")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 10
Profile of buparlisib and its potential in the treatment of breast cancer: evidence to date
Authors Criscitiello C, Viale G, Curigliano G, Goldhirsch A
Received 30 September 2017
Accepted for publication 22 December 2017
Published 30 January 2018 Volume 2018:10 Pages 23—29
DOI https://doi.org/10.2147/BCTT.S134641
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Carmen Criscitiello, Giulia Viale, Giuseppe Curigliano, Aron Goldhirsch
European Institute of Oncology, Milano, Italy
Abstract: Alteration of the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin signaling pathway is key for the growth and survival of several cancers, including breast cancer. In addition, dysregulation of PI3K signaling may contribute to resistance to several anticancer agents. PI3K inhibitors may, therefore, be effective as antineoplastic therapy. Buparlisib is a potent and highly specific oral inhibitor of the pan-class I PI3K family. Buparlisib specifically inhibits class I PIK3 in the PI3K/AKT kinase signaling pathway in an ATP-competitive manner, thus inhibiting the production of the secondary messenger phosphatidylinositol (3,4,5)-trisphosphate and activation of the PI3K signaling pathway. This may induce inhibition of tumor cell growth and survival in susceptible tumor cell populations. Buparlisib is currently under investigation in patients with a variety of solid tumors, including breast cancer. Buparlisib has been validated as a promising anticancer agent, and tremendous efforts have been taken to develop it. However, buparlisib monotherapy has resulted in humble benefit so far. Results from studies combining buparlisib with different anticancer agents – namely, endocrine therapy, anti-HER2 therapy, and chemotherapy – have showed variable efficacy with consistent substantial toxicity.
Keywords: buparlisib, breast cancer, PI3K
Corrigendum for this paper has been published
Introduction
Phosphoinositide 3-kinase (PI3K) pathway, a critical signal transduction system which links oncogenes and several receptor classes to different key cellular functions, is one of the most widely activated signaling pathways in cancer.1
The family of lipid kinases called phosphoinositide 3-kinases (PI3Ks) plays essential regulatory roles in different cellular processes including cell survival, proliferation, differentiation, cytoskeletal organization, and glucose transport.2–4
There are three classes of PI3Ks which differ in their structural characteristics and substrate specificity.5,6 Class I enzymes are the most commonly studied; they are activated directly by cell surface receptors. Furthermore, class I PI3Ks are divided into class IA enzymes, which are activated by RTKs, GPCRs, and some oncogenes such as the small G protein Ras, and class IB enzymes, which are regulated only by GPCRs.
These enzymes – through the formation of the second messenger phosphatidylinositol (3,4,5)-trisphosphate – activate many target proteins, most notably phosphoinositide-dependent kinase-1. The downstream targets of these protein kinases, such as mammalian target of rapamycin (mTOR), BCL2-associated agonist of cell death, and forkhead box O proteins, regulate proliferation, growth, and survival.7
Alteration of the PI3K/AKT/mTOR pathway is key for the growth and survival of several cancers, including breast cancer (BC). Different aberrations in the PI3K signaling pathway, such as PI3K mutation/amplification, loss/mutation of the phosphatase and tensin homologue, AKT overexpression/overactivation, and modulation of tuberous sclerosis protein 1 and 2 tumor suppressors, may be often observed in BC, predominantly in hormone receptor-positive (HR+) tumors.8 Especially, as PI3K is the most proximal component of the pathway, targeting PI3K itself rather than AKT or mTOR with PI3K inhibitors may induce pronounced inhibition of the downstream components within the pathway. Many inhibitors of mTOR have been developed for the treatment of cancers, mostly analogs of rapamycin, which specifically inhibit the activity of the TORC1 complex.9 However, specific inhibition of TORC1 results in a feedback stimulation of AKT, thus providing an inferior therapeutic efficacy compared to inhibition of PI3K and both TORC complexes.10 Moreover, activation of PI3K pathway may be associated with resistance to a variety of antitumor agents.11–13
Targeting PI3K pathway
HR+ tumor is the most frequent subtype of BC, with endocrine therapy (ET)-based regimens being its backbone of treatment.14,15 However, HR+ BC is not homogeneous, but characterized by different genomic alterations that on one hand may affect treatment outcomes and on the other hand may offer many therapeutic opportunities with the use of targeted agents.14,16 Of particular interest, activating PIK3CA mutations (which encode the p110α isoform of PI3K) are frequently detected in HR+ BC and are possibly associated with disease progression and resistance to ET.14,17–19
Roughly, 40% of human epidermal growth factor receptor 2-positive (HER2+) BCs harbor activating mutations in PIK3CA.14 These two oncogenes have different functions and may work together to stimulate tumor growth. Several anti-HER2 agents are approved for the treatment of patients with HER2-positive BC. Nonetheless, both de novo and acquired resistance to anti-HER2 therapies may occur.20 PIK3CA mutations might be implicated in conferring resistance to these therapies.21–23 Given that PIK3CA-mutant BC appears to have distinct tumor biology, development of more individualized targeted therapies based on the PIK3CA genotype is awaited.
