")
Back to Journals » Lung Cancer: Targets and Therapy » Volume 6
Profile of ceritinib in the treatment of ALK+ metastatic non-small-cell lung cancer
Received 7 March 2015
Accepted for publication 16 April 2015
Published 15 May 2015 Volume 2015:6 Pages 35—42
DOI https://doi.org/10.2147/LCTT.S69114
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Pan-Chyr Yang
Mark W Burns, Eric S Kim
Wilmot Cancer Center, University of Rochester, Rochester, NY, USA
Abstract: Lung cancer has become one of the leading causes of death in both men and women in the United States, with approximately 230,000 new cases and 160,000 deaths each year. Approximately 80% of lung cancer patients are diagnosed with non-small-cell lung cancer (NSCLC), a subset of epithelial lung cancers that are generally insensitive to chemotherapy. An estimated 3%–7% of NSCLC patients harbor tumors containing anaplastic lymphoma kinase (ALK) gene rearrangement as an oncogenic driver. Subsequent development of the first-generation tyrosine kinase inhibitor crizotinib demonstrated substantial initial ALK+-tumor regression, yet ultimately displayed resistance in treated patients. The recently approved tyrosine kinase inhibitor ceritinib has been shown to be an effective antitumor agent against crizotinib-naïve and -resistant ALK+-NSCLC patients. In this review, we will provide an overview of biology and management of ALK+-NSCLC with a special focus on clinical development of ceritinib.
Keywords: ceritinib, anaplastic lymphoma kinase, non-small-cell lung cancer
Anaplastic lymphoma kinase and its role in non-small-cell lung cancer
The anaplastic lymphoma kinase (ALK) is a type-I transmembrane tyrosine kinase belonging to the insulin receptor superfamily and is hypothesized to be active in the early development and maintenance of the central and peripheral nervous systems.1 This 729 kbp gene is located on the p-arm of human chromosome 2 and encodes a functioning 220 kDa transmembrane receptor, containing an extracellular ligand-binding domain, a single-pass membrane spanning domain, and a catalytic intracellular kinase domain.2 A significant degree of ALK mRNA expression throughout developing nervous tissues of both embryonic mice and humans has led to the hypothesis that the ALK gene product is involved in the maturation and maintenance of early nervous tissues.3 It has been generally understood that, under normal psychological conditions, binding of a currently unknown mammalian ligand to the extracellular domain induces dimerization of ALK, allowing autophosphorylation and subsequent activation of the intracellular kinase domain.4 The kinase propagates downstream pathways required for cell growth and passage through the cell cycle. Activated ALK proteins are involved in numerous overlapping signaling pathways, specifically the PLC-γ and Ras/ERK1/2 pathways that participate in cell proliferation and the JAK/STAT and PI3K/AKT pathways that propagate cell survival.5 The intensity of ALK mRNA and protein expression subsequently diminishes in mammals after birth, reaching a minimum level approximately 3 weeks after birth and maintained at low levels throughout normal adulthood.6
Considering that normal ALK has very little (if any) activity in mature human tissues, the detection of active ALK domains in tumor samples of non-small-cell lung cancer (NSCLC) patients implicates the oncogenic nature of activated ALK pathways in this subset of metastatic cancers. Despite a vast majority of NSCLCs being associated with mutations induced through tobacco use or exposure, ALK+-NSCLC is most prevalent in nonsmokers.7 Genetic anomalies within the ALK gene through rearrangement, amplification, or mutation produce constitutively active ALK fusion protein domains responsible for the pathogenesis of ALK+-NSCLCs.8 These ALK fusion genes are currently identified in NSCLC patients through tests such as fluorescence in situ hybridization (FISH), immunohistochemistry assay, and reverse transcription-polymerase chain reaction.9 Furthermore, advancements in next-generation genomic sequencing now permit more rapid and reliable detection of ALK+ tumors.10,11
Abnormalities observed in ALK+-NSCLCs are mainly the result of gene rearrangement. Inversions in the p-arm of chromosome 2 lead to the conjunction of the echinoderm microtubule-associated protein-like 4 (EML4) and ALK genes, producing an active fusion protein that is detected in numerous carcinomas. Translocations of the ALK gene have been discovered in numerous carcinomas, including neuroblastomas, anaplastic lymphomas, and inflammatory myofibroblastic tumors,4 but the first instance of EML4–ALK translocation was described in 2007 using tumor cell samples from a 62-year-old NSCLC patient, in which the fusion gene induced the activation of the intracellular kinase domain of ALK.12 Meta-analyses reveal that the EML4–ALK fusion gene is correlated with younger, nonsmoking female populations.12–14 All EML4–ALK fusion genes observed in NSCLCs contain exons 20–29 of ALK (encoding the intracellular domain) and eight different EML4 exons, all of which activate the downstream signals of Ras/ERK1/2 and JAK/STAT.15,16 It has been suggested that the prevalence of both the ALK mRNA and protein (specifically, the intracellular kinase domain) in NSCLC patients is the product of the EML4–ALK translocation and not the intact ALK gene, which is consistent with the view that active EML4–ALK is an oncogenic driver for the growth of tumors in patients harboring this ALK translocation.17 The EML4–ALK fusion protein undergoes auto-phosphorylation of its intracellular ALK kinase domain, constitutively activating the Ras signaling pathway that ultimately results in uncontrolled cell growth.4
