")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Primary Whipple disease of the brain: case report with long-term clinical and MRI follow-up
Authors Peregrin J, Malikova H
Received 9 July 2015
Accepted for publication 11 August 2015
Published 25 September 2015 Volume 2015:11 Pages 2461—2469
DOI https://doi.org/10.2147/NDT.S92066
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Roger Pinder
Jan Peregrin,1 Hana Malikova2,3
1Department of Neurology, 2Department of Radiology, Na Homolce Hospital, 3Second Faculty of Medicine, Institute of Anatomy, Charles University, Prague, Czech Republic
Abstract: Whipple disease (WD) is a rare systemic disorder caused by the bacteria Tropheryma whipplei. In its classic form, it manifests with gastrointestinal problems including diarrhea, abdominal pain, and weight loss. However, various other systems can be affected, including the central nervous system (CNS). Even more rarely, the CNS is primarily affected without gastrointestinal symptoms and with a negative small bowel biopsy. The incidence of primary CNS WD is unknown. We report the case of a young female with the primary CNS form of WD. In this report, we highlight the main clinical features and diagnostic procedures that lead to the diagnosis and comment on the treatment and clinical response. We stress the importance of neuroimaging and brain biopsy. A unique feature of this case is that the patient has been followed up for 12 years. At the time of diagnosis, no neurological manifestations were detected, although a tumor-like lesion in the right temporal lobe and hypothalamic infiltration were present on magnetic resonance imaging (MRI). The first neurological manifestations developed 2 years later despite recommended antibiotic treatment, with cognitive impairment developing more than 10 years later. According to the MRI findings and clinical course, the disease was active for several years when multiple lesions on MRI appeared despite antibiotic therapy. In the discussion, we compare the present case with similar cases previously reported and we elaborate on the similarities and discrepancies in clinical features, diagnostic procedures, results, and treatment options.
Keywords: central nervous system, brain biopsy, antibiotic treatment
Introduction
In 1907, GH Whipple described the case of 36-year-old man with abdominal pain, diarrhea, malabsorption, weight loss, fever, and arthralgia,1 now known as Whipple disease (WD). This rare, chronic, multisystemic infectious disorder is caused by the bacterium Tropheryma whipplei (TW), which is related to the family of Actinomyces.2 WD is characterized predominantly by intestinal involvement, but may affect a variety of other organs and systems including the lymphatic system, heart, joints, as well as the central nervous system (CNS). Approximately 20%–40% of patients suffer from neurological manifestations such as encephalopathy, ophthalmoplegia, myoclonus, ataxia, upper motor neuron manifestations, cognitive impairment, and hypothalamic manifestations.3 CNS involvement carries a poor prognosis, with a mortality rate of approximately 25% within 4 years of diagnosis, and 25% of patients have severe neurological sequelae.4 Approximately 60% of patients with CNS WD improve with antibiotic therapy.3 Thus, prompt diagnosis and treatment are crucial for a favorable outcome. An isolated CNS form of WD without intestinal manifestation and with negative small bowel biopsy is extremely rare, with only a small number of cases previously reported. Thus, each new case has significant potential to expand our understanding of this rare disease.
We report the case of a young woman with primary CNS WD followed for 12 years. Despite tumor-like findings on magnetic resonance imaging (MRI) at the time of diagnosis, the patient did not present any clear neurological manifestations. Neurological manifestations developed several years later, while receiving antibiotic treatment.