Therefore, targeting PI3K may be a valid therapeutic option in these settings. In order to maximize treatment efficacy, it would be crucial to identify those patients with PIK3CA mutations who might derive the greatest benefit from PI3K inhibitors.
Several clinical studies have been conducted – mainly in the setting of HR+/HER2− metastatic disease – to explore the combination of endocrine therapies with agents targeting PI3K/Akt/mTOR, such as PI3K inhibitors (panspecific or specific to the subunit 110 α or δ), AKT inhibitors, mTOR inhibitors, or dual inhibitors of mTOR and PI3K. Among these drugs, everolimus is the only PI3K inhibitor approved in combination with exemestane for the treatment of postmenopausal patients with endocrine-resistant HR+/HER2− BC.24 An exploratory analysis of the BOLERO-2 study showed that the benefit of adding everolimus to ET was consistent regardless of PIK3CA alteration.25 AZD2014 is a promising dual inhibitor of mTORC1 (rapamycin sensitive) and mTORC2 (rapamycin insensitive), which is currently under investigation in a randomized Phase II trial (MANTA).26 Temsirolimus was tested in the Horizon study in combination with letrozole, but no benefit in terms of progression-free survival (PFS) was observed compared to letrozole alone.27 The FERGI trial assessed the combination of pictilisib (GDC-0941), an oral inhibitor of multiple class I PI3K kinase isoforms, and fulvestrant.28 This study demonstrated a nonsignificant benefit in terms of PFS in the pictilisib arm compared with the placebo arm (6.2 vs 3.8 months; hazard ratio, 0.77; 95% CI, 0.50–1.19), independent of PIK3CA mutation status.28 Alternative strategies include specific inhibition of subunit α PI3K (alpelisib) or mutated PI3K (taselisib); initial results from the LORELEI study were presented at European Society for Medical Oncology 2017 and showed a benefit in terms of objective response rate for the addition of taselisib to neoadjuvant letrozole in postmenopausal patients with HR+/HER2− early BC.
Buparlisib is a potent and highly specific oral pan-class I PI3K inhibitor, which is currently under investigation in patients with a variety of solid tumors, including BC. In this article, we focus on the main characteristics of buparlisib and its potential application in the treatment of BC.
Buparlisib has demonstrated preliminary activity in preclinical models, providing a rationale for its use in clinical practice.29 Preclinical data showed that this drug, at high concentrations, might cause cell death in various cellular systems, irrespective of the level of PI3K addiction. Moreover, this agent may interfere with microtubule assembling, inducing cell cycle arrest at G2–M phase. However, at doses and schedules used in clinical settings, these effects may not occur.30
In early-phase clinical studies, buparlisib showed encouraging tolerability profile, but modest clinical efficacy as a single agent.31 As the PI3K pathway is involved in resistance to anticancer treatments, a more promising therapeutic strategy likely consists in combining buparlisib, and PI3K inhibitors in general, with other agents in order to restore sensitivity to such treatments.32–35 Different trials testing the combination of this PI3K inhibitor with various anticancer drugs are ongoing and are summarized in Table 1.
