")
Back to Journals » HIV/AIDS - Research and Palliative Care » Volume 10
Profile of bictegravir/emtricitabine/tenofovir alafenamide fixed dose combination and its potential in the treatment of HIV-1 infection: evidence to date
Authors Hill L , Smith SR, Karris MY
Received 11 July 2018
Accepted for publication 30 September 2018
Published 29 October 2018 Volume 2018:10 Pages 203—213
DOI https://doi.org/10.2147/HIV.S145529
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bassel Sawaya
Lucas Hill,1 Shawn R Smith,1 Maile Young Karris2
1University of California San Diego Skaggs School of Pharmacy and Pharmaceutical Sciences, San Diego, CA, USA; 2Department of Medicine, University of California San Diego, San Diego, CA, USA
Abstract: Modern pharmacologic management of people living with HIV involves the use of fixed dose combinations of antiretrovirals that are simple to take, well tolerated, and highly effective. Specific recent pharmacologic advancements include 1) the second-generation integrase strand transfer inhibitors (dolutegravir and bictegravir) that consistently show less side effects, high tolerability, minimal drug interactions, and rapid rates of HIV viral load decline and 2) tenofovir alafenamide, a prodrug of tenofovir that concentrates in lymphoid tissue and minimizes off target effects. Bictegravir/emtricitabine/tenofovir alafenamide or B/F/TAF is a recently approved fixed dose combination that incorporates these new advancements in the management of HIV. This review focuses on the data supporting the use of B/F/TAF, reviews clinically relevant findings, and highlights the unanswered questions that may limit its clinical utility.
Keywords: Biktarvy, HIV care, review, integrase strand transfer inhibitor
Introduction
Advancements in antiretroviral therapy (ART) broadened the treatment options for people living with HIV (PLWH) who are both naïve to therapy and treatment experienced. Newer classes of medications provide high genetic barriers to resistance, excellent tolerability, and are often co-formulated into fixed dose combinations that simplify the dosing. Recently, the US Food and Drug Administration (FDA) added to the available options for many PLWH with the approval of bictegravir (BIC).
BIC is a novel integrase strand transfer inhibitor (INSTI) approved by the FDA in February 2018 for PLWH who are treatment naïve and for PLWH virologically suppressed on current ART for at least 3 months with no history of prior treatment failure or resistance to any component of the BIC single tablet regimen (STR). BIC has been shown to potently inhibit strand transfer activity at concentrations similar to that of elvitegravir (EV G) and dolutegravir (DTG) with its unique structure minimizing drug–drug interactions, increasing protein binding, and improving solubility. BIC has a favorable pharmacokinetic (PK) profile in that it can be dosed once daily and is not impacted by renal or hepatic impairment. Although BIC is a substrate of CYP3A4 and UGTI1A1, it does not require pharmacologic boosting resulting in minimal drug interactions. In vitro, BIC demonstrates a high genetic barrier to resistance allowing it to maintain activity against variants with INSTI resistance.1–5 Currently, BIC 50 mg daily is available only as a fixed dose combination with tenofovir alafenamide (TAF) 25 mg daily and emtricitabine (FTC) 200 mg daily (B/F/TAF).
In this review, we aimed to 1) review the available literature supporting approved and potential off-label use of BIC, 2) discuss the clinical situations in which it might be used, and 3) review some of the unanswered questions about its place in therapy.
Evidence in treatment-naïve PLWH
BIC first demonstrated promise in a Phase Ib study of 20 HIV-infected ART naïve adults randomized to once daily BIC (5, 25, 50, or 100 mg) or placebo for 10 days.6 BIC showed rapid absorption, had a half-life period supporting once daily therapy, and was well tolerated with no discontinuations due to adverse events. Most importantly, significant reductions in plasma HIV-1 RNA occurred with all BIC doses. Thus BIC proceeded to a Phase II double-blind trial that randomized 98 participants 2:1 to receive oral once daily BIC 75 mg or DTG 50 mg with matching placebo plus the fixed dose combination of 200 mg FTC and 25 mg TAF for 48 weeks.7 Previously untreated PLWH aged ≥18 years with HIV RNA ≥1,000 copies per mL (c/mL), CD4 counts of at least 200 cells/µL, and estimated glomerular filtration (eGFR) rates of at least 70 mL/min were included. Participants were excluded if they were hepatitis B or hepatitis C co-infected, had new AIDS-defining conditions within 30 days of screening, or were pregnant. Interestingly, 97% of participants in the BIC group achieved viral suppression at 24 and 48 weeks, vs 94% at 24 weeks and 91% at 48 weeks in the DTG arm. Treatment-emergent adverse events were reported by 55 (85%) of 65 participants in the BIC group vs 22 (67%) of 33 in the DTG group. The most common adverse events were diarrhea (8 [12%] vs 4 [12%]) and nausea (5 [8%] vs 4 [12%]) in the BIC and DTG groups, respectively. These promising results then led to two Phase III trials of B/F/TAF.
Gallant et al conducted a multicenter non-inferiority study in Europe, Latin America, and North America (Trial 1489) where participants (n=629) were randomly assigned to co-formulated B/F/TAF or co-formulated DTG 50 mg, abacavir 600 mg (ABC), and lamivudine 300 mg (3TC), with matching placebo, once daily for 144 weeks.8 Eligibility criteria included treatment-naïve HIV-1 infected adults, HIV-1 RNA ≥500 c/mL, HLA-B*5701-negative, and eGFR rate of ≥50 mL/min. Patients with hepatitis B infection were excluded. Randomization was stratified by HIV-1 RNA (≤100,000 c/mL,>100,000 to≤400,000 c/mL, or >400,000 c/mL), CD4 count (<50 cells/µL, 50–199 cells/µL, or ≥200 cells/µL), and region (USA or ex-USA). Study participants were predominantly males (90%–91%), white (57%), with median CD4 444–450 cells/µL. Both the arms demonstrated a high level of viral suppression at 48 weeks with 92% of participants in the B/F/TAF arm and 93% of participants in the DTG/ABC/3TC arm demonstrating non-inferiority (P=0.78). The DTG arm had numerically higher suppression rates in certain populations such as patients with viral loads >100,000 c/mL (86% vs 94%), CD4 counts <200 cells/µL (95% vs 100%), and those with poor adherence (84% vs 90%). However, these differences were not statistically significant. Adverse events were less common in the B/F/TAF group vs the DTG/ABC/3TC group (26% vs 40%) driven predominantly by the higher incidence of drug-related nausea in the DTG/ABC/3TC group (5% vs 17%; P<0.001).