Case report
The 33-year-old Caucasian female was first admitted to our hospital in 2003. At that time, the patient had a history of two spontaneous abortions followed by amenorrhea and galactorrhea, and also a history of migratory arthralgia of the large joints. She suffered from nonspecific symptoms including lower back pain accompanied by mild fever. Despite a lack of neurological manifestations, the patient underwent several investigations including brain computerized tomography (CT) and MRI, which surprisingly showed a mass-like lesion in the right temporal lobe with cystic and infiltrative portions that partially enhanced following intravenous (IV) gadolinium administration. Additionally, a smaller lesion was detected in the hypothalamus (Figures 1 and 2). Glioma was suspected based on the imaging results, and stereotactic biopsy of the lesion was performed. Histological analyses revealed granular, foamy, periodic acid–Schiff (PAS)-positive macrophages with no signs of tumor. WD was suspected and then confirmed at a referential histopathological laboratory (Bonn, Germany) using PAS staining technique. For WD confirmation, the patient underwent a thorough gastroenterological evaluation including repeated duodenal biopsy; all biopsies were deep; histology was done with correct PAS staining and the specimens examined by experienced pathologists were found to be negative. The diagnosis of primary CNS WD was postulated and was supported by spinal tap, which showed polymerase chain reaction (PCR)-positive results for WD. Antibiotic treatment was initiated, first with IV ceftriaxone 2 g bid for 2 weeks followed by oral trimethoprim–sulfamethoxazole 160/800 mg bid. The fever subsided with treatment; however, the patient still suffered from lower back pain.
