")
Back to Archived Journals » Research and Reports in Endocrine Disorders » Volume 6
Profile of alglucosidase alfa in the treatment of Pompe disease: safety, efficacy, and patient acceptability
Authors Schneider I, Zierz S
Received 12 August 2015
Accepted for publication 19 October 2015
Published 8 January 2016 Volume 2016:6 Pages 1—9
DOI https://doi.org/10.2147/RRED.S72834
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Mingzhao Xing
Ilka Schneider, Stephan Zierz
Department of Neurology, Martin Luther University Halle-Wittenberg, Halle (Saale), Germany
Abstract: Pompe disease, also referred to as glycogenosis type II, is a rare, autosomal recessive disorder that results from the deficiency of the glycogen-degrading enzyme acid α-glucosidase. The classical form presents shortly after birth with muscle hypotonia, cardiac, and respiratory failure resulting in a fatal outcome. The late onset of Pompe disease has a very variable onset and disease presentation that often causes a delayed diagnosis. Until now enzyme replacement therapy with alglucosidase alfa is the only causative therapy option for Pompe patients that can slow down disease progression. However, uncertainty remains about the efficacy regarding survival and quality of life in Pompe patients under this very cost-intensive treatment. This paper provides a systematic review of the literature stressing different aspects of enzyme replacement therapy in infantile and late onset Pompe patients.
Keywords: lysosomal storage disease, glycogenosis type II, acid maltase deficiency, IOPD, LOPD, enzyme replacement therapy
Introduction
Pompe disease was first described in 1932 by the Dutch pathologist JC Pompe. It is also known as glycogen storage disease type II or acid α-glucosidase deficiency. This orphan disease (OMIM 232300) has an overall incidence which ranges from 1 in 33,000 persons to 1 in 300,000 persons, depending on the geographic region and ethnicity.1–3 Approximately 5,000–10,000 people worldwide are affected. Pathogenetically, the disease is caused by mutations in the GAA gene on the cytogenetic location 17q25.3 (OMIM 606800), resulting in a deficient activity of the enzyme α-1,4-glucosidase that is located in cellular lysosomes involved in the degradation of glycogen. Deposition of lysosomal glycogen takes place in cardiac, skeletal, and respiratory muscle tissue, but also in smooth muscle of the bladder, intestine, esophagus,4 and also the arrector pili muscles of the skin.5 Pompe disease is classified into two clinical forms: The classic infantile onset Pompe disease (IOPD) is related to complete α-glucosidase deficiency that results in a severe disease with onset within the first 6 months after birth. The non-classic form is usually related to partial enzyme deficiency and progresses more slowly with disease onset at any age. It can be sub-categorized as non-classic childhood, juvenile, and adult form (late onset Pompe disease, LOPD).6 Because of the systemic character, clinical features are multiform (Table 1). Cardiomyopathy with heart failure and respiratory insufficiency are the reason for a median age of death between 6 and 9 months in untreated infants7 and a survival rate of 25.7% for the first 12 months.8 In the non-classic form, a progressive limb girdle muscular weakness is the hallmark of the disease, but the broad spectrum of clinical presentations often delays the correct diagnosis in the patients by an average of 5–8.6 years after the first symptoms.9,10 A decline in the pulmonary function is the main cause of death in this form.11–13 Besides the clinical assessment, other diagnostic clues for Pompe disease include the recognition of a myopathic pattern or myotonic discharges without clinical myotonia14,15 in the electromyography (EMG). Muscle magnetic resonance imaging (MRI), or even better whole body MRI, is more sensitive in detecting subclinical affected muscles and in selecting an appropriate location for muscle biopsy.16 Histologically, Pompe disease presents as vacuolar myopathy in the light microscopy (Figure 1). Vacuoles have a high content of glycogen detected by periodic acid Schiff reaction and are strongly reacting for acid phosphatase, indicating that they are secondary lysosomes.17 It is important to keep in mind that in less severely affected muscles, the vacuoles only show in 25%–75%, and clinically unaffected muscle can be spared from pathological changes, especially in the adult disease.18 A relatively reliable tool for