In a second Phase III randomized controlled clinical trial (Trial 1490), Sax et al evaluated B/F/TAF for non-inferiority compared to DTG+F/TAF in 645 participants.9 This study focused on treatment naïve HIV-infected adults with HIV-1 RNA ≥500 c/mL and eGFR ≥30 mL/min, but also allowed chronic hepatitis B virus and hepatitis C co-infection. Similar to Trial 1489, the study population was predominantly male (88%–89%), white (57%–60%), and had a median CD4 440–441 cells/µL. At week 48, 89% of participants in the BIC group and 93% in the DTG group achieved viral suppression showing non-inferiority of BIC regimen to the DTG regimen (P=0.12). Again, study drug–related adverse events were less common in the BIC group (18%) than in the DTG group (26%) with the most common adverse events being headache, diarrhea, and nausea.
A pooled efficacy and resistance analysis of the two Phase III trials demonstrated that treatment with B/F/TAF, DTG/ABC/3TC, or DTG+F/TAF was highly efficacious through week 48 with no development of resistance.10 Pre-existing drug resistance did not affect the efficacy outcomes and no participants developed resistance to study drugs on B/F/TAF or in any other treatment group. Collectively, these studies (summarized in Table 1) confirm the efficacy of B/F/TAF in treatment-naïve patients and lead to the Department of Health and Human Services recommending B/F/TAF as a preferred treatment regimen in treatment-naïve PLWH.4,5
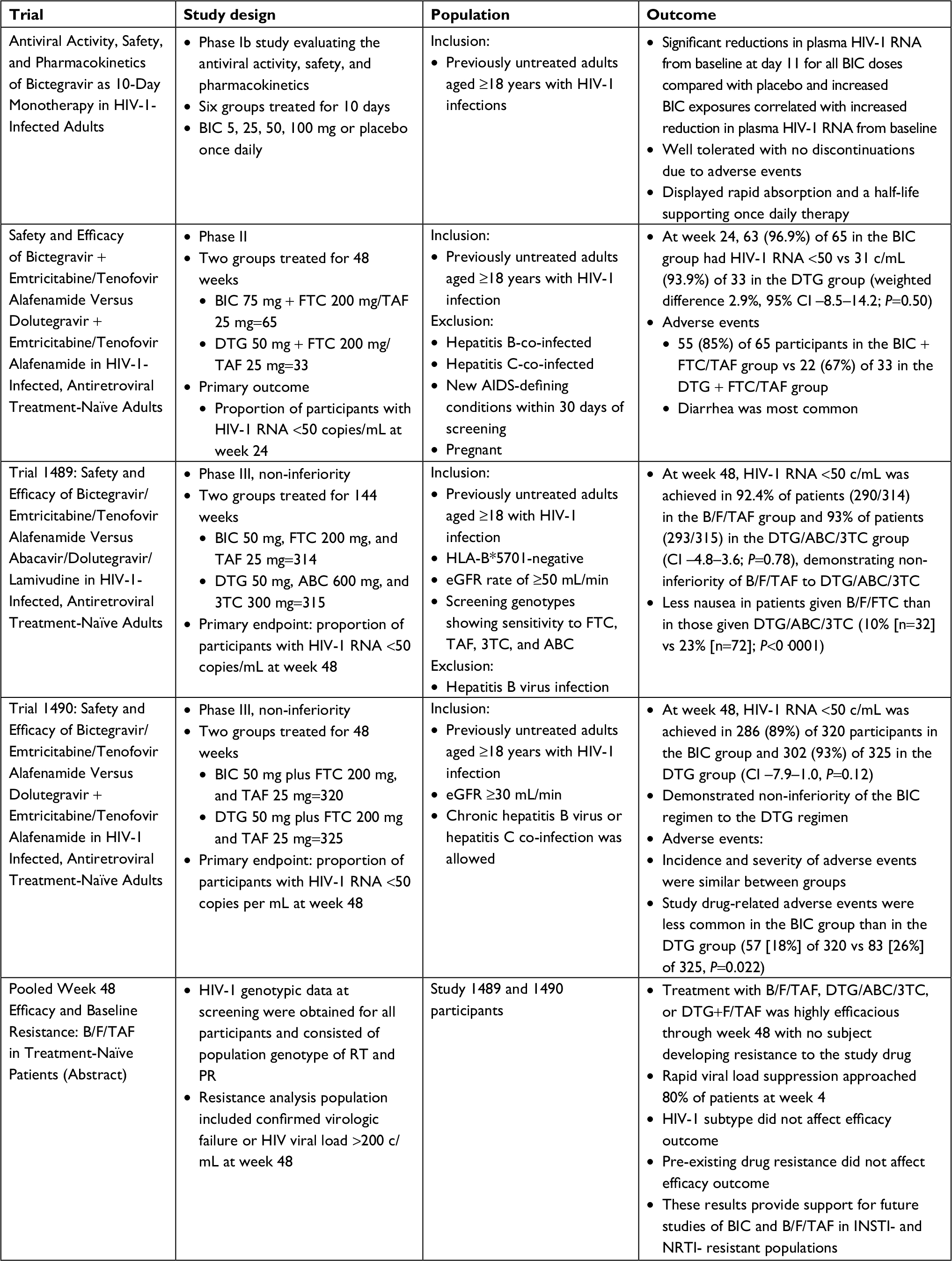 | Table 1 Summary of treatment-naïve studies Abbreviations: 3TC, lamivudine; ABC, abacavir; B/F/TAF, bictegravir/emtricitabine/tenofovir alafenamide single tablet; BIC, bictegravir; DTG, dolutegravir; FTC, emtricitabine; INSTI, integrase strand transfer inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; PR, protease; RT, reverse transcriptase; TAF, tenofovir alafenamide. |
Evidence in treatment-experienced PLWH
BIC use in PLWH who are treatment experienced is limited to studies that evaluate switching ART in PLWH who are virologically suppressed with no known resistance to any component in B/F/TAF (summarized in Table 2). As such, the outcomes included virologic suppression, and other clinically relevant outcomes including kidney function, cholesterol, and other adverse drug effects. Trial 1844, a Phase III, randomized, double-blind, multicenter study evaluated 282 participants who switched from DTG + ABC/3TC or DTG/ABC/3TC to B/F/TAF and 281 who continued DTG + ABC/3TC or DTG/ABC/3TC.11 Patients with active hepatitis B co-infection or eGFR <50 mL/min were excluded from participation. As in previous studies, the participants were predominantly male (88%–90%), white (73%), with median CD4 661–732 cells/µL. In the FDA snapshot analysis at 48 weeks, 1.1% in the BIC arm and 0.4% in the DTG arm had HIV-1 RNA ≥50 c/mL for a difference of 0.7% meeting non-inferiority end-point. Fewer participants in the BIC arm reported drug-related adverse events (8%) as compared to the DTG arm (16%) (P=0.01). The most common adverse events were headache, abnormal dreams, diarrhea, fatigue, flatulence, nausea, and insomnia. A statistically significant difference in the change in eGFR at week 48 was observed, with a 1 mL/min increase in the BIC arm and a 1.8 mL/min decrease in the DTG arm (P=0.001); however, no significant differences in markers of tubular damage were observed (ie, urine albumin:creatinine ratio, retinol binding protein:creatinine ratio, or β-2-microglobulin:creatinine ratio). In addition, there were no significant differences in spine and hip bone mineral density, total cholesterol, LDL, or HDL. The BIC group did have a slight reduction in triglyceride levels of 5 mg/dL compared to a 3 mg/dL increase in the DTG group (P=0.028). Overall, the clinical relevance of these differences remains unknown.