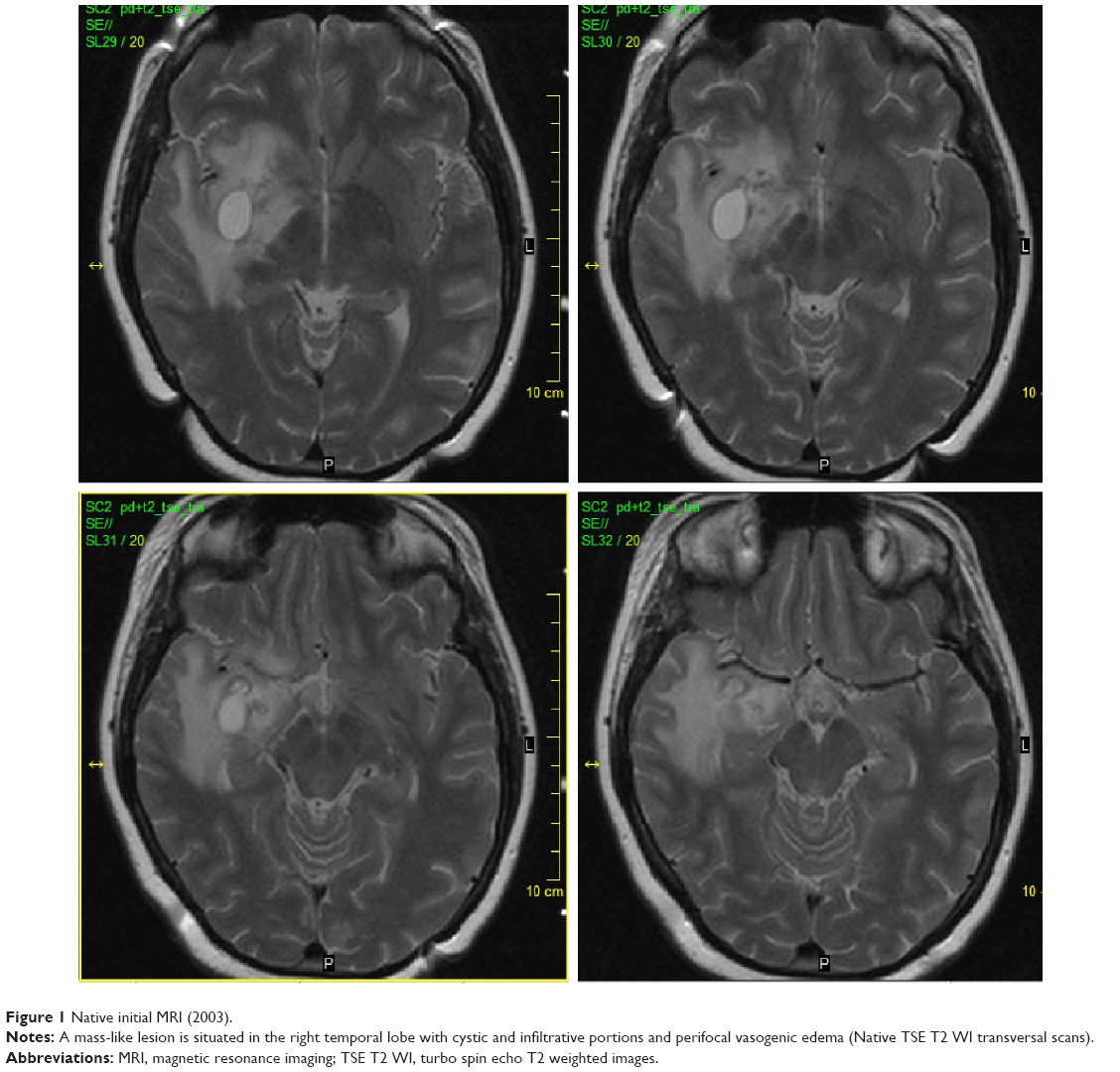 | Figure 1 Native initial MRI (2003). |
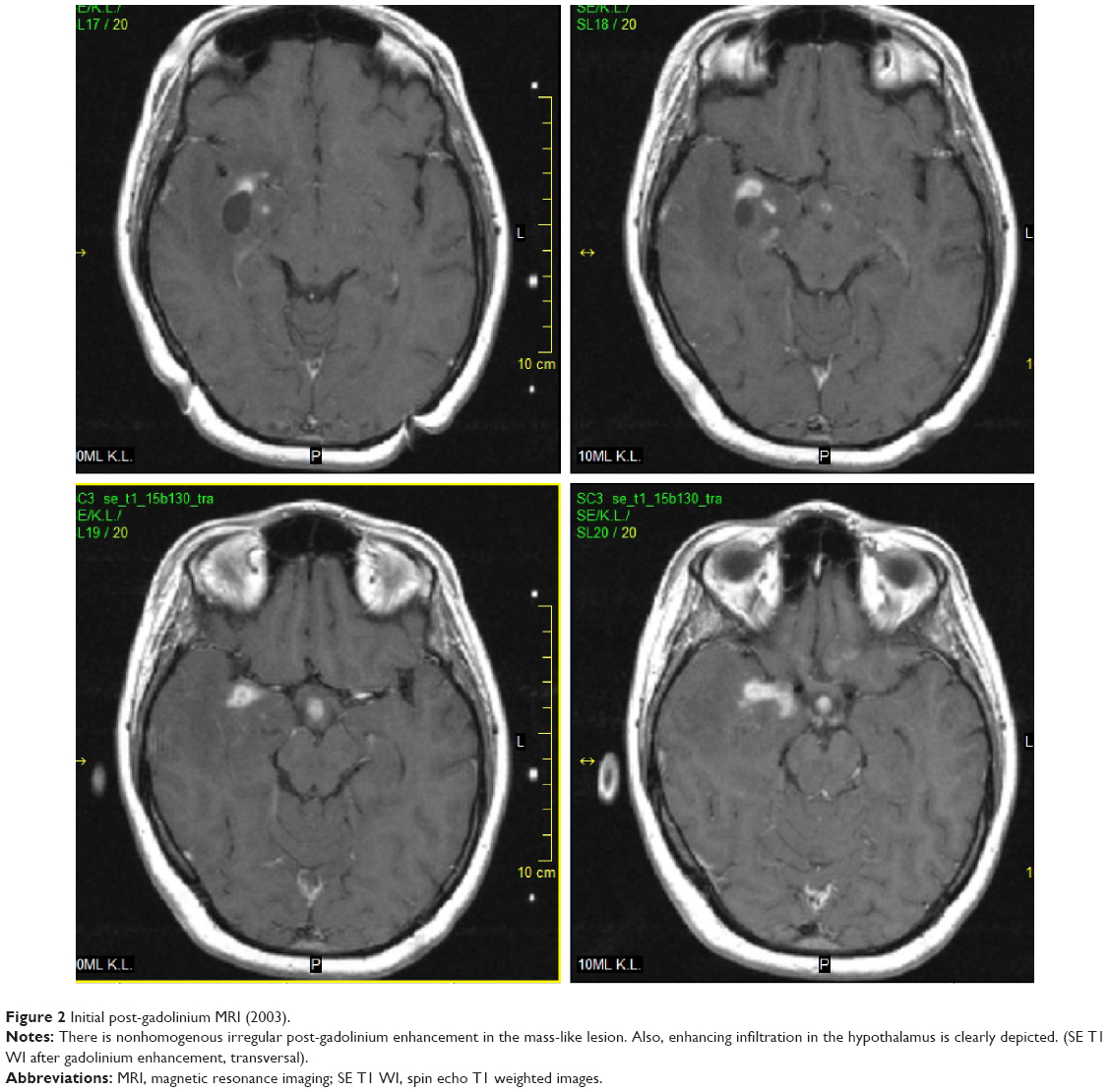 | Figure 2 Initial post-gadolinium MRI (2003). |
In 2005, the patient developed diplopia despite ongoing oral antibiotic treatment. Neurological investigation revealed vertical gaze paresis. Brain MRI was then performed, which showed partial regression of the temporal lesion, no significant change of the hypothalamic lesion, and two new lesions. One small lesion was situated in the right thalamus with discrete, central postgadolinium enhancement (Figure 3). The second new lesion had an infiltrative appearance and surrounded the cerebral aqueduct in the mesencephalon; discrete dot-like enhancement was also apparent (Figure 4). Antibiotic treatment remained unchanged and the patient’s symptoms improved after a few days.
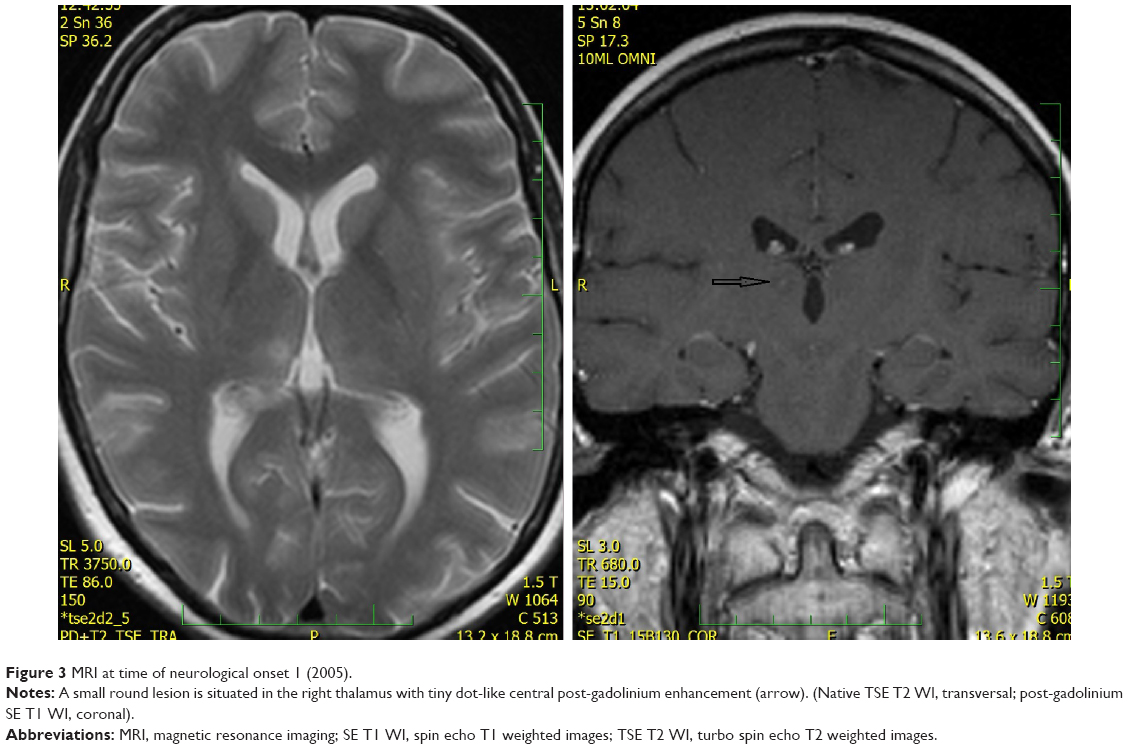 | Figure 3 MRI at time of neurological onset 1 (2005). |