screening for acid α-glucosidase (GAA) deficiency is the assay in blood collected on a filter paper (dried blood spot test [DBS]).19 High-throughput mass spectrometry methods are used for the implementation of newborn screening for lysosomal storage disorders (including Pompe disease) in pilot studies in Taiwan, North America, and Europe.20–23 Currently, the “gold standard” diagnostic for Pompe disease is the GAA assay performed on skin fibroblasts or muscle biopsy followed by DNA analysis24,25 with detection of homozygote or compound heterozygote mutations in the GAA gene. To date more than 300 different mutations have been found,26 without strict genotype–phenotype correlation.27
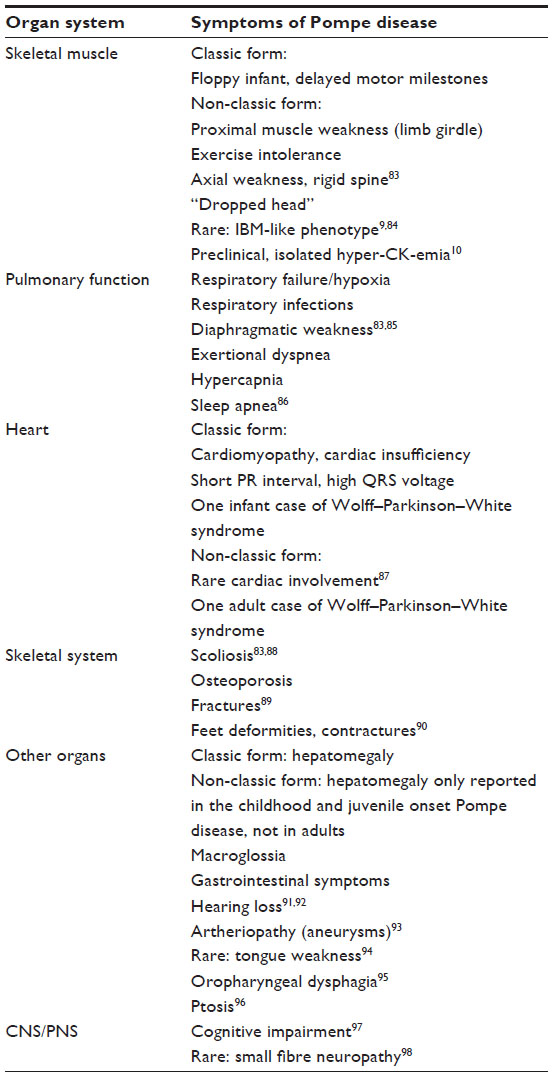 | Table 1 Clinical features of classic and non-classic Pompe patients |
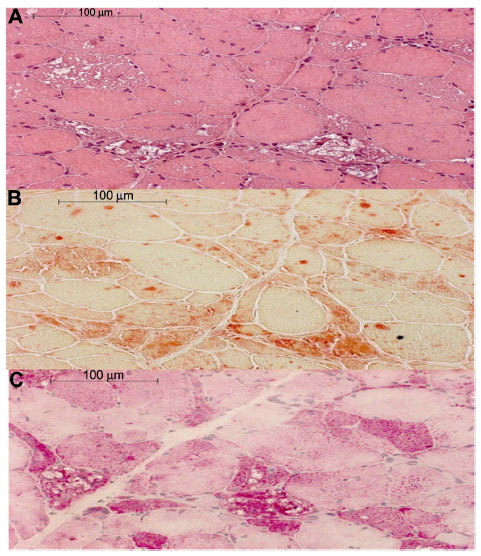 | Figure 1 Histological findings in Pompe disease. |
Enzyme replacement therapy with alglucosidase alfa
The basis for the use of enzyme replacement therapy (ERT) in Pompe disease was built by positive results of intravenously injected precursor forms of recombinant human α-glucosidase (rhGAA) in the Japanese acid maltase-deficient quail that led to reversal of glycogen accumulation in muscle cells.28 Later similar effects were observed with rhGAA in a GAA-knockout mouse model.29 Subsequently, pilot Phase I/II studies of rhGAA from purified Chinese hamster ovarian cell culture30 or transgenic rabbit milk31 significantly prolonged the survival of patients. Since 2006 alglucosidase alfa derived from Chinese hamster ovarian cells (Myozyme®, Lumizyme®; Genzyme Corporation, a Sanofi Company, Cambridge MA, USA) is used for ERT.17,32
ERT in classical form
Multiple studies have demonstrated the impressive effect of ERT for improving the survival in classic-infantile Pompe patients (Table 2). An open-label, multinational, multi-center Phase II study demonstrated safety and efficacy of rhGAA in treatment of infantile onset Pompe disease. While untreated IOPD patients die at a median age of 8.7 months, under ERT the 3-year mortality risk is reduced by 95% compared to the natural course.7,33,34 Especially, the cardiovascular system responds quickly and strikingly to ERT with a significant reversal of cardiac hypertrophy.34–36 However, application of ERT is not a cure but has changed the face of the disease burden from mortality to morbidity. The longer lifespan of infants, who would have died untreated of progressive cardiac involvement, results in manifestation of a more protracted course of muscle weakness, motor speech deficits, sensori-neural and/or conductive hearing loss, osteopenia, gastroesophageal reflux, and dysphagia with aspiration risk and respiratory complications.37 Early treatment before development of clinically noticeable motor weakness appears to have the best results for development of the infants. Therefore, newborn screening, as it is practiced in Taiwan, might further render the prognosis positive in IOPD in the future.38
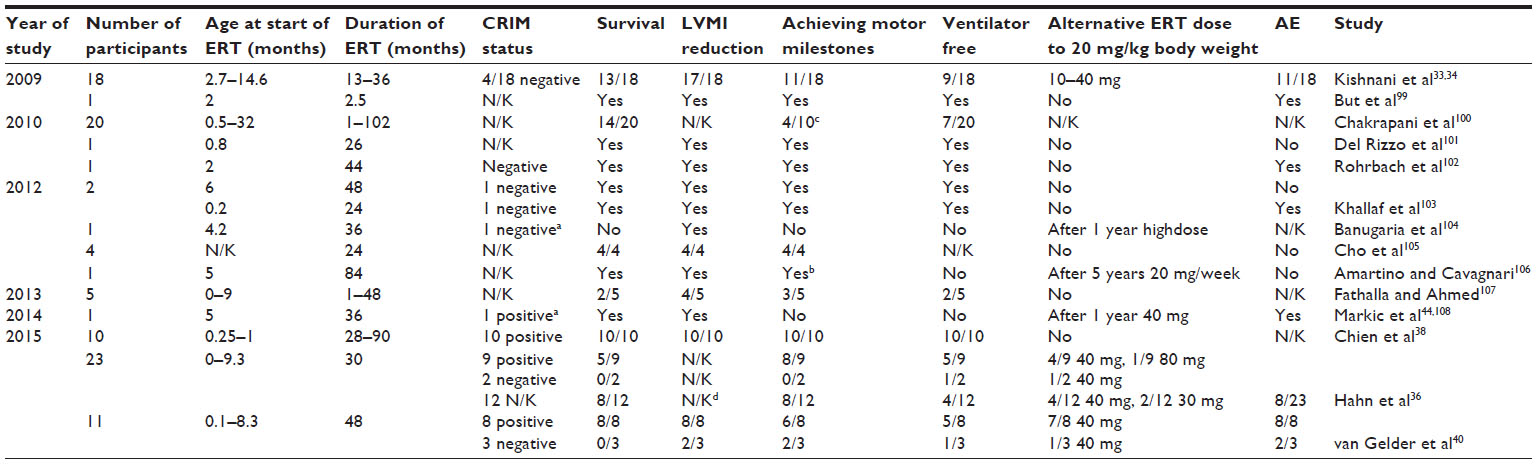 | Table 2 Efficacy of ERT in infantile Pompe disease – study summary |
Antibody formation
Long-term efficacy and safety of ERT in classic-infantile Pompe disease is strongly dependent on antibody formation against alglucosidase alfa (anti-rhGAA-IgG). Pompe patients without any synthesis of GAA protein are labeled as negative for cross-reacting immunological material (CRIM negative). Kishnani et al demonstrated that CRIM negative patients experienced an initial response to ERT, but then entered a phase of devastating clinical decline comparable to the rate observed in untreated infantile patients. After 2 years of ERT, 11/11 CRIM negative patients were deceased or on invasive ventilation support in contrast to only 4/21 CRIM positive patients.39 CRIM negative patients had earlier, higher, and more sustained antibody responses that were associated with a reduced clinical outcome. CRIM positive patients, on the opposite, have detectable amounts of GAA protein, although its function is insufficient or ineffective. These patients are less prone to immune reactions against rhGAA with usually low antibody titer and typically a better clinical outcome.40 However, high doses of therapeutic rhGAA (20–40 mg/kg once a week to biweekly application) can also trigger an immune reaction against ERT in species with only mild mutation of GAA.41 Immunomodulatory therapy is a therapeutic approach to reduce the impact of neutralizing antibodies. Combination