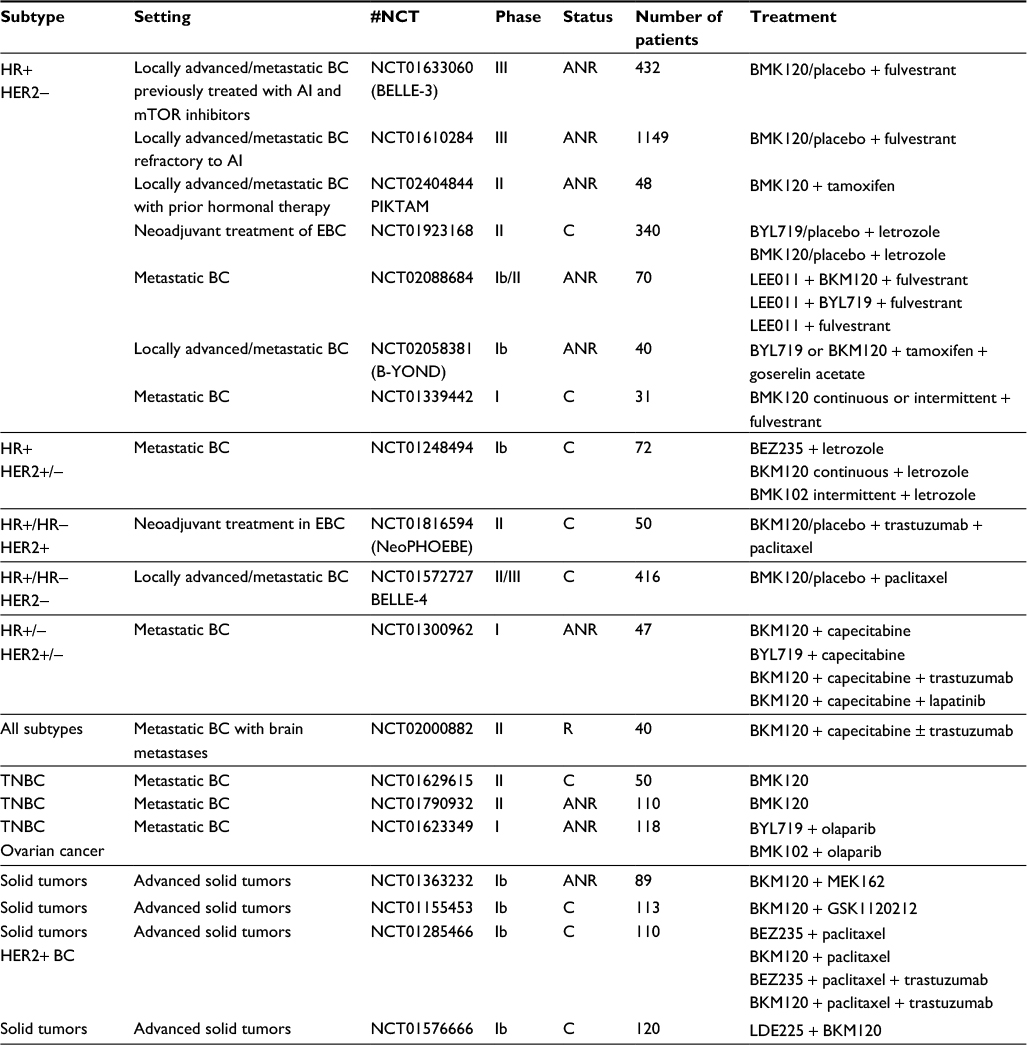 | Table 1 Ongoing trials testing the combination of buparlisib with various anti-cancer drugs Abbreviations: AI, aromatase inhibitors; ANR, active, not recruiting; BC, breast cancer; C, completed; EBC, early breast cancer; HER2, human epidermal growth factor receptor 2; HR, hormone receptor; mTOR, mammalian target of rapamycin; R, recruiting; TNBC, triple-negative breast cancer. |
Materials and methods
We searched PubMed for articles published anytime until September 23, 2017, using the terms “buparlisib” AND “breast cancer”, and identified 45 full-text articles in English. Of these articles, 10 publications were reviews. Five publications reported original data from Phase I clinical trials involving buparlisib in BC, and one publication related to Phase III trial of buparlisib in BC was identified.
