")
Back to Journals » Journal of Blood Medicine » Volume 10
Profile of crizanlizumab and its potential in the prevention of pain crises in sickle cell disease: evidence to date
Received 13 June 2019
Accepted for publication 6 August 2019
Published 30 August 2019 Volume 2019:10 Pages 307—311
DOI https://doi.org/10.2147/JBM.S191423
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin Bluth
Tanya R Riley, Treavor T Riley
Wingate University School of Pharmacy, Hendersonville, NC 28739, USA
Correspondence: Tanya R Riley
Wingate University School of Pharmacy, 805 6th Avenue West; Suite 200, Hendersonville, NC 28739, USA
Tel +1 828 697 0615
Email [email protected]
Abstract: Sickle cell disease (SCD) is one of the most common inherited blood disorders globally. It is a grouping of autosomal recessive genetic disorders identified by a genetic mutation that replaces glutamic acid with valine at the sixth amino acid on the hemoglobin β-globin chain. Millions of people around the world live with a severe genotype of SCD that is often associated with occlusion of the microvasculature resulting in episodes of severe pain and multiple organ system dysfunction. These episodes, commonly categorized as vaso-occlusive crises (VOC), are a distinctive clinical presentation of SCD which represents the majority of SCD morbidity and associated hospitalizations. Though the complete process by which these crises occur is complex and not fully outlined, evidence reveals this process to be multifactorial and heterocellular. For nearly two decades, hydroxyurea was the only FDA-approved therapy for SCD. Evidence to date shows that hydroxyurea treatment significantly reduces the rate of VOC, hospitalizations, and mortality. Despite these benefits, adherence remains problematic due to a variety of adverse effects and interpatient variability connected with hydroxyurea therapy. Crizanlizumab, an adhesion inhibitor of sickled red blood cells, was recently granted breakthrough therapy designation. Results of a phase 2 study have reported a successful reduction in annual rates of vaso-occlusive crisis with a favorable safety profile. This paper reviews the available literature concerning crizanlizumab use in patients with SCD.
Keywords: sickle cell, genotype, microvasculature, endothelium, vaso-occlusive crisis
Introduction
Sickle cell disease (SCD) is one of the most common autosomal recessive genetic disorders globally.1,2 Worldwide, an estimated 20–25 million people live with homozygous SCD (HbS/S). Approximately 300,000 infants are born annually with HbS/S. Areas with a high prevalence of malaria such as sub-Saharan Africa, the Mediterranean basin, Middle East, and India tend to have higher populations of patients affected with SCD.2–4 Within the United States, approximately 100,000 people have SCD accounting for more than 110,000 sickle cell-related hospitalizations annually.3,5,6
SCD is a collection of autosomal recessive genetic disorders denoted by the presence of the sickle hemoglobin (HbS) allele, a genetic mutation resultant of a substitution in the sixth amino glutamic acid with valine on the β-globin chain.1,4,5,7–9 Genotypes of SCD vary based on homozygous and heterozygous inheritance of the HbS allele.2,3 Homozygous SCD (HbS/S), which is the inheritance of one copy of HbS from each parent, is the most common form of SCD.1,9 Heterozygous forms, conditions in which a copy of HbS combines with a copy of another globin mutation such as hemoglobin C (HbS/C), hemoglobin D (HbS/D), hemoglobin E (HbS/E), or hemoglobin β-thalassemia (HbS/β+or β0-thalassemia), are other genetic variations of SCD that can occur.1,4,9 Severe clinical complications are generally associated with HbS/S and HbS/β0-thalassemia genotypes.10 All of the SCD genotypes produce enough HbS for the development of sickle-shaped cells due to cell damage that occurs as a result of polymerization of HbS when deoxygenated.3,8,9 However, there is no connection between the presence of sickle-shaped cells and the clinical severity of the disease.9,11,12