of rituximab, methotrexate, and intravenous immunoglobulin42–44 and also additional treatment with bortezomib, targeting plasma B-cells, successfully reduced antibody titer in infantile Pompe patients and resulted in concomitant sustained clinical improvement.45 Importantly, success to induce immune tolerance toward ERT is more likely when immune modulation is started at the onset off ERT.46 Therefore, the evaluation of CRIM-negative patients with subsequent immune modulation is important for optimizing benefit from ERT in these patients. In approximately 90% CRIM status can be predicted by mutation type in the corresponding GAA gene.47 In uncertain cases, Western blot analysis can be applied for the final CRIM status determination.46
Cost effectiveness and quality of life
Besides the question of therapeutic effectiveness, there is also rising interest in the cost effectiveness of ERT in Pompe disease, especially in the face of tensed health care budgets. In 2014, Kanters et al assessed this topic by a patient simulation model that compared costs, survival, quality of life, and quality-adjusted life years in classic-infantile Pompe disease under ERT and untreated patients. Due to the prolonged survival of treated patients, lifetime incremental quality-adjusted life years were 6.8 with incremental costs-per-life-year-gained estimated to be €0.5 million, of which 95% consisted of treatment costs.48
ERT in non-classic form
In contrast to the substantial effect of ERT for survival in the classic Pompe disease, the data of ERT in LOPD patients are more variable. Only one randomized placebo-controlled trial in the LOPD exists, which is the basis for approval of reimbursement in the treatment of LOPD.49 Since then there have been numerous case reports and observation studies emphasizing the benefit of ERT in non-classic Pompe disease, even though results are not as convincing as in classic Pompe patients (Table 3). Güngor et al demonstrated the positive effect of ERT for survival in 202 ERT-treated versus 81 non-treated adults during a median follow-up of 6 years. In all, 28 (61%) of the 46 deaths occurred in the non-ERT-group at a median age of 59.50 A review of all studies until January 2012 evaluated effectiveness of ERT in 368 patients. It was shown that at least a stabilization of motor and respiratory function was achieved in most patients. Newer studies support the hypothesis that ERT can extend the time until wheelchair or ventilator dependence. Nevertheless, no clear correlation between length of treatment and clinical improvement could be found.51 In contrast, ERT in the British cohort of 59 non-classic Pompe patients suggested a stabilization of pulmonary function, but a motoric decline in the 6-Minute Walk Test after 2 years of treatment.52 Furthermore, it has been found that approximately one-third of patients do not respond to therapy.51,53 The reasons why ERT is less effective in LOPD than in the classic form are various. Firstly, a long-time span until diagnosis is related to advanced muscle degeneration. This might explain why the effect on both motor and pulmonary functions is larger when therapy is started early and baseline muscular function is still high.49 A recent MRI study confirms that mildly affected leg muscles respond better to ERT than severely affected muscles in a 5-year follow-up. However, also severely affected muscle groups can still benefit from ERT and improve in strength.54 Interestingly, in some presymptomatic patients MRI demonstrated already lipomatous muscle alterations despite the absence of clinical symptoms.10,55 Therefore, it may be possible to start ERT at this early time to prevent further worsening with manifestation of muscle weakness. In fact, ERT initiation in five asymptomatic juvenile patients was shown to prevent the development of muscle and respiratory symptoms after 6 years in an Italian cohort.56 However, others have raised ethical concerns regarding this procedure and treatment guidelines recommend beginning of ERT only in patients with objective signs of the disease.57,58 Secondly, effectiveness of ERT is dependent on the rhGAA uptake in the skeletal muscles that is limited by a low expression of the cation-independent mannose-6-phosphate receptor (CI-MPR).59,60 Women seem to benefit more from ERT than males61 in respect to muscle strength because of a higher dose uptake of alglucosidase per gram of muscle fiber due to their smaller fibers with a higher surface-to-volume ratio.