First-generation ALK inhibitor: profile of crizotinib
Because the ALK kinase is required for oncogenic activity in this subdivision of NSCLCs, tyrosine kinase inhibitors (TKIs) have been developed as antitumor therapies. Crizotinib (formerly known as PF-02341066 and marketed as Xalkori®, Pfizer), while originally synthesized as a c-MET inhibitor, demonstrated remarkable antitumor activity in patients harboring ALK+ tumors.18,19 As a TKI, crizotinib acts to competitively inhibit the ATP-binding domain of ALK, effectively preventing its downstream mechanisms of proliferation/replication while simultaneously inducing apoptosis.20 An initial phase I clinical trial investigating crizotinib resulted in a response rate of 57% and a 6-month progression free survival (PFS) rate of 72% with only mild toxicities.21 These clinical results were deemed more favorable than response rates observed in NSCLC patients treated solely with platinum-based chemotherapies. The Food and Drug Administration approved crizotinib for the treatment of ALK+-NSCLC in August 2011. A recently published PROFILE 1014 Phase III trial compared the efficacy and safety profile of crizotinib vs chemotherapy (pemetrexed-cisplatin or pemetrexed-carboplatin) in previously untreated patients with advanced ALK+-NSCLC, and found that the crizotinib arm showed both improved PFS and overall response rate (ORR) (10.9 months and 74%, respectively) over the standard chemotherapy arm (7.0 months and 45%, respectively).22 Patients receiving crizotinib also self-reported more improvements in cancer-related pain and a better quality of life than patients receiving standard chemotherapy.22 Thus, crizotinib has become the standard of care in patients harboring ALK+ tumors.
Despite its initial success in the treatment of ALK+-NSCLC, resistance to crizotinib develops in patients after approximately 10 months of treatment.23 Numerous factors likely contribute to the development of crizotinib resistance. Missense mutations in the kinase domain of the fusion protein that result in either diminished crizotinib binding or increased ATP binding is one such potential mechanism of resistance.24 Currently, seven distinct acquired resistance mutations have been identified in crizotinib-resistant tumors: the two most common being the gatekeeper mutations L1196M and G1269A and the remaining five mutations L1171T, L1152R, C1156Y, G1202R, and S1206Y. Multiple, nonoverlapping resistance mutations of the ALK kinase domain have been observed in some patients.25 Others expressed mechanisms that activated downstream signaling pathways of ALK without mutating the kinase itself,26 and a small number of patients simply failed to respond to crizotinib in the absence of any resistance mutations.19 In vitro analysis also demonstrates that the multiple variants of the EML4–ALK fusion protein may exhibit variable sensitivities to TKIs, consistent with the clinical range of responses to crizotinib treatment.27 Therefore, while some secondary mutations may exhibit continued responses to crizotinib, other mutations may confer resistance to the TKI and ultimately lead to the progression of metastatic ALK+ tumors.
While crizotinib demonstrates substantial antitumor activity in systemic disease, it appears to be less efficacious in targeting CNS metastasis. This could be due to the fact that crizotinib has a poor ability to penetrate the blood–brain barrier. A case report of a 29-year-old patient with ALK+-NSCLC demonstrated cerebrospinal crizotinib concentration of only 0.616 ng/mL, compared to its systemic serum concentration of 237 ng/mL.28 This patient exhibited adequate control of his intrathoracic disease when he developed new progressive disease in his brain.28 Other possible explanations concerning the discrepancy between systemic and CNS crizotinib efficacy, including the development of genetically distinct tumors in the brain that are unaffected by crizotinib and induced secondary mutations in the ALK kinase domain within brain parenchyma that confer resistance to TKIs such as crizotinib, have been suggested.29 With an approximate rate of CNS metastases in NSCLC patients at 30%,30 a novel and improved method for delivering antitumor agents through the blood–brain barrier into the cerebrospinal fluid is of importance for tumor suppression in ALK+-NSCLC patients.