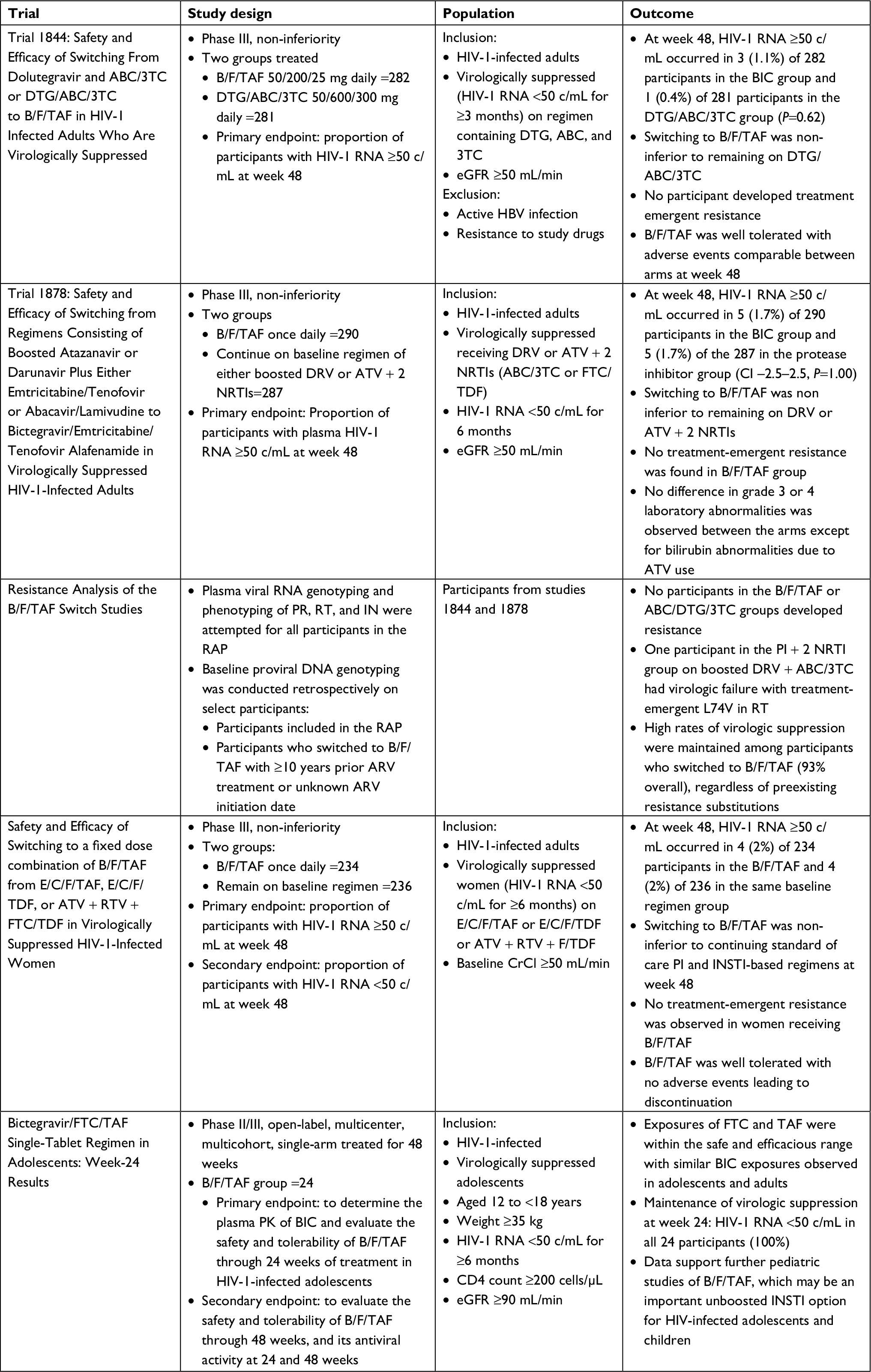 | Table 2 Summary of treatment-experienced switch studies Abbreviations: 3TC, lamivudine; ABC, abacavir; ARV, antiretroviral; ATV, atazanavir; BIC, bictegravir; B/F/TAF, bictegravir/emtricitabine/tenofovir alafenamide single tablet; CrCl, creatinine clearance; DRV, darunavir; DTG, dolutegravir; E/C/F/TAF, elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide; E/C/F/TDF, elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate; FTC, emtricitabine; IN, integrase; INSTI, integrase strand transfer inhibitor; PK, pharmacokinetic; PR, protease; RAP, resistance analysis population; RT, reverse transcriptase; RTV, ritonavir; TAF, bictegravir; TAF, tenofovir alafenamide; PI, protease inhibitor. |
The efficacy of switching from boosted protease inhibitors to B/F/TAF has also been evaluated (Trial 1878) in a Phase III multicenter, randomized, open-label trial.12 Participants on either ritonavir-boosted darunavir (DRV) or atazanavir (ATV) in combination with two nucleoside reverse transcriptase inhibitors (NRTIs) were randomized to stay on their current regimen (n=287) or switch to B/F/TAF (n=290). Again, the study included primarily white males with well controlled HIV. A slightly higher proportion of participants were on a DRV-based regimen (54%–57%) than on an ATV-based regimen (43%–46%) but most were combined with a NRTI backbone of tenofovir disoproxil fumarate (TDF)/FTC. The primary outcome was the proportion of patients with HIV-1 RNA ≥50 c/mL at week 48 using the FDA snapshot analysis with a non-inferiority margin of 4%. At week 48, 1.7% in both the arms had HIV-1 RNA ≥50 c/mL demonstrating non-inferiority. In the BIC arm, 92.1% were virologically suppressed at week 48 compared to 88.9% in the boosted protease inhibitor (PI) arm demonstrating a non-statistically significant difference. Again, no treatment-emergent resistance occurred in the participants switched to B/F/TAF. In terms of safety, 12% in the BIC arm and 4% in the boosted PI arm reported an adverse event, with the difference driven primarily by headache (12% vs 4%); however, the majority were mild in severity. Overall, discontinuations due to adverse events were similar between the two arms. The median change in eGFR in the BIC arm was –4.3 mL/min vs 0.2 mL/min in the boosted PI arm (P<0.001). This observation was not anticipated given the demonstrated improvement in urine albumin to creatinine ratio when switching from TDF to a TAF-based regimen in this study (10% vs –2% respectively). Thus, the authors hypothesized that eGFR differences were likely due to the inhibition of creatinine secretion by BIC. No differences in change from baseline in total cholesterol, LDL, or HDL cholesterol were observed between the two arms; however, switching to the BIC regimen resulted in a median change of –6 mg/dL vs 4 mg/dL in triglycerides (P=0.002) and a median change of –0.2 verses 0 in the total cholesterol: HDL ratio (P=0.033).
