. Haematologica. 2008;93:1095–1096. | ||
Roghi A, Cappellini MD, Wood JC, et al. Absence of cardiac siderosis despite hepatic iron overload in Italian patients with thalassemia intermedia: an MRI T2* study. Ann Hematol. 2010;89:585–589. | ||
Taher AT, Musallam KM, Wood JC, Cappellini MD. Magnetic resonance evaluation of hepatic and myocardial iron deposition in transfusion-independent thalassemia intermedia compared to regularly transfused thalassemia major patients. Am J Hematol. 2010;85(4):288–290. | ||
Taher AT, Viprakasit V, Musallam KM, Cappellini MD. Treating iron overload in patients with non-transfusion-dependent thalassemia. Am J Hematol. 2013;88(5):409–415. | ||
Musallam KM, Cappellini MD, Wood JC, et al. Elevated liver iron concentration is a marker of increased morbidity in patients with b-thalassemia intermedia. Haematologica. 2011;96:1605–1612. | ||
Borgna-Pignatti C, Garani MC, Forni GL, et al. Hepatocellular carcinoma in thalassaemia: an update of the Italian Registry. Br J Haematol. 2014;167(1):121–126. | ||
Musallam KM, Motta I, Salvatori M, et al. Longitudinal changes in serum ferritin levels correlate with measures of hepatic stiffness in transfusion-independent patients with β-thalassemia intermedia. Blood Cells Mol Dis. 2012;49(3–4):136–139. | ||
Taher A, Vichinsky E, Musallam K, et al. Guidelines for the management of non transfusion dependent thalassaemia (NTDT). Thalassaemia International Federation. 2013. Available from: http://www.thalassaemia.org.cy/wp-content/uploads/eBook-publications/guidelines_for_the_management_of_non_transfusion_dependent_thalassaemia/#/0. Accessed March 14, 2014. | ||
Taher A, El Rassi F, Isma’eel H, et al. Correlation of liver iron concentration determined by R2 magnetic resonance imaging with serum ferritin in patients with thalassemia intermedia. Haematologica. 2008;93:1584–1586. | ||
Pakbaz Z, Fischer R, Fung E, et al. Serum ferritin underestimates liver iron concentration in transfusion independent thalassemia patients as compared to regularly transfused thalassemia and sickle cell patients. Pediatr Blood Cancer. 2007;49:329–332. | ||
Taher AT, Porter JB, Viprakasit V, et al. Defining serum ferritin thresholds to predict clinically relevant liver iron concentrations for guiding deferasirox therapy when MRI is unavailable in patients with non-transfusion-dependent thalassaemia. Br J Haematol. 2015;168(2):284–290. | ||
Novartis. Desferal (deferoxamine): prescribing information 2011. Available from: https://www.pharma.us.novartis.com/product/pi/pdf/desferal.pdf. Accessed July 27, 2015. | ||
ApoPharma Inc. Ferriprox (deferiprone): prescribing information and medication guide. 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000236/WC500022050.pdf. Accessed July 27, 2015. | ||
Ma Y, Zhou T, Kong X, Hider RC. Chelating agents for the treatment of systemic iron overload. Curr Med Chem. 2012;19:2816–2827. | ||
Delea TE, Sofrygin O, Thomas SK, Baladi JF, Phatak PD, Coates TD. Cost effectiveness of once-daily oral chelation therapy with deferasirox versus infusional deferoxamine in transfusion-dependent thalassaemia patients. US healthcare system perspective. Pharmacoeconomics. 2007;25:329–342. | ||
Delea TE, Edelsberg J, Sofrygin O, et al. Consequences and costs of noncompliance with iron chelation therapy in patients with transfusion-dependent thalassemia: a literature review. Transfusion. 2007;47:1919–1929. | ||
Daar S, Pathare AV. Combined therapy with desferrioxamine and deferiprone in beta thalassemia major patients with transfusional iron overload. Ann Hematol. 2006;85:315–319. | ||