The acute presentation of SCD is often associated with occlusion of the microvasculature resulting in episodes of severe pain, acute chest syndrome, hepatic and splenic sequestration, and priapism.13 These episodes, commonly categorized as vaso-occlusive crises (VOC), is a hallmark clinical presentation of SCD which represents the majority of SCD morbidity and associated hospitalizations.5,14 Though the entire mechanism by which these crises occur is complex and not fully explained, evidence reveals this process to be multifactorial and heterocellular.8,15 Studies have shown that complex interactions between receptors on erythrocytes in combination with other blood cells and abnormal endothelial interactions can lead to obstruction of the vasculature.4,8,11,12,15,16 Postcapillary adhesion of low-density sickle cells and reticulocytes to the endothelium secondarily traps older abnormally shaped dense sickle cells causing decreased blood flow and cellular movement through the microvasculature.12,15 These vascular endothelium interactions can lead to upregulation of endothelial adhesive molecules such as E-selectin and P-selectin.15 Another factor that contributes to VOC development is the adhesion of heterocellularly formed combinations of leukocytes and dense sickled cells to the endothelium.8,12,15 These interactions may precipitate local hypoxia which can lead to increased endothelial cell surface expression of P-selectin, increased formation of HbS molecules, and subsequent VOC generation.8,12,15,17 Other potential VOC triggers are increased circulation of inflammatory cytokines, neutrophil movement through gap junctions of the endothelium which contribute to inflammation of the microvasculature, and dysregulation of nitric oxide (NO).8,15,16
Crizanlizumab
Crizanlizumab, a humanized monoclonal antibody, inhibits adhesion of sickled red blood cells by binding to P-selectin and preventing interaction with P-selectin glycoprotein ligand 1.13 Dosing of crizanlizumab is currently weight-based and must be administered as a 30-min intravenous infusion. The safety and efficacy of crizanlizumab in SCD was evaluated in a single, multi-center, randomized, placebo-controlled phase 2 study. There were 198 participants between 16 and 65 years of age, with a history of 2–10 sickle cell crises in the 12 months prior to enrollment. Those receiving hydroxyurea prior to enrollment were required to have received at least 6 months of continuous therapy, with their dose being stable for the last 3 months. Participants not previously receiving hydroxyurea were not allowed initiation of the drug during the study period. Patients receiving long-term red blood cell transfusion therapy were excluded.4,13
Study participants were randomized into three groups: high-dose crizanlizumab (5 mg/kg), low-dose crizanlizumab (2.5 mg/kg), and placebo. All participants received a loading dose of the drug or placebo in two divided doses, 2 weeks apart. Maintenance dosing was then administered every 4 weeks for 50 weeks, for a total of 14 doses. The primary efficacy endpoint of the study was the annual rate of sickle cell pain crises, defined as VOCs resulting in a visit to a medical facility and requiring treatment with oral or parenteral narcotic analgesics or with parenteral nonsteroidal anti-inflammatory agents. Secondary efficacy endpoints included the annual rate of days hospitalized, times to first and second VOC, and the annual rate of uncomplicated VOC (defined as a crisis other than acute chest syndrome, hepatic sequestration, splenic sequestration, or priapism).13
Patients were enrolled between August 2013 through January 2015 from over 60 sites in the United States, Brazil, and Jamaica. Of the 198 patients randomized, only 129 completed the trial. Following the 52-week treatment phase, the median crisis rate per year was 1.63 in the high-dose group compared with 2.98 in the placebo group, accounting for a 45.3% lower rate in the intention-to-treat population (P=0.01). Likewise, when comparing the per-protocol population, the median crisis rate per year was 1.04 in the high-dose group and 2.18 in the placebo group, indicating a statistically significant 52.3% lower rate. There was no significance found when comparing the low-dose group to placebo in either the intention-to-treat or per-protocol population.13