Chemistry and pharmacology of ceritinib
Ceritinib (formerly known as LDK378 and marketed as Zykadia™, synthesized from the compound TAE684 by Novartis Pharmaceuticals) is a small, orally administered ALK inhibitor prescribed to patients harboring ALK+-NSCLC that has become resistant to crizotinib therapy. The synthesized molecule has a formula C28H36ClN5O3S, an International Union of Pure and Applied Chemistry (IUPAC) name 5-chloro-2-N-(5-methyl-4-piperidin-4-yl-2-propan-2-yloxyphenyl)-4-N-(2-propan-2-ylsulfonylphenyl)pyrimidine-2,4-diamine, and a molecular weight 557.22 g/mol (Figure 1).31 Molecular ceritinib has the capacity to accept seven hydrogen bonds and can donate three hydrogen atoms for hydrogen bonding, while also maintaining nine rotatable bonds in physiological conditions. Ceritinib acts as a powerful inhibitor of ALK, yet does not demonstrate c-MET inhibition as does its predecessor crizotinib.8
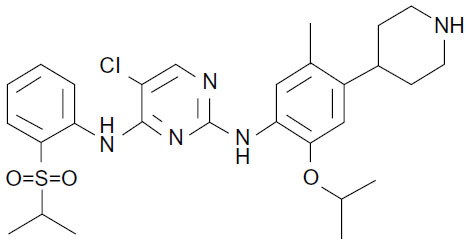 | Figure 1 Molecular structure of ceritinib. |
Molecular docking simulations of the reported crystal structure of ceritinib have revealed the mechanism by which the molecule interacts with the kinase domain of ALK. Acting as a ligand, ceritinib binds via hydrogen bonds with amino acids of the kinase’s hinge area through both the amino nitrogen atoms and the central pyrimidine ring of the inhibitor. The ligand-active site interaction is further stabilized by a salt bridge formed between the terminal piperidine of the inhibitor with Glu1210 of the kinase domain.31 While molecular ceritinib contains almost double the amount of rotatable bonds compared to crizotinib (ceritinib maintains nine, crizotinib maintains five), the former exhibits potency toward ALK 20-fold higher than the latter.32
Ceritinib has exhibited cellular specificity to tumors harboring the ALK translocated gene through assays determining 50% of maximal inhibition of tumor cell proliferation (GI50 assays) in multiple tumor samples expressing various oncogenic drivers. Ceritinib demonstrated potency in two ALK+-NSCLC cell lines (H3112 and H2228), but failed to show significant GI50s in tumor cells expressing the other oncogenic drivers EGFR, HER2, KRAS, or PI3K.32 These findings highlight the specificity of ceritinib in targeting ALK+-NSCLC cells.
Ceritinib specifically binds to the intracellular kinase domain of the EML4–ALK fusion protein, inhibiting its phosphorylation and subsequent activation of its downstream pathways.32 This mechanism of kinase inhibition involves competitive binding in the fusion kinase domain, preventing induced structural changes that result in activated downstream pathways involved in cell growth and proliferation.32 By selectively inhibiting this oncogenic process at the initial step of irregular ALK activation, ceritinib exhibits significant antitumor activity in NSCLC patients harboring ALK translocations. Figure 2 represents a schematic diagram of ceritinib inhibiting phosphorylation of the EML4–ALK kinase and deactivating downstream growth pathways.