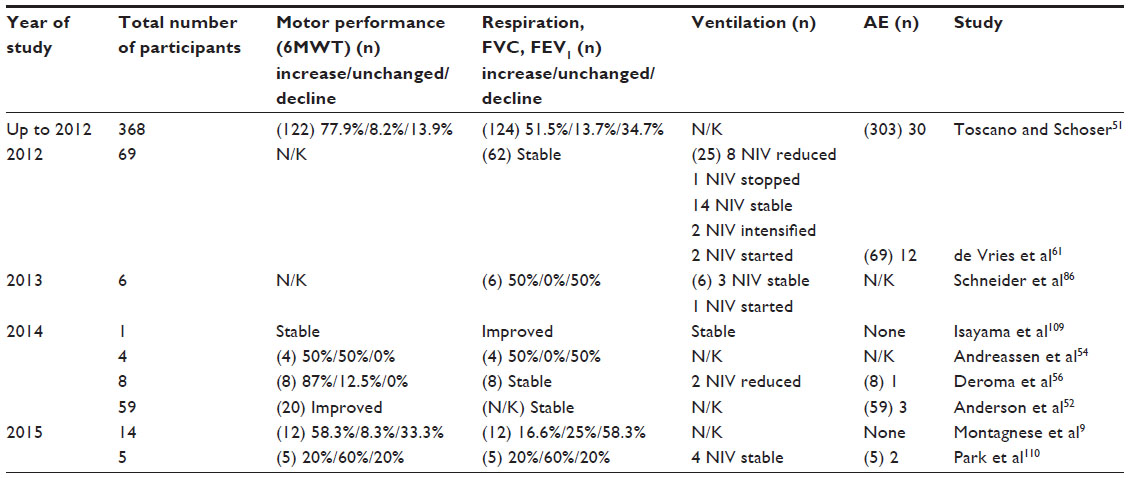 | Table 3 Efficacy of ERT in non-classic Pompe disease – study summary |
Antibody formation in non-classic Pompe disease
As in infants, the late onset Pompe patients usually develop IgG anti-rhGAA within the first 3 months under ERT.53,62 Data from our own cohort, as well as the data from Patel et al, attest that peaks of anti-rhGAA IgG antibody titers can occur even after several years of ERT.63 However, a clear correlation between anti-rhGAA IgG titer and clinical outcome under ERT cannot be found. Interestingly, 3/10 patients had undetectable or borderline anti-rhGAA-IgG titers and remained stable under ERT after 4 years compared to deterioration in some patients with antibodies.64
Quality of life
A sufficient tool for assessment of Quality of Life (QoL) is the Medical Outcomes Survey Short Form-36 Health Survey (SF-36), which can be divided further into a mental and a physical component score. The international Pompe survey revealed that in 210 untreated adult Pompe patients, the QoL was lower in almost all SF-36 domains relative to the general population. Interestingly, a longer disease duration was associated with a better score in the “role functioning physical” and “mental health” domains, most likely due to coping mechanisms. The 10-year international follow-up data suggest a positive effect of ERT on QoL and on participation in daily life activities in adults, by prolonging the natural course of the disease decline.65 However, only 8.3% of 156 patients under ERT reported an overall improvement in the QoL-scores in contrast to the objective physical improvement of motor and respiratory performance.51
Safety of ERT
ERT is unusually well tolerated by the patients and estimated as a safe therapy. Infusion-associated reactions can occur, such as sweating, headaches, elevated temperature, or hypotension, during infusion of rhGAA. Adverse events, involving at least two of three body systems, are reported in 14% of all patients with ERT. Anaphylactic shock and/or cardiac arrest requiring life-support measures have been registered in 1% of patients (Table 4). Steroids or anti-histaminic treatment can be administered to prevent the occurrence of adverse events during the infusions.32
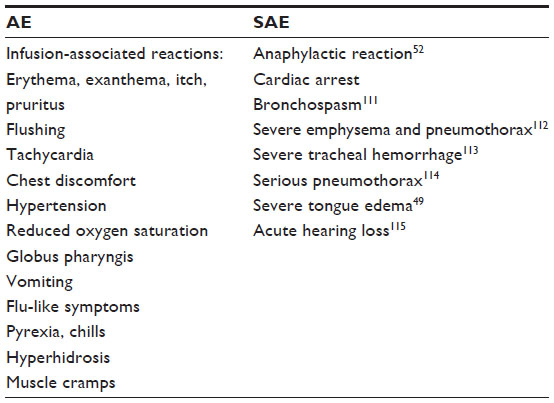 | Table 4 Adverse events (AE) and severe adverse events (SAE) reported under ERT |
Other therapeutic strategies
A lot of effort has been undertaken to further improve the efficacy of ERT. Insufficient rhGAA muscle uptake is likely due to sub-optimal levels of mannose-6-phosphate (M6P), a carbohydrate that binds CI-MPR at the cell surface, resulting in enzyme internalization and lysosomal targeting. Therefore, the design of novel, so-called next-generation forms of rhGAA with optimized muscle uptake is in progress. Results are awaited for a completed Phase I study known as NEO1 and an ongoing extension study (NEO-ext), evaluating the safety and efficacy of neoGAA with improvement of the CI-MPR-dependent muscle uptake66 in adults with LOPD.67 In 2010, BioMarin (BioMarin Pharmaceutical, San Rafael, CA, USA; ClinicalTrials.gov identifier: NCT01230801) started clinical studies with GAA ligated with insulin-like growth factor 2 (BMN 701), which allows the enzyme to attach to the surface of the muscle cell more tightly than the normal acid α-glucosidase. Others aim to design ERT with a significantly higher M6P content compared to the alglucosidase alfa (ATB200).68 Enzyme enhancers might also increase the CI-MPR expression and thereby improved rhGAA uptake as it was successfully achieved by the selective β2 agonists Albuterol in GAA-/- mice, as well as in humans.69 Notably, Albuterol-reduced muscular glycogen even independently from receptor-mediated GAA uptake in the murine model.70 Another strategy is the use of small molecules known as pharmacological chaperones, which bind to the mutant enzyme and recover its lysosomal activity by correcting the protein folding.71,72 However, the presence of a misfolded enzyme with recoverable function is essential for an effect of this class of drug. A potential candidate for pharmacological chaperone therapy is the imino sugar N-butyldeoxynojirimycin, which was shown to increase more than 1.85-fold in α-glucosidase activities in 11 of 13 patients treated with ERT in combination with this chaperone.73 Promising future therapies also involve injection of recombinant adeno-associated virus vectors, as this therapy has been demonstrated to augment cardiac, respiratory, and motorneuron function in GAA-/- knockout mice.74–78 Importantly, the degree of restoration once again seems to depend on the severity of disease. Therefore, an early therapy appears to be the most effective.79 First clinical trials of gene transfer treatment in five ventilator-dependent Pompe patients demonstrated acceptable safety outcomes and longer periods of unassisted breathing.80,81 Genome editing via CRISPR/Cas982 is another revolutionary approach that might tremendously change the therapeutic options for Pompe patients by modulating disease-causing alleles. However, this approach is yet far away from clinical trials.
Acknowledgments
We thank Professor Gisela Stoltenburg-Didinger for supervising histological analysis and Kathleen Zietz and Thekla Wangemann, Neurological Department Halle, for their skillfull technical assistance in the muscular laboratory. We thank Dr Torben Schneider for proof-reading the article.
Disclosure
IS and SZ received honoraria for lectures and compensation for travel expenses and accommodation from Genzyme Corporation. The authors have no other conflicts of interest in this work.