Of the patients randomized to high-dose crizanlizumab, 63% received concomitant hydroxyurea. Similarly, 62% of the placebo group also received hydroxyurea. The median annual crisis rate in this subgroup was 2.43 in the high-dose crizanlizumab group and 3.58 in the placebo group which was a 32% lower rate. In those not receiving concomitant hydroxyurea, median annual crisis rates were 1.00 and 2.00, respectively, which was a 50% lower rate in the high-dose group. When stratifying patients based on the number of crises in the 12 months prior to study enrollment, 63% of the patients in both groups had 2 to 4 crises, while the remaining 37% had 5 to 10 crises. In the 2–4 crises subgroup, the annual crisis rate was 43% lower in the high-dose group compared to placebo. In the 5–10 crises sub-group, the rates were 1.97 and 5.32, respectively, which was a 63% lower rate in the high-dose group. Similar results were also found when groups were stratified based on SCD genotype. Forty-seven patients in each group had the genotype HbS/S, the median yearly crises rates were 1.97 for high-dose and 3.01 for placebo. Those expressing other genotypes had a 50.5% lower median crisis rate compared to placebo.13
There were no significant differences in median rate of days hospitalized (P=0.45). The time to first crisis was significantly longer in the high-dose group at 4.07 months compared to 1.38 months with placebo (P=0.001). Time to second crisis was also longer at 10.32 months versus 5.09 months, respectively (P=0.02). When comparing low-dose crizanlizumab to placebo, there was no difference reported for any secondary endpoints. The incidence of complicated crises in the trial were rare with a median rate of 0.00 for all groups. The rate of uncomplicated crisis was 62.5% lower in the high-dose group compared with placebo (P=0.02). Patient quality of life scores based on the Brief Pain Inventory questionnaire reported no significant changes from baseline.13
The two most serious adverse effects seen with crizanlizumab were pyrexia (2 patients in the high-dose group) and influenza (3 patients in the low-dose group). Common adverse effects occurring in at least 10% of the patients receiving crizanlizumab included arthralgia, back pain, diarrhea, headache, musculoskeletal pain, nausea, vomiting, upper and lower extremity pain, urinary and upper respiratory tract infections, fever, pruritus, and chest pain. Five patients died during the study period. Two patients in the high-dose group died from sepsis and endocarditis, respectively. In the low-dose group, one patient died from a combination of acute chest syndrome, aspiration, respiratory failure, and progressive vascular congestion. Also, two patients receiving placebo died during the study. One suffered from right ventricular failure, while the other suffered from VOC, ischemic stroke, coma, sepsis, and venous thrombosis of the right lower limb. There were no significant differences observed in any measurable variables related to hemolysis between groups.13
Yu and colleagues, upon evaluating the study, questioned the lack of synergistic effect when hydroxyurea was combined with high-dose crizanlizumab.18 Patients in the high-dose group receiving hydroxyurea had a 32.1% lower median rate than placebo while those with no hydroxyurea had a 50% difference.18 When used as monotherapy, hydroxyurea has been shown to reduce median annual rates of pain crisis by 44% primarily through increases in HbF levels in red blood cells.18–21 Since crizanlizumab inhibits cell adhesion via P-selectin inhibition, additional reduction in the annual rate of VOCs is expected. However, the level of reduction of VOCs in patients receiving hydroxyurea was greater in the low-dose crizanlizumab group (44.1% lower median rate than placebo), as compared to the 32.1% rate with the high-dose group, suggesting there may be a concentration-related interaction leading to the reduced effect.13,18 Ataga and colleagues, while emphasizing that the trial was not designed to detect differences between subgroups, acknowledge that patients with more clinically severe disease were more likely to have received hydroxyurea therapy, potentially influencing the results.13,18,22