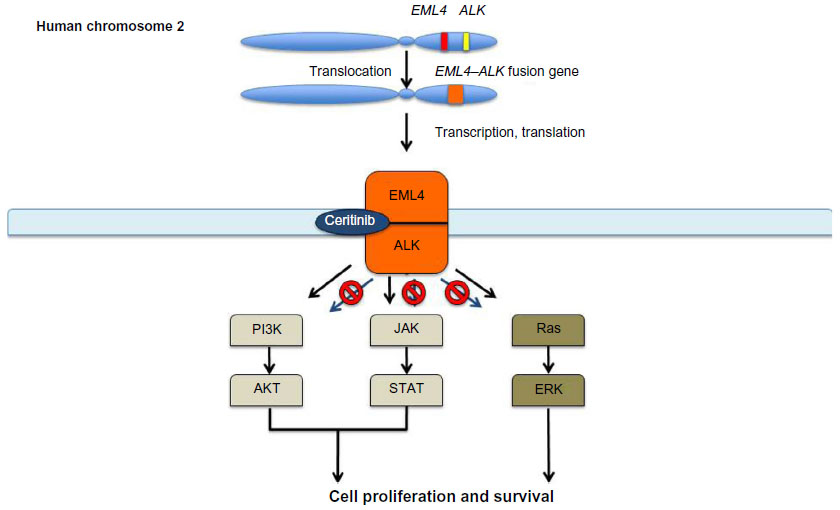 | Figure 2 Inappropriate activation of the ALK pathway results in uncontrolled tumor growth and proliferation. |
Ceritinib maintains a remarkable pharmacokinetic profile in mammals with only mild to few severe side effects in human patients. The maximum tolerated dose (MTD) of ceritinib is 750 mg on an empty stomach, with antitumor results observable in patients treated with 400 mg daily. The maximum human plasma concentration of ceritinib is realized 4–6 hours after receiving the MTD.33
Clinical profile of ceritinib
Ceritinib has demonstrated significant activity in crizotinib-naïve ALK+-NSCLC models. In vitro enzymatic assays reveal that ceritinib is more potent than crizotinib at inhibiting the oncogenic ALK fusion protein.32 Ceritinib also inhibits the phosphorylation of both the ALK fusion protein and the downstream signaling pathways ERK and PI3K/AKT at much lower doses than crizotinib.32 In vivo H2228 xenograft models demonstrated the long-lasting antitumor properties of ceritinib. Tumor-bearing animals harboring ALK+ tumors were treated with either crizotinib (100 mg/kg) or ceritinib (25–50 mg/kg) daily for 2 weeks, with the latter experiencing both stronger antitumor suppression and longer durations of responses than the former.32 Additional assays demonstrated that ceritinib potently inhibited cell proliferation in cell lines with one of four resistance mutations in EML4–ALK amino acid sequencing (L1196M, G1269A, I1171T, and S1206Y), yet the five EML4–ALK resistance mutants exhibiting one of five amino acid sequence mutations (C1156Y, L1152R, F1174C, G1202R, and 1151T) demonstrated insignificant suppression of growth (albeit with stronger antitumor activity than the same mutant lines treated with crizotinib). From a structural basis of ligand-kinase domain docking, variation in ceritinib efficacy can be explained by the structural change in the kinase’s active site via point mutation. As previously mentioned, the mutations L1196M and G1269A confer resistance to crizotinib but remain sensitive to ceritinib – changes that result in steric interference of crizotinib with the EML4–ALK catalytic domain but do not affect ceritinib binding.32 However, the G1202R mutation results in significant steric hindrance in the active site, which disallows binding of both ceritinib and crizotinib. While currently unknown in their effects on TKI inhibition, the resistance mutations C1156Y, L1152R, F1174C, and 1151T have been predicted to either alter that conformation of the entire kinase or decrease the Km of ceritinib and crizotinib – both possibilities significantly impacting the TKI ability to act as an ATP-competitive agent.32 Thus, ceritinib shows efficacy in tumor cells harboring the common crizotinib-induced resistance mutations, with less impressive results in treating tumors harboring the rarer crizotinib-induced resistance mutations.
In ASCEND-1, a Phase I study investigating ceritinib’s safety and efficacy in ALK+-NSCLC patients, a total of 246 ALK+-NSCLC patients were treated with ceritinib at the established maximal tolerated dose of 750 mg/daily.8,34 A total of 163 (66%) patients had received prior crizotinib and 83 (34%) patients were ALK inhibitor-naïve. The ORR was 58.5%. Among 163 patients previously treated with crizotinib, an ORR of 54.6% as well as a PFS of 6.9 months was achieved. Among the remaining 83 patients naïve to crizotinib treatment, an ORR of 66.3% was achieved. At the time of data cutoff, a median PFS could not be estimated for crizotinib-naïve patients.34 This study concluded that ceritinib at 750 mg/day demonstrated potent antitumor activity in ALK+-NSCLC regardless of previous therapy status.