HR+/HER2– BC
The combination of a PI3K inhibitor and endocrine treatment was tested in a Phase IB study of buparlisib plus letrozole in patients with HR+/HER2− metastatic BC whose disease was refractory to ET.36 In this study, 46 patients were evaluable for response and 1 achieved complete response, 1 partial response, and 25 patients had stable disease. Clinical activity did not correlate with PIK3CA mutational status, thus suggesting that other alterations in the pathway might be responsible for PI3K pathway dependence.36
In the Phase III randomized, double-blind, placebo-controlled, multicenter BELLE-2 study, postmenopausal patients with HR+/HER2− metastatic BC, who had progressed on/after aromatase inhibitor therapy, were randomized 1:1 to receive either buparlisib (100 mg/day) or placebo, starting on day 15 of cycle 1, plus fulvestrant (500 mg) on days 1 and 15 of cycle 1, and thereafter on day 1 of 28-day cycles.37
PI3K pathway activation status (activated vs non-activated vs and unknown) and visceral disease status (present vs absent) were stratification factors. The objective of the trial was to assess the predictive value of single PI3K–AKT–mTOR pathway alterations on the clinical response of buparlisib. The primary endpoints were PFS in the overall population, in patients with known (either activated or non-activated) PI3K pathway status, and in patients with PI3K pathway activation.37
One thousand one hundred forty-seven patients were included in this trial; 576 received buparlisib plus fulvestrant and 571 received placebo plus fulvestrant.37 Median PFS was 6.9 vs 5.0 months in the overall population, receiving either buparlisib or placebo, respectively (p=0.00021). Patients with known PI3K status had a median PFS of 6.8 months when treated with buparlisib and 4.5 months with placebo (p=0.0033). In patients with PI3K pathway activation, median PFS was 6.8 vs 4.0 months in the buparlisib and placebo groups, respectively (p=0.014). Patients receiving buparlisib experienced more frequently grade 3–4 increased alanine aminotransferase (25% vs 1%), increased aspartate aminotransferase (18% vs 3%), hyperglycemia (15% vs <1%), and rash (8% vs none).37 Twenty-three percent of patients treated with buparlisib had serious adverse events as compared to 16% of patients who received placebo. Finally, this study showed that PI3K inhibition combined with ET is effective in patients with HR+/HER2− metastatic BC, but the toxicity associated with this combination is not negligible.37
PI3K pathway activation in preclinical models of BC is associated with tumor growth and resistance to anticancer therapies, including paclitaxel. Furthermore, buparlisib has shown synergistic activity when combined with paclitaxel in preclinical and clinical models. Therefore, the double-blind, placebo-controlled, adaptive Phase II/III BELLE-4 trial randomized (1:1) patients with untreated metastatic HER2− BC to receive either buparlisib or placebo with paclitaxel.38 PI3K pathway activation and HR status were the stratification factors. The primary endpoint was PFS in the overall and PI3K pathway-activated populations. An adaptive interim analysis following the Phase II part of the study was planned to decide whether to go or not to Phase III (in the overall or PI3K pathway-activated population).38 Four hundred sixteen patients were entered in the trial. At adaptive interim analysis, PFS did not increase with buparlisib over placebo, either in the overall or in the PI3K pathway-activated population. Therefore, the study was stopped for futility.38
HER2+ BC
The PI3K/AKT/mTOR pathway is often activated in HER2+ BC.39 The Phase III BOLERO-1 trial combined trastuzumab and paclitaxel with or without everolimus in this setting and showed no benefit in terms of PFS in the overall population. Nevertheless, in the subgroup of patients with HR−/HER2+, there was an improvement – although not statistically significant – in median PFS of 7.2 months.40 Moreover, in the Phase III BOLERO-3 trial, addition of everolimus to trastuzumab and vinorelbine led to an increased median PFS (from 5.78 to 7.00 months) in women with HER2+ metastatic BC.41 A combined analysis of BOLERO-1 and BOLERO-3 demonstrated a strong positive correlation between PI3K pathway activation and PFS increase.42 These findings pushed the investigation of PI3K inhibitors in HER+ BC.
In the CLEOPATRA study, which assessed the combination of pertuzumab, trastuzumab, and docetaxel in first-line setting,43 PIK3CA mutation was established as a negative prognostic factor in HER2+ BC.44 Patients with PIK3CA mutant tumors had a significantly worse PFS than those with PIK3CA non-mutant tumors.44
A Phase IB/II study has investigated the combination of buparlisib and trastuzumab in HER2+ BC, who previously progressed on trastuzumab.45 In this study, buparlisib and trastuzumab combination was well tolerated, with preliminary signs of clinical activity.45
The Phase II NeoPHOEBE neoadjuvant study was designed to evaluate the combination of buparlisib with trastuzumab and paclitaxel in HER2+ BC in two independent cohorts by PIK3CA mutation status; furthermore, each cohort was stratified by HR status.46 The main objective of the trial was to assess whether PIK3CA mutation status may predict a benefit from combining anti-HER2 therapy and a PI3K inhibitor.46 The trial was designed to enroll 256 patients. However, after enrollment of the first 50 patients, recruitment was stopped due to liver toxicity.46 A trend toward higher objective response rate (68.8% vs 33.3%; p=0.053) and a significant decrease in Ki67 (75% vs 26.7%; p=0.021) was observed with buparlisib vs placebo in the HR+ subgroup.46 Unfortunately, such a treatment strategy does not appear to be feasible.