References
Martiniuk F, Chen A, Mack A, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet. 1998;79(1):69–72. | |
Ausems MG, Verbiest J, Hermans MP, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999;7(6):713–716. | |
Chien YH, Chiang SC, Zhang XK, et al. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics. 2008;122(1):e39–e45. | |
Hobson-Webb LD, Proia AD, Thurberg BL, Banugaria S, Prater SN, Kishnani PS. Autopsy findings in late-onset Pompe disease: a case report and systematic review of the literature. Mol Genetics Metab. 2012;106(4):462–469. | |
Katona I, Weis J, Hanisch F. Glycogenosome accumulation in the arrector pili muscle in Pompe disease. Orphanet J Rare Dis. 2014;9:17. | |
Di Rocco M, Buzzi D, Taro M. Glycogen storage disease type II: clinical overview. Acta Myol. 2007;26(1):42–44. | |
Kishnani PS, Hwu WL, Mandel H, et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148(5):671–676. | |
Kishnani PS, Howell RR. Pompe disease in infants and children. J Pediatr. 2004;144(Suppl 5):S35–S43. | |
Montagnese F, Barca E, Musumeci O, et al. Clinical and molecular aspects of 30 patients with late-onset Pompe disease (LOPD): unusual features and response to treatment. J Neurol. 2015;262(4):968–978. | |
Musumeci O, la Marca G, Spada M et al. LOPED study: looking for an early diagnosis in a late-onset Pompe disease high-risk population. Journal of neurology, neurosurgery, and psychiatry. Epub 2015 Mar 17. | |
Winkel LP, Hagemans ML, van Doorn PA, et al. The natural course of non-classic Pompe’s disease; a review of 225 published cases. J Neurol. 2005;252(8):875–884. | |
van der Beek NA, van Capelle CI, van der Velden-van Etten KI, et al. Rate of progression and predictive factors for pulmonary outcome in children and adults with Pompe disease. Mol Genetics Metab. 2011;104(1–2):129–136. | |
Van der Beek NA, Hagemans ML, Reuser AJ, et al. Rate of disease progression during long-term follow-up of patients with late-onset Pompe disease. Neuromuscul Disord. 2009;19(2):113–117. | |
Hanisch F, Kronenberger C, Zierz S, Kornhuber M. The significance of pathological spontaneous activity in various myopathies. Clin Neurophysiol. 2014;125(7):1485–1490. | |
Kassardjian CD, Engel AG, Sorenson EJ. Electromyographic findings in 37 patients with adult-onset acid maltase deficiency. Muscle Nerve. 2015;51(5):759–761. | |
Horvath JJ, Austin SL, Case LE, et al. Correlation between quantitative whole-body muscle magnetic resonance imaging and clinical muscle weakness in pompe disease. Muscle Nerve. 2015;51(5):722–730. | |
Lim JA, Li L, Raben N. Pompe disease: from pathophysiology to therapy and back again. Front Aging Neurosci. 2014;6:177. | |
Vissing J, Lukacs Z, Straub V. Diagnosis of Pompe disease: muscle biopsy vs blood-based assays. JAMA Neurol. 2013;70(7):923–927. | |
Camelier M, De Mari J, Burin M, Civallero G, Giugliani R. Extended use of dried-leukocytes impregnated in filter paper samples for detection of Pompe, Gaucher, and Morquio A diseases. Clin Chim Acta Int J Clin Chem. 2015;446:218–220. | |
Spacil Z, Tatipaka H, Barcenas M, Scott CR, Turecek F, Gelb MH. High-throughput assay of 9 lysosomal enzymes for newborn screening. Clin Chem. 2013;59(3):502–511. | |
Mechtler TP, Stary S, Metz TF, et al. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. Lancet. 2012;379(9813):335–341. | |
Paciotti S, Persichetti E, Pagliardini S, et al. First pilot newborn screening for four lysosomal storage diseases in an Italian region: identification and analysis of a putative causative mutation in the GBA gene. Clin Chim Acta Int J Clin Chem. 2012;413(23–24):1827–1831. | |
Chiang SC, Hwu WL, Lee NC, Hsu LW, Chien YH. Algorithm for Pompe disease newborn screening: results from the Taiwan screening program. Mol Genetics Metab. 2012;106(3):281–286. | |
Pompe Disease Diagnostic Working Group, Winchester B, Bali D, et al. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. Mol Genetics Metab. 2008;93(3):275–281. | |
Oba-Shinjo SM, da Silva R, Andrade FG, et al. Pompe disease in a Brazilian series: clinical and molecular analyses with identification of nine new mutations. J Neurol. 2009;256(11):1881–1890. | |
Kroos M, Hoogeveen-Westerveld M, Michelakakis H, et al. Update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. Hum Mutat. 2012;33(8):1161–1165. | |
De Filippi P, Saeidi K, Ravaglia S, et al. Genotype–phenotype correlation in Pompe disease, a step forward. Orphanet J Rare Dis. 2014;9:102. | |
Kikuchi T, Yang HW, Pennybacker M, et al. Clinical and metabolic correction of pompe disease by enzyme therapy in acid maltase-deficient quail. J Clin Invest. 1998;101(4):827–833. | |
Raben N, Danon M, Gilbert AL, et al. Enzyme replacement therapy in the mouse model of Pompe disease. Mol Genetics Metab. 2003;80(1–2):159–169. | |
Amalfitano A, Bengur AR, Morse RP, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med Off J Am Coll Med Genet. 2001;3(2):132–138. | |
Winkel LP, Van den Hout JM, Kamphoven JH, et al. Enzyme replacement therapy in late-onset Pompe’s disease: a three-year follow-up. Ann Neurol. 2004;55(4):495–502. | |
FULL PRESCRIBING INFORMATION MYOZYME® (alglucosidase alfa); Genzyme Corporation, a Sanofi company. MZ-US-P090-03-14. Available from: https://www.myozyme.com. Accessed December 7, 2015. | |
Kishnani PS, Nicolino M, Voit T, et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149(1):89–97. | |