A subgroup analysis between Asian and Caucasian patients (82 and 156 patients, respectively) in the ASCEND-1 sought to determine differences in efficacy and adverse reactions among the two distinct populations.35 Data from 173 patients demonstrated ORRs of 69% in Asian patients and 57% in Caucasian patients. Median durations of responses in the Asian and Caucasian patients were 10.1 months and 6.9 months, respectively.35 Observable variances in ceritinib treatment for these two populations could not be explained by previous ALK inhibitor therapies.
Adverse reaction data (Table 1) collected from 255 ALK+-NSCLC patients treated with 750 mg daily ceritinib were mainly gastrointestinal, manifested by diarrhea, vomiting, constipation, nausea, and abdominal pain.8 Additional serious side effects include hypophosphatemia, hyperglycemia, anemia, elevated liver and pancreatic enzymes, interstitial lung disease, and QT interval prolongation.8,33 These adverse events are often reversible after discontinuation of ceritinib treatment.
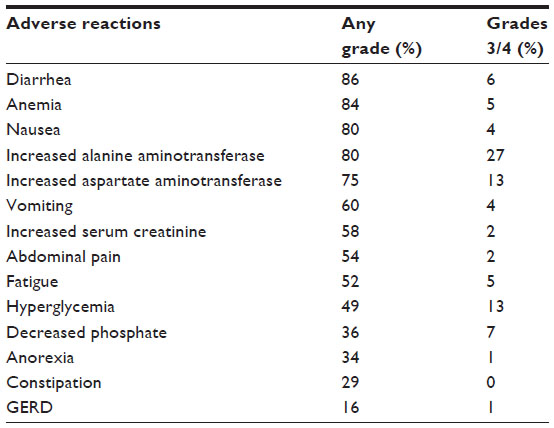 | Table 1 The most common adverse reactions observed in 255 ALK+-NSCLC patients treated with ≥400 mg daily ceritinib |
A total of 19 crizotinib-resistant NSCLC patients underwent tumor biopsy prior to beginning ceritinib treatment. Biopsies from all 19 patients tested positive for ALK rearrangement through FISH: two of the 19 patients exhibited ALK amplification, five patients exhibited secondary resistance mutations in the tyrosine kinase domain of the EML4–ALK fusion protein, and the remaining 12 patients solely exhibited the original oncogenic rearrangement of ALK.8 Treatment of these cases with the daily maximum dosage of ceritinib resulted in notable antitumor activity in all 19 patients. Significant responses were observed in six of the seven patients harboring either ALK amplification or resistance mutations as well as in seven of the 12 patients harboring no other genetic anomalies besides the original oncogenic alteration. These results demonstrate the efficacy of ceritinib in the tumor regression of crizotinib-resistant ALK+-NSCLC patients, regardless of the various genetic mechanisms leading to crizotinib resistance.8
As with crizotinib, patients treated with ceritinib also develop progression of disease. While ceritinib has been observed to overcome the resistance mutations L1196M, G1269A, S1206Y, and L1171T induced through crizotinib treatment, secondary mutations remain that are unable to be overcome by ceritinib. In a small study of 11 biopsied ceritinib-resistant tumors (two of the 11 obtained from different tumors in the same patient), five samples were found to harbor either F1174C or G1202R, two mutations that were also observed in crizotinib-resistant tumor samples. The patient who underwent biopsies at two different sites harbored two distinct resistance mutations, highlighting the inter-tumoral heterogeneity of resistance mechanisms.32 Better understanding of mechanisms of acquired ceritinib resistance is necessary to maximize the clinical efficacy of ceritinib in targeting ALK+-NSCLCs.
Ceritinib in the treatment of brain metastases
In contrast to crizotinib, ceritinib has demonstrated clinical responses in untreated CNS metastases in both crizotinib-naïve and -resistant ALK+-NSCLC patients. In the ASCEND-1 study of 246 ALK+-NSCLC patients, 124 patients harboring brain metastases were treated with ceritinib. An ORR of 54.0% and a median PFS of 6.9 months were observed in these patients.36 Tumor response was seen in 50.0% of patients with brain metastases who received prior crizotinib compared to 69.2% of patients who were crizotinib naïve.36 Shaw et al also reported that 64 patients harboring ALK+ brain metastases maintain a median PFS similar to 50 patients harboring only systemic tumors (6.9 months vs 7.0 months, with all patients receiving >400 mg of ceritinib daily).8
It appears that ceritinib has a tentative advantage over crizotinib regarding antitumor activity in ALK+-NSCLC patients with brain metastases. While its penetration mechanism remains unknown, its capacity to cross the blood–brain barrier and offer tumor shrinkage in the CNS marks a significant milestone in the treatment of ALK+-NSCLC patients.