In these two studies, participants with known resistance to the components of B/F/TAF were excluded; however, no genotype testing was done at screening and historical genotypes were only collected if available. To better evaluate if pre-existing HIV resistance to any components in B/F/TAF impacted study outcome, a pooled HIV resistance analysis of participants in these studies was conducted.13 The resistance analysis population included confirmed virologic failures (defined as two consecutive visits with HIV-1 RNA ≥50 c/mL and ≥200 c/mL at the confirmation visit or having a HIV-1 RNA ≥200 c/mL at week 48), that also had a phenotype, genotype, or proviral DNA genotype (archived genotype) and all participants who switched to B/F/TAF with ≥10 years of ARV experience or unknown ARV start date who had a proviral DNA genotype. This resulted in 405 participants in the B/F/TAF arm, 125 patients in the PI + 2 NRTI arm, and 138 in the ABC/3TC/DTG arm. In the total resistance cohort of 405 patients, 13% had NRTI resistance, 18% had non nucleoside reverse transcriptase inhibitor (NNRTI)resistance, and 6% had PI resistance at baseline. Only one participant in the B/F/TAF arm demonstrated primary (may have impact on INSTI susceptibility) INSTI resistance at baseline (T97A) but 51% out of 170 evaluated had secondary (unlikely to impact INSTI susceptibility) INSTI resistance. The important take away from the analysis is that no participants in the B/F/TAF arm developed resistance, and 35/36 (97%) of patients with archived FTC or TAF resistance (5/5 with K65R, 18/18 with M184V/I/T, 12/13 with M184V + other NRTI resistance, and 4/4 with ≥2 TAMs) maintained viral suppression through week 48.
Two other treatment-experienced studies involving B/F/TAF have been conducted. The first (study 1961) included virologically suppressed women on elvitegravir/cobicistat/FTC/TAF (E/C/F/TAF), elvitegravir/cobicistat/FTC/TDF (E/C/F/TDF), or TDF/FTC + ATV/ritonavir (ATV/r) and randomized them to stay on their current regimen (n=236) or switch to B/F/TAF (n=234).14 The primary endpoint was HIV-1 RNA ≥50 c/mL at week 48 using the FDA snapshot algorithm with a 4% non-inferiority margin. The median age of the female participants was 39–40 years, 37% were black or African descent, 16% Hispanic/Latino with median CD4 T-cell counts of 667–704 cells/µL. In terms of ARV regimen at randomization, 53% were on E/C/F/TAF, 42% were on E/C/F/TDF, and 5% were on TDF/FTC + ATV/r. For the primary outcome, 1.7% in each arm had HIV-1 RNA ≥50 at week 48 demonstrating non-inferiority of B/F/TAF. Again, no treatment-emergent resistance was observed in patients receiving B/F/TAF. Similar rates of drug-related adverse events, 9% vs 6%, and serious adverse events, 5% vs 6%, were seen in the B/F/TAF and continuation of baseline regimen arms, respectively. Switching from a TDF- to a TAF-based regimen resulted in significant improvement in retinol binding protein:creatinine ratio and beta-2-microglobulin:creatinine ratio which are tubular biomarkers suggesting improved kidney function. In addition, there was no difference in median change in eGFR between BIC arm compared to continuation of baseline arm (−1.8% vs –2.7%). The only significant difference in lipid parameters was a 10 mg/dL decrease in triglycerides in the BIC arm vs a 4 mg/dL increase in the continuation arm (P<0.001).