Buparlisib has also been studied in combination with lapatinib in a Phase IB/II trial including patients with HER2+, trastuzumab-resistant, metastatic BC.47 In the Phase IB part, the PI3K pathway activation status was assessed in a retrospective exploratory analysis, and evidence of PI3K pathway activation was an inclusion criterion for Phase II. Twenty-four patients were treated at five dose levels. Main drug-related adverse events included diarrhea, nausea, skin rash, asthenia, depression, anxiety, and increase in transaminases. Disease control rate was 79% (one patient with complete response and six patients with stable disease for ≥24 weeks).47 Finally, this trial showed preliminary evidence of antitumor activity in this heavily pretreated population with manageable safety profile.
Discussion
The PI3K pathway represents both an opportunity and a challenge for cancer treatment. In this review, we have highlighted both major issues and recent progress made in the understanding of the PI3K pathway. We have analyzed both the challenges and promises for the therapeutic development of PI3K pathway inhibitors in BC.
PI3K inhibitors have been validated as promising anticancer targets, and tremendous efforts have been made to develop this class of agents for cancer therapy. However, results of these drugs used as monotherapy have been modest thus far.
Overall, results from studies combining PI3K inhibition with different anticancer agents – although with inconsistent efficacy – showed consistent substantial toxicity; hence, no further studies are being pursued.
A potential role for PI3K-targeted therapy may warrant further investigation with better-tolerated second-generation PI3K inhibitors.
Several controversies remain regarding the best treatment strategy for PI3K inhibition between pan-PI3K and isoform-selective inhibitors. Different agents/class of agents may be more active in tumors with defined molecular characteristics.
Further research is needed to assess biomarkers predictive of response to PI3K inhibitor treatment. No correlation between PI3K/AKT/mTOR pathway aberrations and clinical response has been found to date.
Finally, the potential mechanisms of PI3K inhibitor resistance are still unknown and the complexity of PI3K/AKT/mTOR pathway with extensive crosstalk with other signaling provides general possibility to overcome Pi3K inhibition. A deeper understanding of resistance mechanisms will enable rational design of combination regimens.
With the recent knowledge regarding the divergent roles of PI3K isoforms in different types of cancer, isoform-selective PI3K inhibitors have attracted increasing interest for precise cancer therapy, and over a dozen inhibitors are undergoing clinical evaluation (Table 1). Among them, CAL101 has been approved by the US Food and Drug Administration for patients with relapsed chronic lymphocytic leukemia and indolent lymphoma.
Interestingly, the PI3K pathway may be considered – at the same time – a dramatic therapeutic opportunity and a big challenge for cancer therapy with no doubt. Drugs targeting PI3K (or AKT or mTOR) are under investigation at different stages; there are potential issues associated with their toxicity and resistance. Oncogenic changes in PI3K pathway components may render cancer cells resistant to PI3K inhibition. Hence, it is important to identify new therapeutic targets for the development of agents that may either replace PI3K inhibitors or enhance the efficacy of PI3K inhibitors.
Further investigation is required to determine the best setting and the best combinations.
Disclosure
The authors report no conflicts of interest in this work.
References
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644. | ||
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619. | ||
Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5(12):921–929. | ||
Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. | ||
Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. | ||
Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. | ||
Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008;8(3):187–198. | ||
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. | ||
Chan S. Targeting the mammalian target of rapamycin (mTOR): a new approach to treating cancer. Br J Cancer. 2004;91(8):1420–1424. | ||
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. | ||
Fekete M, Santiskulvong C, Eng C, Dorigo O. Effect of PI3K/Akt pathway inhibition-mediated G1 arrest on chemosensitization in ovarian cancer cells. Anticancer Res. 2012;32(2):445–452. | ||
Shi Y, Chen L, Li J, et al. Prognostic and predictive values of pERK1/2 and pAkt-1 expression in non-small cell lung cancer patients treated with adjuvant chemotherapy. Tumour Biol. 2011;32(2):381–390. | ||
Carden CP, Stewart A, Thavasu P, et al. The association of PI3 kinase signaling and chemoresistance in advanced ovarian cancer. Mol Cancer Ther. 2012;11(7):1609–1617. | ||
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. | ||