Future perspectives
Success with crizotinib in the treatment of ALK+-NSCLC patients has instigated continuous research and development of more potent ALK inhibitors. Ceritinib has demonstrated efficacy in the treatment of ALK+-NSCLC in both crizotinib-naïve and crizotinib-refractory patients. In addition, there are several ongoing trials as well as scheduled trials further investigating ceritinib (see Table 2 for details).
 | Table 2 Current list of ongoing trials of ceritinib (LDK378) |
With the EML4–ALK fusion gene accounting for only 3%–7% of NSCLC patients, the method of precisely determining ALK positivity in tumors is a crucial step in the treatment of patients harboring ALK+ tumors. While FISH is currently the primary mechanism for ALK+ detection, investigations into more cost-effective and accurate diagnostic tests could pave the road for improvements in the molecular identification of ALK positivity as well as the acquired resistance mechanisms resulting from TKI treatment.
There are other novel ALK inhibitors under development that have significantly demonstrated antitumor activity in advanced ALK+-NSCLC patients. The novel inhibitor alectinib is one such ALK inhibitor. In an ongoing Phase II study of 46 patients, 43 (93.5%) of them achieved overall responses (two complete, 41 partial response) with only mild reportable side effects.37 Alectinib also demonstrated significant anti-tumor response in CNS.38 Optimal treatment strategies for ALK+ NSCLC will continue to evolve with further investigations.
Finally, combination therapy involving ALK inhibitors and immune checkpoint inhibitors such as nivolumab, an anti-PD1 antibody that is currently approved for melanoma and squamous cell carcinoma of lung, may result in more effective and prolonged anti-tumor responses by optimizing anti-tumor T-cell responses. The study of safety and efficacy of ceritinib in combination with nivolumab in patients with ALK-positive NSCLC is being planned (NCT02393625).
Conclusion
In the recent past, tyrosine kinases have become an important area of cancer research, both in the understanding of their role in malignant growth pathways and the mechanisms necessary to inhibit their oncogenic activities in cancer patients. The ALK is one such target, having been identified as an oncogenic driver in a small subset (3%–7%) of NSCLC. The advent of molecularly targeted therapies for NSCLCs has allowed for significantly improved clinical outcomes in ALK+ patients. After limitations concerning treatment with the first-generation TKI crizotinib surfaced, ceritinib received Food and Drug Administration approval in April 2014 for treating crizotinib-resistant ALK+-NSCLC patients. Ceritinib appears to be a potent successor to the first-generation TKI crizotinib in its ability to suppress untreated ALK+ tumors as well as tumors harboring resistance mutations that render crizotinib ineffective. Several trials are currently underway.
Disclosure
The authors indicated no conflicts of interest in this work.