The final switch study was conducted in children (age 6–11 years) and adolescents (age 12–17 years) who were suppressed on a regimen of two NRTIs plus a third agent.15 The primary endpoint was steady-state PK parameters at week 2 or 4, but also included safety through week 24, and the secondary endpoint was viral suppression at week 24 and 48. In the interim analysis in 24 adolescents, 100% maintained virologic suppression at week 24. There was only one adverse event (grade 1 vomiting) attributed to B/F/TAF suggesting good tolerability in this small population of adolescents.
Other ongoing studies in treatment-experienced PLWH include a Phase III, randomized, double-blind, multicenter study in HIV-suppressed adults on DTG plus F/TDF or F/TAF randomized to stay on their current regimen or switch to B/F/TAF. In this study, participants with any NRTI, NNRTI, or PI resistance will be included; however, they must have no documented INSTI resistance or confirmed virologic failure while on an INSTI regimen. In addition, there is a Phase III, multicenter, open-label, single-arm study in HIV-suppressed adults aged ≥65 years who will be switched to B/F/TAF. Results for these studies are not yet available but will further inform the utility of B/F/TAF in treatment-experienced PLWH.
PKs and drug interactions
The PK of BIC and the other components of the single-tablet regimen allow for use in a broad population of patients. BIC has a plasma half-life period of 17.3 hours allowing for once daily dosing in combination with FTC and TAF which have intracellular half-lives of 39 and 150 hours, respectively.4 BIC is not renally eliminated and therefore does not have meaningful changes in plasma concentrations in patients with severe renal impairment. The PKs of TAF are also not significantly changed in patients with a creatinine clearance (CrCl) down to 15 mL/min; however, due to the renal elimination of FTC, B/F/TAF is not recommended in patients with a CrCl <30 mL/min.1,4,16 BIC and TAF have been studied in patients with moderate hepatic impairment (Child-Pugh B), but not in patients with severe hepatic impairment, and due to lack of data, they are not recommended in this population. FTC is not hepatically metabolized and is expected to be safe to use in patients with hepatic impairment.1,4,16
BIC is metabolized by UGT1A1 and CYP3A4 and may be susceptible to drug interactions with strong inducers or inhibitors of these enzymes. One important drug interaction is with rifampin, a strong CYP3A4 inducer. Studies suggest that when rifampin is provided concurrently with B/F/TAF, BIC exposure is 60% lower with twice daily dosing and 80% lower when dosed once daily.17 Therefore, the use of B/F/TAF in combination with rifampin, rifabutin, or rifapentin as well as other potent inducers such as the anticonvulsants carbamazepine, oxcarbazepine, and phenytoin is not recommended. Of note, the use of TAF with rifampin is still being evaluated as TAF is a p-glycoprotein (P-gp) substrate and rifampin is a P-gp inducer. A recent study suggested that although plasma concentrations of tenofovir are lowered (47% reduction) when TAF is given with rifampin, the intracellular concentrations of tenofovir diphosphate were still 82% higher as compared to the intracellular concentrations achieved with TDF without the use of rifampin.18
Another important interaction to consider is the impact of polyvalent cations on the absorption of BIC. When BIC was given with antacids containing aluminum, magnesium, or calcium simultaneously in a fasted state, BIC exposure decreased almost 80%, and when given 2 hours after the antacid, it decreased to 52%. However, if BIC was given 2 hours before the antacid, the area under the curve (AUC) of BIC was decreased by only 13%. When given with calcium or iron, BIC concentrations are not significantly altered even when given simultaneously as long as it is in a fed state. Therefore, BIC should be given 2 hours prior to aluminum, magnesium, or calcium containing antacids or simultaneously with calcium or iron as long as taken with food.4
Finally, BIC is an inhibitor of the drug transporters OCT2 and MATE1 that may impact other medications that are substrates for these transporters. In previous PK studies with DTG, also an OCT2 and MATE1 inhibitor, the combination of DTG and metformin resulted in an increase of about 80% in metformin exposure which is clinically significant.19 As compared to placebo, co-administration with BIC increased metformin AUC by 39% when studied in 32 healthy subjects. Metformin-induced increases in lactate were similar between placebo and BIC group, suggesting this interaction may not be clinically significant.20
Comparison of available integrase inhibitors