Cardoso F, Costa A, Senkus E, et al. 3rd ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 3). Ann Oncol. 2017;28(Suppl 12). | ||
Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. | ||
Miller TW, Hennessy BT, Gonzalez-Angulo AM, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120(7):2406–2413. | ||
Bosch A, Li Z, Bergamaschi A, et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med. 2015;7(283):283ra51. | ||
Sanchez CG, Ma CX, Crowder RJ, et al. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011;13(2):R21. | ||
Garrett JT, Arteaga CL. Resistance to HER2-directed antibodies and tyrosine kinase inhibitors: mechanisms and clinical implications. Cancer Biol Ther. 2011;11(9):793–800. | ||
Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. | ||
Chandarlapaty S, Sakr RA, Giri D, et al. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin Cancer Res. 2012;18(24):6784–6791. | ||
Esteva FJ, Guo H, Zhang S, et al. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol. 2010;177(4):1647–1656. | ||
Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–529. | ||
Hortobagyi GN, Chen D, Piccart M, et al. Correlative analysis of genetic alterations and everolimus benefit in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from BOLERO-2. J Clin Oncol. 2016;34(5):419–426. | ||
Jordan NJ, Dutkowski CM, Barrow D, et al. Impact of dual mTORC1/2 mTOR kinase inhibitor AZD8055 on acquired endocrine resistance in breast cancer in vitro. Breast Cancer Res. 2014;16(1):R12. | ||
Wolff AC, Lazar AA, Bondarenko I, et al. Randomized Phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J Clin Oncol. 2013;31(2):195–202. | ||
Krop IE, Mayer IA, Ganju V, et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17(6):811–821. | ||
Maira SM, Pecchi S, Huang A, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11(2):317–328. | ||
Brachmann SM, Kleylein-Sohn J, Gaulis S, et al. Characterization of the mechanism of action of the pan class I PI3K inhibitor NVP-BKM120 across a broad range of concentrations. Mol Cancer Ther. 2012;11(8):1747–1757. | ||
Rodon J, Brana I, Siu LL, et al. Phase I dose-escalation and-expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2014;32(4):670–681. | ||
LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11(1–2):32–50. | ||
West KA, Castillo SS, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist Updat. 2002;5(6):234–248. | ||
Massacesi C, Di Tomaso E, Urban P, et al. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Onco Targets Ther. 2016;9:203–210. | ||
Burris HA 3rd. Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. 2013;71(4):829–842. | ||
Mayer IA, Abramson VG, Isakoff SJ, et al. Stand up to cancer phase Ib study of pan-phosphoinositide-3-kinase inhibitor buparlisib with letrozole in estrogen receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2014;32(12):1202–1209. | ||
Baselga J, Im SA, Iwata H, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(7):904–916. | ||
Martin M, Chan A, Dirix L, et al. A randomized adaptive phase II/III study of buparlisib, a pan-class I PI3K inhibitor, combined with paclitaxel for the treatment of HER2-advanced breast cancer (BELLE-4). Ann Oncol. 2017;28(2):313–320. | ||
Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13(6):224. | ||
Hurvitz SA, Andre F, Jiang Z, et al. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): a phase 3, randomised, double-blind, multicentre trial. Lancet Oncol. 2015;16(7):816–829. | ||
Andre F, O’Regan R, Ozguroglu M, et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014;15(6):580–591. | ||
Andre F, Hurvitz S, Fasolo A, et al. Molecular Alterations and everolimus efficacy in human epidermal growth factor receptor 2-overexpressing metastatic breast cancers: combined exploratory biomarker analysis from BOLERO-1 and BOLERO-3. J Clin Oncol. 2016;34(18):2115–2124. | ||
Swain SM, Kim SB, Cortes J, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA study): overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013;14(6):461–471. | ||
Baselga J, Cortes J, Im SA, et al. Biomarker analyses in CLEOPATRA: a phase III, placebo-controlled study of pertuzumab in human epidermal growth factor receptor 2-positive, first-line metastatic breast cancer. J Clin Oncol. 2014;32(33):3753–3761. | ||
Saura C, Bendell J, Jerusalem G, et al. Phase Ib study of buparlisib plus trastuzumab in patients with HER2-positive advanced or metastatic breast cancer that has progressed on Trastuzumab-based therapy. Clin Cancer Res. 2014;20(7):1935–1945. | ||
Loibl S, de la Pena L, Nekljudova V, et al. Neoadjuvant buparlisib plus trastuzumab and paclitaxel for women with HER2+ primary breast cancer: a randomised, double-blind, placebo-controlled phase II trial (NeoPHOEBE). Eur J Cancer. 2017;85:133–145. | ||
Guerin M, Rezai K, Isambert N, et al. PIKHER2: A phase IB study evaluating buparlisib in combination with lapatinib in trastuzumab-resistant HER2-positive advanced breast cancer. Eur J Cancer. 2017;86:28–36. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.