References
Roskoski R Jr. Anaplastic lymphoma kinase (ALK): structure, oncogenic activation, and pharmacological inhibition. Pharmacol Res. 2013;68:68–94. | |
Morris SW, Naeve C, Mathew P, et al. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene. 1997;14:2175–2188. | |
Reiff T, Huber L, Kramer M, Delattre O, Janoueix-Lerosey I, Rohrer H. Midkine and Alk signaling in sympathetic neuron proliferation and neuroblastoma predisposition. Development. 2011;138: 4699–4708. | |
Shaw AT, Solomon B. Targeting anaplastic lymphoma kinase in lung cancer. Clin Cancer Res. 2011;17:2081–2086. | |
Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008; 8:11–23. | |
Iwahara T, Fujimoto J, Wen D, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14:439–449. | |
Subramanian J, Govindan R. Lung cancer in never smokers: a review. J Clin Oncol. 2007;25:561–570. | |
Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189–1197. | |
Jurmeister P, Lenze D, Berg E, et al. Parallel screening for ALK, MET and ROS1 alterations in non-small cell lung cancer with implications for daily routine testing. Lung Cancer. 2015;87:122–129. | |
Peled N, Palmer G, Hirsch FR, et al. Next-generation sequencing identifies and immunohistochemistry confirms a novel crizotinib-sensitive ALK rearrangement in a patient with metastatic non-small-cell lung cancer. J Thorac Oncol. 2012;7:e14–e16. | |
Ou SH, Soo RA, Kubo A, Kawaguchi T, Ahn MJ. Will the requirement by the US FDA to simultaneously co-develop companion diagnostics (CDx) delay the approval of receptor tyrosine kinase inhibitors for RTK-rearranged (ROS1-, RET-, AXL-, PDGFR-alpha-, NTRK1-) non-small cell lung cancer globally? Front Oncol. 2014;4:58. | |
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. | |
Wong DW, Leung EL, So KK, et al; University of Hong Kong Lung Cancer Study Group. The EML4–ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009;115:1723–1733. | |
Inamura K, Takeuchi K, Togashi Y, et al. EML4–ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol. 2009;22:508–515. | |
Sanders HR, Li HR, Bruey JM, et al. Exon scanning by reverse transcriptase-polymerase chain reaction for detection of known and novel EML4–ALK fusion variants in non-small cell lung cancer. Cancer Genet. 2011;204:45–52. | |
Li Y, Ye X, Liu J, Zha J, Pei L. Evaluation of EML4–ALK fusion proteins in non-small cell lung cancer using small molecule inhibitors. Neoplasia. 2011;13:1–11. | |
Guo Y, Ma J, Lyu X, et al. Non-small cell lung cancer with EML4–ALK translocation in Chinese male never-smokers is characterized with early-onset. BMC Cancer. 2014;14:834. | |
Gozdzik-Spychalska J, Szyszka-Barth K, Spychalski L, et al. C-MET inhibitors in the treatment of lung cancer. Curr Treat Options Oncol. 2014;15:670–682. | |
Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368: 2385–2394. | |
Steuer CE, Ramalingam SS. ALK-positive non-small cell lung cancer: mechanisms of resistance and emerging treatment options. Cancer. 2014;120:2392–2402. | |
Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. | |
Solomon BJ, Mok T, Kim DW, et al; PROFILE 1014 Investigators. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371:2167–2177. | |
Shaw AT, Yeap BY, Solomon BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12:1004–1012. | |
Bresler SC, Weiser DA, Huwe PJ, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. 2014;26:682–694. | |
Kim S, Kim TM, Kim DW, et al. Heterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancer. J Thorac Oncol. 2013;8:415–422. | |
Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–1482. | |
Heuckmann JM, Balke-Want H, Malchers F, et al. Differential protein stability and ALK inhibitor sensitivity of EML4–ALK fusion variants. Clin Cancer Res. 2012;18:4682–4690. | |
Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol. 2011;29:e443–e445. | |
Choi YL, Soda M, Yamashita Y, et al; ALK Lung Cancer Study Group. EML4–ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–1739. | |
Ma S, Xu Y, Deng Q, Yu X. Treatment of brain metastasis from non-small cell lung cancer with whole brain radiotherapy and Gefitinib in a Chinese population. Lung Cancer. 2009;65:198–203. | |
Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulf onyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem. 2013;56: 5675–5690. | |
Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4:662–673. | |
Zykadia™ (ceritinib) capsules[package insert]. Novartis, 2015. Available from: http://www.pharma.us.novartis.com/product/pi/pdf/zykadia.pdf. Accessed April 29, 2015. | |
Kim DW, Filep E, Mehra R, et al. Efficacy and safety of ceritinib in patients with advanced anaplastic lymphoma kinase (ALK)-rearranged (ALK+) non-small cell lung cancer (NSCLC): an update of ASCEND-1. Presented at: 2014 ESMO Annual Congress; 2014. | |
Tan DSW, Mehra R. Ceritinib in Asian versus Caucasian patients with advanced lymphoma kinase (ALK)-rearranged (ALK+) NSCLC: subgroup analysis of the ASCEND-1 trial. J Clin Oncol. 2014;32:5s. | |
Kim DW, Mehra R, Tan DSW. Ceritinib in advanced anaplastic lymphoma kinase (ALK)-rearranged (ALK+) non-small cell lung cancer (NSCLC): results of the ASCEND-1 trial. J Clin. 2014;32:5s. [Abstract # 8003 2014]. | |
Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1–2 study. Lancet Oncol. 2013;14:590–598. | |
Gadgeel SM, Gandhi L, Riely GJ, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014;15:1119–1128. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.