Origa R, Bina P, Agus A. Combined therapy with deferiprone and desferrioxamine in thalassemia major. Haematologica. 2005;90:1309–1314. | ||
Ricchi P, Ammirabile M, Spasiano A, et al. Combined chelation therapy in thalassemia major with deferiprone and desferrioxamine: a retrospective study. Eur J Haematol. 2010;85:36–42. | ||
Pepe A, Meloni A, Rossi G, et al. Cardiac and hepatic iron and ejection fraction in thalassemia major: multicentre prospective comparison of combined deferiprone and deferoxamine therapy against deferiprone or deferoxamine monotherapy. J Cardiovasc Magn Reson. 2013;15(1):1–11. | ||
Taher A, Hershko C, Cappellini MD. Iron overload in thalassaemia intermedia: reassessment of iron chelation strategies. Br J Haematol. 2009;147(5):634–640. | ||
Calvaruso G, Vitrano A, Di Maggio R, Lai E, Colletta G, Quota A. Deferiprone versus deferoxamine in thalassemia intermedia: Results from a 5-year long-term Italian multicenter randomized clinical trial. Am J Hematol. 2015;90(7):634–638. | ||
Stumpf JL. Deferasirox. Am J Health Syst Pharm. 2007;64:606–616. | ||
Vanorden HE, Hagemann TM. Deferasirox – an oral agent for chronic iron overload. Ann Pharmacother. 2006;40:1110–1117. | ||
Porter J, Galanello R, Saglio G, et al. Relative response of patients with myelodysplastic syndromes and other transfusion-dependent anemias to deferasirox (ICL670): a 1-yr prospective study. Eur J Haematol. 2008;80(2):168–176. | ||
Lee JW, Yoon SS, Shen ZX, et al. Hematologic responses in patients with aplastic anemia treated with deferasirox: a post hoc analysis from the EPIC study. Haematologica. 2013;98(7):1045–1048. | ||
Tunc B, Tavil B, Karakurt N, et al. Deferasirox therapy in children with Fanconi aplastic anemia. J Pediatr Hematol Oncol. 2012;34(4):247–251. | ||
Pietrangelo A, Brissot P, Bonkovsky H, et al. A phase I/II, open-label, dose-escalation trial using the once-daily oral chelator deferasirox to treat iron overload in HFE-related hereditary hemochromatosis. Hepatology. 2010;52(5):1671–1679. | ||
Saliba AN, Harb AR, Taher AT. Iron chelation therapy in transfusion-dependent thalassemia patients: current strategies and future directions. J Blood Med. 2015;6:197–209. | ||
Taher AT, Temraz S, Cappellini MD. Deferasirox for the treatment of iron overload in non-transfusion-dependent thalassemia. Expert Rev Hematol. 2013;6(5):495–509. | ||
Shirley M, Plosker GL. Deferasirox: a review of its use for chronic iron overload in patients with non-transfusion-dependent thalassaemia. Drugs. 2014;74(9):1017–1027. | ||
Miyazawa K, Ohyashiki K, Urabe A, et al. A safety, pharmacokinetic and pharmacodynamic investigation of deferasirox (Exjade, ICL670) in patients with transfusion-dependent anemias and iron-overload: a Phase I study in Japan. Int J Hematol. 2008;88(1):73–81. | ||
Novartis Pharmaceuticals. EXJADE (deferasirox) US Prescribing Information (2013). Available from: www.pharma.us.novartis.com/product/pi/pdf/exjade.pdf. Accessed July 27, 2015. | ||
Voskaridou E, Plata E, Douskou M, et al. Treatment with deferasirox (Exjade) effectively decreases iron burden in patients with thalassaemia intermedia: results of a pilot study. Br J Haematol. 2010;148:332–334. | ||
Ladis V, Berdousi H, Gotsis E, Kattamis A. Deferasirox administration for the treatment of non-transfusional iron overload in patients with thalassaemia intermedia. Br J Haematol. 2010;151:504–508. | ||
Taher AT, Porter J, Viprakasit V, et al. Deferasirox significantly reduces iron overload in non-transfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood. 2012;120:970–977. | ||