Kutlar and colleagues provide a more in-depth analysis of subgroups from the SUSTAIN study population comparing only high-dose crizanlizumab to placebo.23 When considering patients with 2–4 VOC events in the year prior to study enrollment, crizanlizumab-treated patients were 1.67 times more likely to be VOC event-free at 52 weeks (40.5% vs 24.4%, respectively). In those with 5–10 VOC events prior to enrollment, crizanlizumab-treated patients were 6.7 times more likely to be VOC event-free at 52 weeks (28% vs 4.2%, respectively). Similar results were found when comparing SCD genotypes. In patients with HbS/S, those receiving crizanlizumab were 1.9 times more likely to be VOC event-free at 52 weeks (31.9% vs 17%, respectively). Likewise, in those with non-HbS/S genotypes, 45% of the crizanlizumab-treated patients were VOC event-free at study completion compared with 16.7% in the placebo group.23
When considering both hydroxyurea use and the number of VOC events prior to study enrollment, crizanlizumab was also superior to placebo. Patients with 2–4 VOC events prior to enrollment and hydroxyurea use were 1.76 times more likely to be VOC event-free at 52 weeks with crizanlizumab treatment (44% vs 25%, respectively). In the 5–10 VOC plus hydroxyurea group, 17.6% of crizanlizumab-treated patients were event-free compared to 6.3% receiving placebo. Crizanlizumab also significantly delayed median time to first VOC in each subgroup evaluated. In those with 2–4 VOC events prior to study initiation, median time to first event was 4.76 months in the treatment group compared to 1.61 months for placebo. Similarly, in the 5–10 VOC event subgroup, median time to first event was 2.43 months and 1.03 months, respectively. Patients with the HbS/S genotype treated with crizanlizumab had a median time to first VOC of 4.07 months compared to 1.12 months for placebo. In those with non-HbS/S genotypes, median time to first event was 6.90 months and 3.09 months, respectively. Similarly, median time to first VOC event was significantly longer in crizanlizumab-treated patients irrespective of concomitant hydroxyurea use (2.43 vs 1.15 in hydroxyurea treated patients; 5.68 vs 2.86 in non-hydroxyurea patients).23 This analysis of subgroups further supports the efficacy of crizanlizumab in patients with varying severities of SCD, genotypes, and hydroxyurea utilization. While there remains a question as to whether low-dose crizanlizumab would be more beneficial than high-dose treatment in patients stable on hydroxyurea, the existing data support the use of high-dose treatment in all presentations of SCD.23
Conclusion
Between 1998 and 2017, hydroxyurea was the only FDA-approved therapy for SCD.4 By increasing both fetal hemoglobin concentrations in erythrocytes and circulating NO levels, hydroxyurea treatment significantly reduces the rate of VOC, subsequent hospitalizations, and mortality in a range of patients.5,21,24 Despite these benefits, adherence remains problematic. Adverse effects such as bone marrow suppression, large inter-patient variability in pharmacokinetics and pharmacodynamics, and variations in the maximum tolerable dose are all obstacles clinicians must face with initiating therapy.24
With VOCs being the major contributing factor to hospitalizations and reduced patient quality of life, greater understanding of the pathophysiology of VOC is essential. Adhesion interactions triggered by inflammation or hemolysis, release of reactive oxidants secondary to ischemic episodes, platelet and complement activation all contribute to vascular obstruction.25 Studies involving various agents targeting each of these areas have been conducted over the past decade with sporadic results. The results of this recent crizanlizumab study are encouraging, displaying consistent benefit in the prevention of VOCs in diverse patient groups. There are more studies underway, addressing the pharmacokinetics of the drug, along with its utilization in specific patient groups (Table 1).