Kishnani PS, Corzo D, Leslie ND, et al. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res. 2009;66(3):329–335. | |
Levine JC, Kishnani PS, Chen YT, Herlong JR, Li JS. Cardiac remodeling after enzyme replacement therapy with acid alpha-glucosidase for infants with Pompe disease. Pediatr Cardiol. 2008;29(6):1033–1042. | |
Hahn A, Praetorius S, Karabul N, et al. Outcome of patients with classical infantile pompe disease receiving enzyme replacement therapy in Germany. JIMD Rep. 2015;20:65–75. | |
Prater SN, Banugaria SG, DeArmey SM, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med Off J Am Coll Med Genet. 2012;14(9):800–810. | |
Chien YH, Lee NC, Chen CA, et al. Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J Pediatr. 2015;166(4):985–991, e981–e982. | |
Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genetics Metab. 2010;99(1):26–33. | |
van Gelder CM, Hoogeveen-Westerveld M, Kroos MA, Plug I, van der Ploeg AT, Reuser AJ. Enzyme therapy and immune response in relation to CRIM status: the Dutch experience in classic infantile Pompe disease. J Inher Metab Dis. 2015;38(2):305–314. | |
Nayak S, Doerfler PA, Porvasnik SL, et al. Immune responses and hypercoagulation in ERT for Pompe disease are mutation and rhGAA dose dependent. PloS One. 2014;9(6):e98336. | |
Mendelsohn NJ, Messinger YH, Rosenberg AS, Kishnani PS. Elimination of antibodies to recombinant enzyme in Pompe’s disease. New Engl J Med. 2009;360(2):194–195. | |
Messinger YH, Mendelsohn NJ, Rhead W, et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med Off J Am Coll Med Genet. 2012; 14(1):135–142. | |
Markic J, Polic B, Kuzmanic-Samija R, et al. Immune modulation therapy in a CRIM-positive and IgG antibody-positive infant with pompe disease treated with alglucosidase alfa: a case report. JIMD Rep. 2012;2:11–15. | |
Banugaria SG, Prater SN, McGann JK, et al. Bortezomib in the rapid reduction of high sustained antibody titers in disorders treated with therapeutic protein: lessons learned from Pompe disease. Genet Med Off J Am Coll Med Genet. 2013;15(2):123–131. | |
Banugaria SG, Prater SN, Patel TT, et al. Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic material-negative classic infantile pompe disease: a step towards improving the efficacy of ERT. PloS One. 2013;8(6):e67052. | |
Bali DS, Goldstein JL, Banugaria S, et al. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet C Semin Med Genet. 2012;160C(1):40–49. | |
Kanters TA, Hoogenboom-Plug I, Rutten-Van Molken MP, Redekop WK, van der Ploeg AT, Hakkaart L. Cost-effectiveness of enzyme replacement therapy with alglucosidase alfa in classic-infantile patients with Pompe disease. Orphanet J Rare Dis. 2014;9:75. | |
van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. New Engl J Med. 2010;362(15):1396–1406. | |
Gungor D, Kruijshaar ME, Plug I, et al. Impact of enzyme replacement therapy on survival in adults with Pompe disease: results from a prospective international observational study. Orphanet J Rare Dis. 2013;8:49. | |
Toscano A, Schoser B. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol. 2013;260(4):951–959. | |
Anderson LJ, Henley W, Wyatt KM, et al. Effectiveness of enzyme replacement therapy in adults with late-onset Pompe disease: results from the NCS-LSD cohort study. J Inher Metab Dis. 2014;37(6):945–952. | |
Regnery C, Kornblum C, Hanisch F, et al. 36 months observational clinical study of 38 adult Pompe disease patients under alglucosidase alfa enzyme replacement therapy. J Inher Metab Dis. 2012;35(5):837–845. | |
Andreassen CS, Schlutter JM, Vissing J, Andersen H. Effect of enzyme replacement therapy on isokinetic strength for all major muscle groups in four patients with Pompe disease – a long-term follow-up. Mol Genetics Metab. 2014;112(1):40–43. | |
Gruhn KM, Heyer CM, Güttsches A-K, et al. Muscle imaging data in late-onset Pompe disease reveal a correlation between the pre-existing degree of lipomatous muscle alterations and the efficacy of long-term enzyme replacement therapy. Mol Genetics Metab Rep. 2015;3(0):58–64. | |
Deroma L, Guerra M, Sechi A, et al. Enzyme replacement therapy in juvenile glycogenosis type II: a longitudinal study. Eur J Pediatr. 2014;173(6):805–813. | |
Echaniz-Laguna A, Carlier RY, Laloui K, et al. SHOULD patients with asymptomatic pompe disease be treated? A nationwide study in france. Muscle Nerve. 2015;51(6):884–889. | |
Schoser B, Laforêt P, Kruijshaar ME, Toscano A, van Doorn PA, van der Ploeg AT. 208th ENMC International Workshop: Formation of a European Network to develop a European data sharing model and treatment guidelines for Pompe disease Naarden, The Netherlands, 26–28 September 2014. Neuromusc Disorders. 25(8):674–678. | |
Raben N, Lu N, Nagaraju K, et al. Conditional tissue-specific expression of the acid alpha-glucosidase (GAA) gene in the GAA knockout mice: implications for therapy. Human Mol Genetics. 2001;10(19):2039–2047. | |
Schoser B, Hill V, Raben N. Therapeutic approaches in glycogen storage disease type II/Pompe Disease. Neurotherapeutics. 2008;5(4):569–578. | |
de Vries JM, van der Beek NA, Hop WC, et al. Effect of enzyme therapy and prognostic factors in 69 adults with Pompe disease: an open-label single-center study. Orphanet J Rare Dis. 2012;7:73. | |
de Vries JM, van der Beek NA, Kroos MA, et al. High antibody titer in an adult with Pompe disease affects treatment with alglucosidase alfa. Mol Genetics Metab. 2010;101(4):338–345. | |