The introduction of the integrase inhibitor class in 2007 was a significant advancement in the treatment of HIV as it provided a new class of medications with a novel mechanism of action, and treatment options for PLWH with acquired drug resistance to multiple classes of ARVs.21 Literature extensively documents that INSTIs are very well tolerated and, with the exception of EVG which requires boosting by cobicistat or ritonavir, carry the advantage of limited drug interactions.22–27 The first-generation INSTIs include raltegravir (RAL) and EVG. In addition to limitations that require twice daily dosing of RAL and issues with drug interactions with boosted EVG, the first-generation INSTIs have a low genetic barrier to resistance.28 DTG, a second-generation INSTI has significant advantages over the first-generation INSTIs in that it can be dosed once daily in patients without documented or inferred INSTI resistance, has a high genetic barrier to resistance, limited drug interactions, and can be used in PLWH with INSTI resistance at doses of 50 mg twice daily.29 BIC carries similar advantages to DTG in that it is dosed once daily and has limited drug interactions. In vitro data evaluating BIC against 47 clinical isolates with INSTI resistance mutations suggests a high genetic barrier to resistance which is discussed below in more detail.30 BIC is also the only unboosted integrase inhibitor that is available co-formulated with TAF/FTC and is the smallest tablet size among the INSTI-based single-tablet regimens available. The primary limitation with BIC as compared to the other INSTI is that studies with BIC have been limited to either the treatment-naïve population or PLWH who are already suppressed on their current ARV regimen and switching to BIC. Unlike the other INSTIs, BIC has not been studied in treatment-experienced populations (Table 3). In addition, although BIC shows in vitro activity against INSTI-resistant strains, clinical trial data are absent to guide use in PLWH with documented or suspected INSTI resistance, as compared to DTG which carries an indication for PLWH with INSTI resistance.31
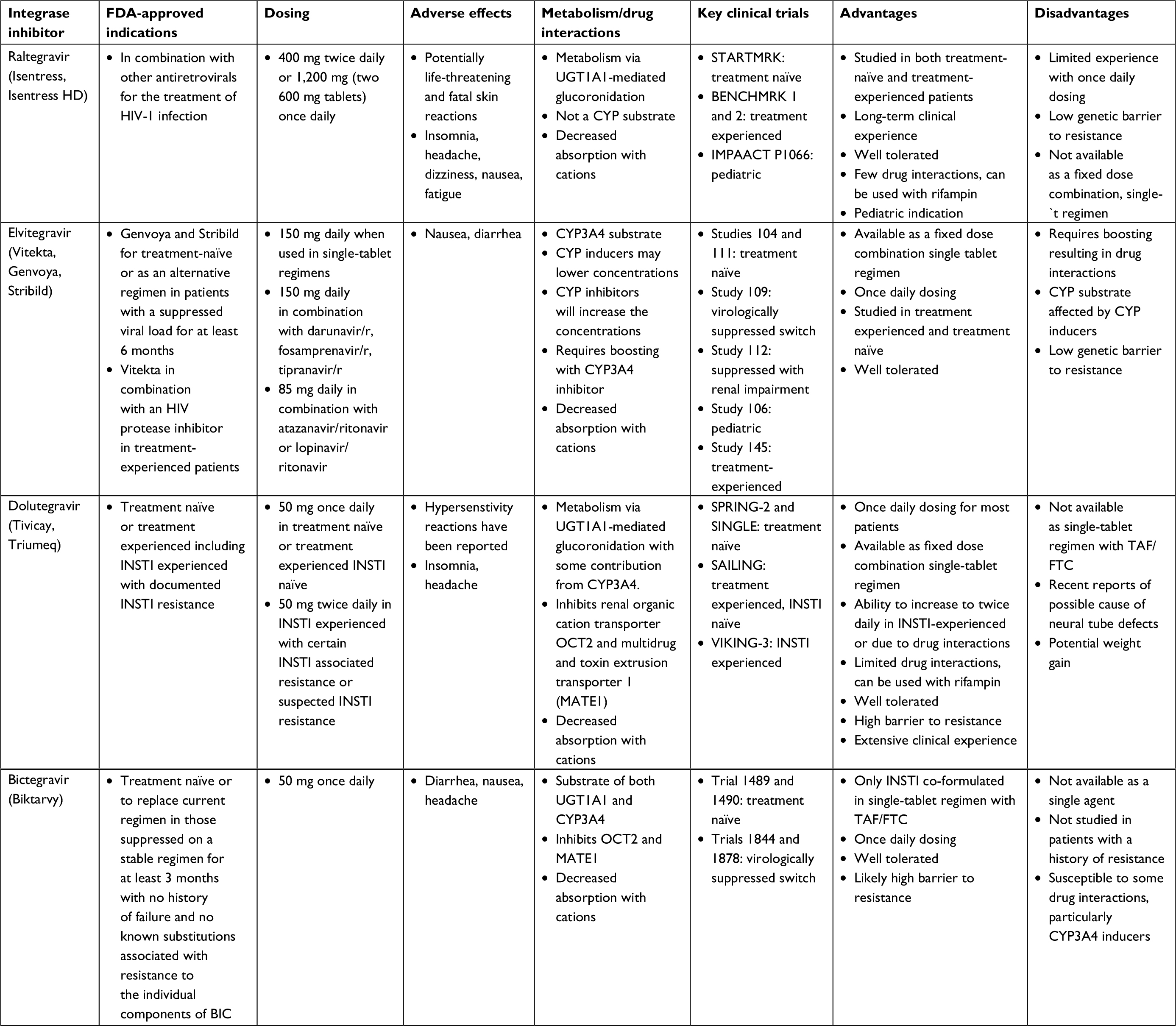 | Table 3 Comparison of available integrase inhibitors Abbreviations: FDA, The Food and Drug Administration; INSTI, integrase strand transfer inhibitor; r, ritonavir. |
Unanswered questions
Despite significant clinical trial data in both treatment-naïve and virologically suppressed treatment-experienced populations, unanswered questions about the use of B/F/TAF in certain clinical situations still remain. The first being in PLWH with documented integrase resistance, or suspected integrase resistance based on virologic failure while on an integrase-based regimen. To date, this represents a relatively small population of patients; however, the use of integrase inhibitors is growing at a rapid pace and the potential for increasing class resistance (including transmitted resistance) is a real possibility. When tested against 47 patient-derived isolates with INSTI resistance mutations, BIC showed a reduced fold change as compared to wild type when compared to DTG, EVG, and RAL. Specifically BIC demonstrated <2.5-fold change in half maximal effective concentration in 70% of the clinical isolates vs 49% for DTG.30 However, the only clinical data that exist is the observation that one participant in a treatment-naïve study with transmitted INSTI (G140S and Q148H) and reverse transcriptase (RT) mutations (K70R and K103N) achieved viral suppression by week 4 with B/F/TAF that was maintained through week 72.10 Significantly more clinical trial data exist for the use of DTG in PLWH with documented or suspected integrase resistance. For example, in the VIKING-3 study, DTG at 50 mg twice daily was studied in 183 subjects who failed an RAL- or EVG-containing regimen with documented INSTI resistance. Using DTG with an optimized background, regimen resulted in 69% of subjects achieving a HIV viral load of <50 c/mL at week 24.29
It also remains unknown if B/F/TAF can attain and maintain viral suppression in PLWH with NRTI resistance, particularly in PLWH starting B/F/TAF with a M184V mutation that confers resistance to 3TC or FTC. In switch studies, B/F/TAF did maintain viral suppression in participants with an isolated M184V; however, these numbers were relatively small and participants were virologically suppressed at the time of exposure to B/F/TAF.13 Related to this is the question of the impact of drug interactions on the role of B/F/TAF in salvage regimens. As stated previously, BIC is a substrate of UGT1A1 but also CYP3A4. The impact of CYP3A4 inducers such as rifampin has been studied with BIC, with a significant decrease in BIC concentrations; however, BIC has not been studied with most other antiretrovirals.4 Specifically, the clinical impact of drug interactions with other antiretrovirals such as DRV in combination with ritonavir or cobicistat (which would be expected to increase BIC concentrations) or etravirine (which would be expected to decrease in BIC concentrations) remains unanswered. In addition, there are ongoing efforts to identify strategies to reduce medication exposure while maintaining viral suppression, and it is unclear how BIC may fit into this strategy, particularly since it is only available as a three-drug fixed dose combination.