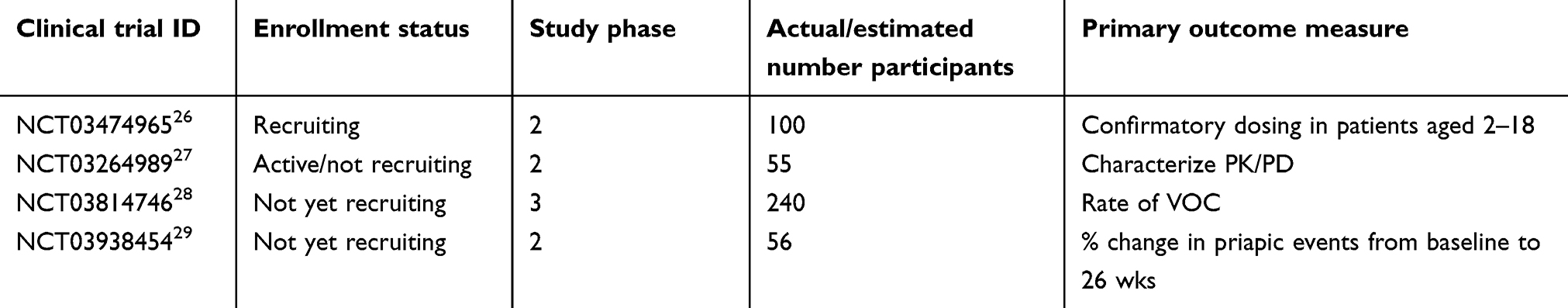 |
Table 1 Pending crizanlizumab studies |
In January 2019, the United States Food and Drug Administration granted crizanlizumab Breakthrough Therapy designation for the prevention of VOCs in SCD.30 This designation applies to all genotypes of SCD and indicates that crizanlizumab demonstrates substantial improvement over existing therapies on at least one efficacy endpoint based on the available preliminary clinical evidence.30 Though still awaiting approval, crizanlizumab may be a vital component of therapy for the prevention of VOC in SCD patients.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (Hb S) Allele and Sickle cell disease: a HuGE review. Am J Epidemiol. 2000;151(9):839–845. doi:10.1093/oxfordjournals.aje.a010288
2. Saraf SL, Molokie RE, Nouraie M, et al. Differences in the clinical and genotypic presentation of sickle cell disease around the world. Paediatr Respir Rev. 2014;15(1):4–12.
3. Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;376(16):1561–1573. doi:10.1056/NEJMra1510865
4. Riley TR, Boss A, McClain D, Riley TT. Review of medication therapy for the prevention of sickle cell crisis. Pharm Ther. 2018;43(7):417–437.
5. Okam MM, Shaykevich S, Ebert BL, Zaslavsky AM, Ayanian JZ. National trends in hospitalizations for sickle cell disease in the United States following the FDA approval of hydroxyurea, 1998-2008. Med Care. 2014;52(7):612–618. doi:10.1097/MLR.0000000000000143
6. Steiner CA, Miller JL. Sickle cell disease patients in U.S. hospitals, 2004. HCUP Statistical Brief #21. Agency for Healthcare Research and Quality; December 2006. Available from: www.hcup-us.ahrq.gov/reports/statbriefs/sb21.pdf.
7. Serjeant GR. Sickle-cell disease. Lancet. 1997;350(9079):725–730. doi:10.1016/S0140-6736(97)07330-3
8. Stuart MJ, Nagel RL. Sickle cell disease. Lancet. 2004;364(9442):1343–1360. doi:10.1016/S0140-6736(04)17192-4
9. Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017;390(10091):311–323. doi:10.1016/S0140-6736(17)30193-9
10. Serjeant GR. The natural history of sickle cell disease. Cold Spring Harb Perspect Med. 2013;3(10):a011783. doi:10.1101/cshperspect.a015511
11. Hebbel RP, Yamada O, Moldow CF, Jacob HS, White JG, Eaton JW. Abnormal adherence of sickle erythrocytes to cultured vascular endothelium: possible mechanism for microvascular occlusion in sickle cell disease. J Clin Invest. 1980;65(1):154–160. doi:10.1172/JCI109646
12. Kaul DK, Finnegan E, Barabino GA. Sickle red cell-endothelium interactions. Microcirculation. 2009;16(1):97–111. doi:10.1080/10739680802279394
13. Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376(5):429–439. doi:10.1056/NEJMoa1611770
14. Uwaezuoke SN, Ayuk AC, Ndu IK, Eneh CI, Mbanefo NR, Ezenwosu OU. Vaso-occlusive crisis in sickle cell disease: current paradigm on pain management. J Pain Res. 2018;11:3141–3150. doi:10.2147/JPR.S185582
15. Manwani D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood. 2013;122(24):3892–3898. doi:10.1182/blood-2013-05-498311