Patel TT, Banugaria SG, Case LE, Wenninger S, Schoser B, Kishnani PS. The impact of antibodies in late-onset Pompe disease: a case series and literature review. Mol Genetics Metab. 2012;106(3):301–309. | |
Schneider I, Deschauer M, Hanisch F. Enzyme replacement therapy and antibodies in late-onset Pompe disease. Mol Genetics Metab Reports. 2014;1(0):232–234. | |
van der Meijden JC, Gungor D, Kruijshaar ME, Muir AD, Broekgaarden HA, van der Ploeg AT. Ten years of the international Pompe survey: patient reported outcomes as a reliable tool for studying treated and untreated children and adults with non-classic Pompe disease. J Inher Metab Dis. 2015;38(3):495–503. | |
Zhu Y, Jiang JL, Gumlaw NK, et al. Glycoengineered acid alpha-glucosidase with improved efficacy at correcting the metabolic aberrations and motor function deficits in a mouse model of Pompe disease. Mol Ther J Am Soc Gene Therapy. 2009;17(6):954–963. | |
Sanofi, Genzyme aSC. Safety and Efficacy Evaluation of Repeat neoGAA Dosing in Late Onset Pompe Disease Patients. Available from: https://clinicaltrials.gov/ct2/show/NCT01898364. Identifier: NCT01898364. Accessed November 24, 2015. | |
Lun Y, Xu S, Gotschall R et al. Histological Examination of the Effect of a Highly Phosphorylated Proprietary Recombinant Human Acid α-Glucosidase on Glycogen Reduction in Disease-relevant Muscles of Pompe Mice. Available from: http://files.shareholder.com/downloads/AMTX/3343666038x0x808164/DAAA784E-E5E0-4642-A9C7-582953571737/Lun_et_al_Histological_Examination_of_Effect_of_Highly_Phosphorylated_Proprietary_Recombinant_Human_Acid_Alfa_Glucosidase.pdf. Accessed November 24, 2015. | |
Koeberl DD, Austin S, Case LE, et al. Adjunctive albuterol enhances the response to enzyme replacement therapy in late-onset Pompe disease. FASEB J. 2014;28(5):2171–2176. | |
Farah BL, Madden L, Li S, et al. Adjunctive beta2-agonist treatment reduces glycogen independently of receptor-mediated acid alpha-glucosidase uptake in the limb muscles of mice with Pompe disease. FASEB J. 2014;28(5):2272–2280. | |
Okumiya T, Kroos MA, Vliet LV, Takeuchi H, Van der Ploeg AT, Reuser AJ. Chemical chaperones improve transport and enhance stability of mutant alpha-glucosidases in glycogen storage disease type II. Mol Genetics Metab. 2007;90(1):49–57. | |
Flanagan JJ, Rossi B, Tang K, et al. The pharmacological chaperone 1- deoxynojirimycin increases the activity and lysosomal trafficking of multiple mutant forms of acid alpha-glucosidase. Hum Mutat. 2009;30(12):1683–1692. | |
Parenti G, Fecarotta S, la Marca G, et al. A chaperone enhances blood alpha-glucosidase activity in Pompe disease patients treated with enzyme replacement therapy. Mol Ther J Am Soc Gene Therapy. 2014;22(11):2004–2012. | |
Elmallah MK, Falk DJ, Nayak S, et al. Sustained correction of motoneuron histopathology following intramuscular delivery of AAV in pompe mice. Mol Ther J Am Soc Gene Therapy. 2014;22(4):702–712. | |
Falk DJ, Mah CS, Soustek MS, et al. Intrapleural administration of AAV9 improves neural and cardiorespiratory function in Pompe disease. Mol Ther J Am Soc Gene Therapy. 2013;21(9):1661–1667. | |
Qiu K, Falk DJ, Reier PJ, Byrne BJ, Fuller DD. Spinal delivery of AAV vector restores enzyme activity and increases ventilation in Pompe mice. Mol Ther J Am Soc Gene Therapy. 2012;20(1):21–27. | |
Byrne BJ, Falk DJ, Pacak CA, et al. Pompe disease gene therapy. Human Mol Genetics. 2011;20(R1):R61–R68. | |
Falk DJ, Soustek MS, Todd AG, et al. Comparative impact of AAV and enzyme replacement therapy on respiratory and cardiac function in adult Pompe mice. Mol Ther Methods Clin Dev. 2015;2:15007. | |
Todd AG, McElroy JA, Grange RW, et al. Correcting Neuromuscular Deficits With Gene Therapy in Pompe Disease. Annals of Neurology. 2015;78(2):222–234. | |
Smith BK, Collins SW, Conlon TJ, et al. Phase I/II trial of adeno-associated virus-mediated alpha-glucosidase gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Hum Gene Ther. 2013;24(6):630–640. | |
Byrne PI, Collins S, Mah CC, et al. Phase I/II trial of diaphragm delivery of recombinant adeno-associated virus acid alpha-glucosidase (rAAaV1-CMV-GAA) gene vector in patients with Pompe disease. Hum Gene Ther Clin Dev. 2014;25(3):134–163. | |
Savic N, Schwank G. Advances in therapeutic CRISPR/Cas9 genome editing. Transl Res. 2015. Epub 2015 Sep 26. | |
Schuller A, Wenninger S, Strigl-Pill N, Schoser B. Toward deconstructing the phenotype of late-onset Pompe disease. Am J Med Genet C Semin Med Genet. 2012;160C(1):80–88. | |
Bandyopadhyay S, Wicklund M, Specht CS. Novel presentation of pompe disease: Inclusion body myositis-like clinical phenotype. Muscle Nerve. 2015;52(3):466–467. | |
Wens SC, Ciet P, Perez-Rovira A, et al. Lung MRI and impairment of diaphragmatic function in Pompe disease. BMC Pulm Med. 2015; 15:54. | |
Schneider I, Hanisch F, Muller T, Schmidt B, Zierz S. Respiratory function in late-onset Pompe disease patients receiving long-term enzyme replacement therapy for more than 48 months. Wien Medizinisc Wochensch. 2013;163(1–2):40–44. | |
Soliman OI, van der Beek NA, van Doorn PA, et al. Cardiac involvement in adults with Pompe disease. J Inter Med. 2008;264(4):333–339. | |
Roberts M, Kishnani PS, van der Ploeg AT, et al. The prevalence and impact of scoliosis in Pompe disease: lessons learned from the Pompe Registry. Mol Genetics Metab. 2011;104(4):574–582. | |