A final unanswered question centers around the recent warning issued for DTG in which a higher prevalence of neural tube defects (0.9%, 4/426 patients) was observed as compared to the background prevalence (0.1%) in women in Botswana receiving DTG either during contraception or in the first trimester of pregnancy.32 This information is relatively new and still being investigated to determine if this is a true effect of the medication. DTG and BIC are similar molecules, and until more information is available, a similar concern for increased risk of neural tube defects should be considered for BIC.
Conclusion
The approval of BIC introduces the first unboosted integrase inhibitor in a fixed dose combination with TAF/FTC. The evidence for use of B/F/TAF in treatment-naïve and virologically suppressed treatment-experienced patients without resistance is strong, with the clinical trials demonstrating an equivalent rate of virologic suppression as compared to the most frequently used modern ART regimens. In addition, there was no documented resistance to BIC or any component of the STR that occurred in the clinical trials, suggesting a high barrier to resistance. Confidence in the use of BIC in treatment-experienced patients with documented or presumed resistance is yet to be established; however, it is expected that more data will accumulate over time for this specific population of PLWH.
Disclosure
MYK receives funding to the institution from Gilead Sciences and ViiV healthcare and has served on Gilead Sciences advisory board. LH has served on a ViiV advisory board.The other author reports no conflicts of interest in this work.
References
Zhang H, Custodio JM, Wei X, et al. Clinical pharmacology of the HIV integrase strand transfer inhibitor bictegravir. Paper presented at: The Conference on Retroviruses and Opportunistic Infections (CROI); 2017; Seattle, WA. Available from: http://www.croiconference.org/sessions/clinical-pharmacology-hiv-integrase-strand-transfer-inhibitor-bictegravir. Accessed May 29, 2018. | ||
Lazerwith SE, Cai R, Chen X, et al. Discovery of GS-9883, an HIV-1 integrase strand transfer inhibitor (INSTI) with improved pharmaco-kinetics and in vitro resistance profile. Paper presented at: ASM Microbe; 2016; Boston, MA. Available from: http://www.natap.org/2016/HIV/062016_05.htm. Accessed May 29, 2018. | ||
Tsiang M, Jones GS, Goldsmith J, et al. Antiviral activity of bictegravir (GS-9883), a novel potent HIV-1 integrase strand transfer inhibitor with an improved resistance profile. Antimicrob Agents Chemother. 2016; 60(12):7086–7097. | ||
Biktarvy [package insert]. Foster City, CA: Gilead Sciences Inc.; 2018. | ||
US Department of Health and Human Services. Department of Health and Human Services Adults and Adolescents Antiretroviral Guidelines Panel classifies a fixed-dose combination product of bictegravir/tenofovir alafenamide/emtricitabine as one of the recommended initial regimens for most people with HIV. AIDSinfo. 2018. Available from: https://aidsinfo.nih.gov/news/2044/department-of-health-and-human-services-adults-and-adolescents-antiretroviral-guidelines-panel-classifies-a-fixed-dose-combination-product-of-bictegravir-tenofovir-alafenamide-emtricitabine-as-one-of-the-recommended-initial-regimens-for. Accessed May 29, 2018. | ||
Gallant JE, Thompson M, Dejesus E, et al. Antiviral activity, safety, and pharmacokinetics of bictegravir as 10-Day monotherapy in HIV-1-infected adults. J Acquir Immune Defic Syndr. 2017;75(1):61–66. | ||
Sax PE, Dejesus E, Crofoot G, et al. Bictegravir versus dolutegravir, each with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection: a randomised, double-blind, phase 2 trial. Lancet HIV. 2017;4(4):e154–e160. | ||
Gallant J, Lazzarin A, Mills A, et al. Bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir, abacavir, and lamivudine for initial treatment of HIV-1 infection (GS-US-380-1489): a double-blind, multicentre, phase 3, randomised controlled non-inferiority trial. Lancet. 2017;390(10107):2063–2072. | ||
Sax PE, Pozniak A, Montes ML, et al. Coformulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection (GS-US-380-1490): a randomised, double-blind, multicentre, phase 3, non-inferiority trial. Lancet. 2017;390(10107):2073–2082. | ||
White K, Kulkarni R, Willkim M, et al. Pooled week 48 efficacy and baseline resistance: B/F/TAF in treatment-naive patients. Paper presented at: The Conference on Retroviruses and Opportunistic Infections (CROI); 2018; Boston, MA. Available from: http://www.croiconference.org/sessions/pooled-week-48-efficacy-and-baseline-resistance-bftaf-treatment-naive-patients. Accessed May 29, 2018. | ||
Molina JM, Ward D, Brar I, et al. Switch to bictegravir/F/TAF from DTG and ABC/3TC. Paper presented at: The Conference on Retroviruses and Opportunistic Infections (CROI); 2018; Boston, MA. Available from: http://www.natap.org/2018/CROI/croi_09.htm. Accessed May 29, 2018. | ||
Daar E, Dejesus E, Ruane P, et al. Phase 3 randomized, controlled trial of switching to fixed-dose bictegravir/emtricitabine/tenofovir alafenamide (B/F/TAF) from boosted protease inhibitor-regimes in virologically suppressed Adults: Week 48 results. Paper presented at: ID Week; 2017; San Diego, CA. Available from: https://idsa.confex.com/idsa/2017/webprogram/Paper67504.html. Accessed May 29, 2018. | ||