Taher AT, Porter JB, Viprakasit V, et al. Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann Hematol. 2013;92(11):1485–1493. | ||
Taher AT, Porter JB, Viprakasit V, et al. Deferasirox demonstrates a dose-dependent reduction in liver iron concentration and consistent efficacy across subgroups of non-transfusion-dependent thalassemia patients. Am J Hematol. 2013;88(6):503–506. | ||
Karimi M, Arandi N, Haghpanah S, et al. Efficacy of Deferasirox (Exjade®) in modulation of iron overload in patients with β-thalassemia intermedia. Hemoglobin. 2015;26:1–3. | ||
Riva A. Comment on: a record number of fatalities in many categories of patients treated with deferasirox: loopholes in regulatory and marketing procedures undermine patient safety and misguide public funds? Expert Opin Drug Saf. 2013;12(5):793–795. | ||
Taher AT, Porter JB, Viprakasit V, et al. Approaching low liver iron burden in chelated patients with non-transfusion-dependent thalassemia: the safety profile of deferasirox. Eur J Haematol. 2014;92(6):521–526. | ||
No authors listed. Efficacy and safety study of deferasirox in patients with non-transfusion dependent thalassemia (THETIS). Available from: https://clinicaltrials.gov/ct2/show/NCT01709838?term=deferasirox+ntdt&rank=1. Accessed July 27, 2015. | ||
Mallat NS, Mallat SG, Musallam KM, Taher AT. Potential mechanisms for renal damage in beta-thalassemia. J Nephrol. 2013;26(5):821–828. | ||
Ziyadeh FN, Musallam KM, Mallat NS, et al. Glomerular hyperfiltration and proteinuria in transfusion-independent patients with β-thalassemia intermedia. Nephron Clin Pract. 2012;121(3–4):c136–c143. | ||
Origa R, Cazzola M, Mereu E, et al. Differences in the erythropoiesis-hepcidin-iron store axis between hemoglobin H disease and β-thalassemia intermedia. Haematologica. 2015;100(5):e169–e171. | ||
Taher A, Musallam KM, El Rassi F, et al. Levels of non-transferrin-bound iron as an index of iron overload in patients with thalassaemia intermedia. Br J Haematol. 2009;146:569–572. | ||
Porter JB, El-Alfy M, Viprakasit V, et al. Utility of labile plasma iron and transferrin saturation in addition to serum ferritin as iron overload markers in different underlying anemias before and after deferasirox treatment. Eur J Haematol. Epub 2015 Feb 18. | ||
Daar S, Pathare A, Nick H, et al. Reduction in labile plasma iron during treatment with deferasirox, a once-daily oral iron chelator, in heavily iron-overloaded patients with beta-thalassaemia. Eur J Haematol. 2009;82(6):454–457. | ||
Casale M, Citarella S, Filosa A, et al. Endocrine function and bone disease during long-term chelation therapy with deferasirox in patients with β-thalassemia major. Am J Hematol. 2014;89(12):1102–1106. | ||
Deugnier Y, Turlin B, Ropert M, et al. Improvement in liver pathology of patients with β-thalassemia treated with deferasirox for at least 3 years. Gastroenterology. 2011;141(4):1202–1211. | ||
Musallam KM, Taher AT, Cappellini MD, Sankaran VG. Clinical experience with fetal hemoglobin induction therapy in patients with β-thalassemia. Blood. 2013;121(12):2199–2212. | ||
Suragani RN, Cawley SM, Li R, et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine beta-thalassemia. Blood. 2014;123(25):3864–3872. | ||
Dussiot M, Maciel TT, Fricot A, et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in beta-thalassemia. Nat Med. 2014;20(4):398–407. | ||
Rivella S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl 1):S12–S15. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.