Bertoldo F, Zappini F, Brigo M, et al. Prevalence of asymptomatic vertebral fractures in late-onset Pompe disease. J Clin Endocrinol Metab. 2015;100(2):401–406. | |
Haaker G, Forst J, Forst R, Fujak A. Orthopedic management of patients with Pompe disease: a retrospective case series of 8 patients. Sci World J. 2014;2014:963861. | |
van Capelle CI, Goedegebure A, Homans NC, Hoeve HL, Reuser AJ, van der Ploeg AT. Hearing loss in Pompe disease revisited: results from a study of 24 children. J Inher Metab Dis. 2010;33(5):597–602. | |
Hanisch F, Rahne T, Plontke SK. Prevalence of hearing loss in patients with late-onset Pompe disease: Audiological and otological consequences. Int J Audiol. 2013;52(12):816–823. | |
Laforet P, Petiot P, Nicolino M, et al. Dilative arteriopathy and basilar artery dolichoectasia complicating late-onset Pompe disease. Neurology. 2008;70(22):2063–2066. | |
Dubrovsky A, Corderi J, Lin M, Kishnani PS, Jones HN. Expanding the phenotype of late-onset Pompe disease: tongue weakness: a new clinical observation. Muscle Nerve. 2011;44(6):897–901. | |
Hobson-Webb LD, Jones HN, Kishnani PS. Oropharyngeal dysphagia may occur in late-onset Pompe disease, implicating bulbar muscle involvement. Neuromuscul Disord. 2013;23(4):319–323. | |
Slingerland NW, Polling JR, van Gelder CM, van der Ploeg AT, Bleyen I. Ptosis, extraocular motility disorder, and myopia as features of pompe disease. Orbit. 2011;30(2):111–113. | |
Spiridigliozzi GA, Heller JH, Kishnani PS. Cognitive and adaptive functioning of children with infantile Pompe disease treated with enzyme replacement therapy: long-term follow-up. Am J Med Genet C Semin Med Genet. 2012;160C(1):22–29. | |
Hobson-Webb LD, Austin SL, Jain S, Case LE, Greene K, Kishnani PS. Small-fiber neuropathy in pompe disease: first reported cases and prospective screening of a clinic cohort. Am J Case Rep. 2015;16:196–201. | |
But WM, Lee SH, Chan AO, Lau GT. Enzyme replacement therapy for infantile Pompe disease during the critical period and identification of a novel mutation. Hong Kong Med J. 2009;15(6):474–477. | |
Chakrapani A, Vellodi A, Robinson P, Jones S, Wraith JE. Treatment of infantile Pompe disease with alglucosidase alpha: the UK experience. J Inher Metab Dis. 2010;33(6):747–750. | |
Del Rizzo M, Fanin M, Cerutti A, et al. Long-term follow-up results in enzyme replacement therapy for Pompe disease: a case report. J Inher Metab Dis. 2010;33(Suppl 3):S389–S393. | |
Rohrbach M, Klein A, Kohli-Wiesner A, et al. CRIM-negative infantile Pompe disease: 42-month treatment outcome. J Inher Metab Dis. 2010;33(6):751–757. | |
Khallaf HH, Propst J, Geffrard S, Botha E, Pervaiz MA. CRIM-negative pompe disease patients with satisfactory clinical outcomes on enzyme replacement therapy. JIMD Rep. 2013;9:133–137. | |
Banugaria SG, Patel TT, Mackey J, et al. Persistence of high sustained antibodies to enzyme replacement therapy despite extensive immunomodulatory therapy in an infant with Pompe disease: need for agents to target antibody-secreting plasma cells. Mol Genetics Metab. 2012;105(4):677–680. | |
Cho A, Kim SJ, Lim BC, et al. Infantile Pompe disease: clinical and genetic characteristics with an experience of enzyme replacement therapy. J Child Neurol. 2012;27(3):319–324. | |
Amartino HM, Cavagnari BM. [Enzyme replacement therapy in the infantile form of Pompe disease: Argentinean experience in a seven-year follow up case]. Arch Argent Pediatr. 2012;110(4):323–327. Spanish. | |
Fathalla W, Ahmed E. The Changing Face of Infantile Pompe Disease: A Report of Five Patients from the UAE. JIMD Rep. 2013;8:7–10. | |
Markic J, Polic B, Stricevic L, et al. Effects of immune modulation therapy in the first Croatian infant diagnosed with Pompe disease: a 3-year follow-up study. Wien Klin Wochenschr. 2014;126(3–4):133–137. | |
Isayama R, Shiga K, Seo K, et al. Sixty six-month follow-up of muscle power and respiratory function in a case with adult-type Pompe disease treated with enzyme replacement therapy. J Clin Neuromusc Dis. 2014;15(4):152–156. | |
Park JS, Kim HG, Shin JH, Choi YC, Kim DS. Effect of enzyme replacement therapy in late onset Pompe disease: open pilot study of 48 weeks follow-up. Neurol Sci Off J Italian Neurol Soc Italian Soc Clin Neurophysiol. 2015;36(4):599–605. | |
Angelini C, Semplicini C, Ravaglia S, et al. Observational clinical study in juvenile-adult glycogenosis type 2 patients undergoing enzyme replacement therapy for up to 4 years. J Neurol. 2012;259(5):952–958. | |
Furusawa Y, Mori-Yoshimura M, Yamamoto T, et al. Effects of enzyme replacement therapy on five patients with advanced late-onset glycogen storage disease type II: a 2-year follow-up study. J Inher Metab Dis. 2012;35(2):301–310. | |
Orlikowski D, Pellegrini N, Prigent H, et al. Recombinant human acid alpha-glucosidase (rhGAA) in adult patients with severe respiratory failure due to Pompe disease. Neuromuscul Disord. 2011;21(7):477–482. | |
Bembi B, Pisa FE, Confalonieri M, et al. Long-term observational, non-randomized study of enzyme replacement therapy in late-onset glycogenosis type II. J Inher Metab Dis. 2010;33(6):727–735. | |
Strothotte S, Strigl-Pill N, Grunert B, et al. Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2:12-month results of an observational clinical trial. J Neurol. 2010;257(1):91–97. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.