Andreatta K, Willkom M, Martin R, et al. Resistance analysis of bictegravir/emtricitabine/tenofovir alafenamide switch studies. Paper presented at: The Conference on Retroviruses and Opportunistic Infections (CROI); 2018; Boston, MA. Available from: http://www.croiconference.org/sessions/resistance-analyses-bictegraviremtricitabinetenofovir-alafenamide-switch-studies. Accessed May 29, 2018. | ||
Kityo C, Hagins D, Koenig E, et al. Switching to bictegravir/emtricitabine/tenofovir alafenamide in women. Paper presented at: The Conference on Retroviruses and Opportunistic Infections (CROI); 2018; Boston, MA. Available from: http://www.croiconference.org/sessions/switching-bictegraviremtracitabinetenofovir-alafenimide-bftaf-women. Accessed May 29, 2018. | ||
Gaur A, Rodriguez C, Mcgrath E, et al. Bictegravir/FTC/TAF single-tablet regimen in adolescents: Week-24 results. Paper presented at: The Conference on Retroviruses and Opportunistic Infections (CROI); 2018; Boston, MA. Available from: http://www.croiconference.org/sessions/bictegravirftctaf-single-tablet-regimen-adolescents-week-24-results. Accessed May 29, 2018. | ||
Zhang H, Shao Y, Garner W, et al. The effect of hepatic or renal impairment on bictegravir pharmacokinetics. Paper presented at: The 18th International Workshop on Clinical Pharmacology of Antiviral Therapy; 2017; Chicago, IL. Available from: http://www.natap.org/2017/Pharm/Pharm_31.htm. Accessed May 29, 2018. | ||
Custodio J, West S, Vu A, et al. Pharmacokinetics of bictegravir administered twice daily with rifampin. Paper presented at: The Conference on Retroviruses and Opportunistic Infections (CROI); 2018; Boston, MA. Available from: http://www.croiconference.org/sessions/pharmacokinetics-bictegravir-administered-twice-daily-combination-rifampin. Accessed May 29, 2018. | ||
Cerrone M, Alfarisi O, Neary M, et al. Rifampin effect on tenofovir alafenamide (TAF) plasma/intracellular pharmacokinetics. Paper presented at: The Conference on Retroviruses and Opportunistic Infections (CROI); 2018; Boston, MA. Available from: http://www.croiconference.org/sessions/rifampin-effect-tenofovir-alafenamide-taf-plasmaintracellular-pharmacokinetics. Accessed May 29, 2018. | ||
Zong J, Borland J, Jerva F, Wynne B, Choukour M, Song I. The effect of dolutegravir on the pharmacokinetics of metformin in healthy subjects. J Int AIDS Soc. 2014;17(4 Suppl 3):19584. | ||
Custodio J, West S, Vu A, et al. Lack of clinically relevant effect of bictegravir on metformin pharmacokinetics and pharmacodynamics. Paper presented at: 18th International Workshop on Clinical Pharmacology of Antiviral Therapy; 2017; Chicago, IL. Available from: http://www.natap.org/2017/Pharm/Pharm_32.htm. Accessed May 29, 2018. | ||
Eron JJ, Cooper DA, Steigbigel RT, et al. Efficacy and safety of raltegravir for treatment of HIV for 5 years in the BENCHMRK studies: final results of two randomised, placebo-controlled trials. Lancet Infect Dis. 2013;13(7):587–596. | ||
Rockstroh JK, Dejesus E, Lennox JL, et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naive HIV-1-infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr. 2013;63(1):77–85. | ||
Lennox JL, Landovitz RJ, Ribaudo HJ, et al. Efficacy and tolerability of 3 nonnucleoside reverse transcriptase inhibitor-sparing antiretroviral regimens for treatment-naive volunteers infected with HIV-1: a randomized, controlled equivalence trial. Ann Intern Med. 2014;161(7):461–471. | ||
Sax PE, Dejesus E, Mills A, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet. 2012;379(9835):2439–2448. | ||
Dejesus E, Rockstroh JK, Henry K, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet. 2012;379(9835):2429–2438. | ||
Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. 2013;369(19):1807–1818. | ||
Molina J-M, Clotet B, van Lunzen J, et al. Once-daily dolutegravir versus darunavir plus ritonavir for treatment-naive adults with HIV-1 infection (FLAMINGO): 96 week results from a randomised, open-label, phase 3b study. Lancet HIV. 2015;2(4):e127–e136. | ||
Quashie PK, Mesplède T, Wainberg MA. Evolution of HIV integrase resistance mutations. Curr Opin Infect Dis. 2013;26(1):43–49. | ||
Castagna A, Maggiolo F, Penco G, et al. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the phase III VIKING-3 study. J Infect Dis. 2014;210(3):354–362. | ||
Jones G, Goldsmith J, Mulato A, et al. Bictegravir (GS-9883), a novel HIV-1 integrase strand transfer inhibitor (INSTI) with optimized in vitro resistance profile. Paper presented at: ASM Microbe; 2016; Boston, MA. Available from: http://www.natap.org/2016/HIV/062016_03.htm. Accessed May 29, 2018. | ||
Dolutegravir [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2013. | ||
FDA. FDA Drug Safety Communication: FDA to evaluate potential risk of neural tube birth defects with HIV medicine dolutegravir (Juluca, Tivicay, Triumeq); September 2018. Available from: https://www.fda.gov/Drugs/DrugSafety/ucm608112.htm. Accessed May 29, 2018. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.