16. Morris CR. Mechanisms of vasculopathy in sickle cell disease and thalassemia. Hematol Am Soc Hematol Educ Program. 2008;2008:177–185. doi:10.1182/asheducation-2008.1.177
17. Gutsaeva DR, Parkerson JB, Yerigenahally SD, et al. Inhibition of cell adhesion by anti-P-selectin aptamer: a new potential therapeutic agent for sickle cell disease. Blood. 2011;117(2):727–735. doi:10.1182/blood-2010-05-285718
18. Yu Z, Blankenship L, Jaiyesimi I. Crizanlizumab in sickle cell disease. N Engl J Med. 2017;376(18):1795–1796. doi:10.1056/NEJMc1703162
19. Heeney MM, Ware RE. Hydroxyurea for children with sickle cell disease. Pediatr Clin North Am. 2008;55(2):483–501, x. doi:10.1016/j.pcl.2008.02.003
20. Brandow AM, Panepinto JA. Hydroxyurea use in sickle cell disease: the battle with low prescription rates, poor patient compliance and fears of toxicities. Expert Rev Hematol. 2010;3(3):255–260. doi:10.1586/ehm.10.22
21. Siklos (hydroxyurea) [package insert]. Bryn Mawr, Pennsylvania: Medunik USA, Inc; 2017.
22. Ataga KI, Kutlar A, Kanter J. Crizanlizumab in sickle cell disease. N Engl J Med. 2017;376(18):1795–1796. doi:10.1056/NEJMc1703162
23. Kutlar A, Kanter J, Liles DK, et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: a SUSTAIN study analysis. Am J Hematol. 2019;94(1):55–61. doi:10.1002/ajh.25308
24. Ware RE, Despotovic JM, Mortier NA, et al. Pharmacokinetics, pharmacodynamics, and pharmacogenetics of hydroxyurea treatment for children with sickle cell anemia. Blood. 2011;118(18):4985–4991. doi:10.1182/blood-2011-07-364190
25. Sins JWR, Mager DJ, Davis SCAT, Biemond BJ, Fijnvandraat K. Pharmacotherapeutical strategies in the prevention of acute, vaso-occlusive pain in sickle cell disease: a systematic review. Blood Adv. 2017;1(19):1598–1616. doi:10.1182/bloodadvances.2017007211
26. ClinicalTrials.gov. Study of dose confirmation and safety of crizanlizumab in pediatric sickle cell disease patients. NCT03474965. Available from: https://clinicaltrials.gov/ct2/show/NCT03474965.
27. ClinicalTrials.gov. Pharmacokinetics and pharmacodynamics study of SEG101 (Crizanlizumab) in sickle cell disease (SCD) patients with vaso- occlusive crisis (VOC). NCT03264989. Available from: https://clinicaltrials.gov/ct2/show/NCT03264989.
28. ClinicalTrials.gov. Study of two doses of crizanlizumab versus placebo in adolescent and adult sickle cell disease patients (STAND). NCT03814746. Available from: https://clinicaltrials.gov/ct2/show/NCT03814746.
29. ClinicalTrials.gov. A Study to Evaluate the Safety and Efficacy of Crizanlizumab in Sickle Cell Disease Related Priapism (SPARTAN). NCT03938454. Available from: https://clinicaltrials.gov/ct2/show/NCT03938454.
30. Novartis. Press release: Novartis investigational therapy crizanlizumab (SEG101) receives FDA breakthrough therapy designation for the prevention of vaso-occlusive crises in sickle cell disease. January 8, 2019. Available from: https://www.novartis.com/news/media-releases/novartis-investigational-therapy-crizanlizumab-seg101-receives-fda-breakthrough-therapy-designation-prevention-vaso-occlusive-crises-sickle-cell-disease.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.