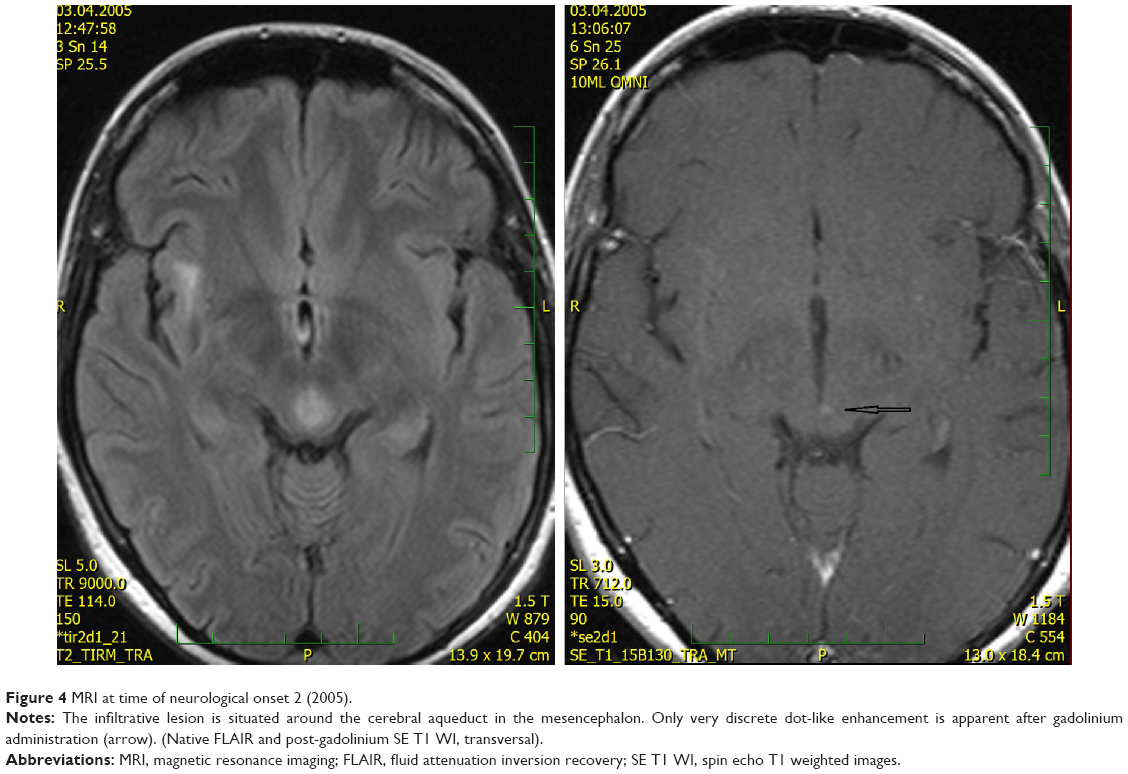 | Figure 4 MRI at time of neurological onset 2 (2005). |
In 2006, the patient suffered from recurrent diplopia and paresthesia of the right arm. Vertical gaze palsy again was observed, and additionally, paresis of the left abducens nerve was also detected. Brain MRI showed a new mass-like lesion in the brain stem with small ring-like, central, postgadolinium enhancement (Figures 5 and 6). The lesion previously observed in the right thalamus as well as hypothalamic infiltration had completely regressed. Remarkable regression of the initial mass-like lesion in the right temporal lobe was also noted. Spinal tap was again performed, and although the cerebrospinal fluid (CSF) was PCR-negative for WD, no other cause was found. Antibiotic treatment therefore remained unchanged (oral trimethoprim–sulfamethoxazole 160/800 mg bid).
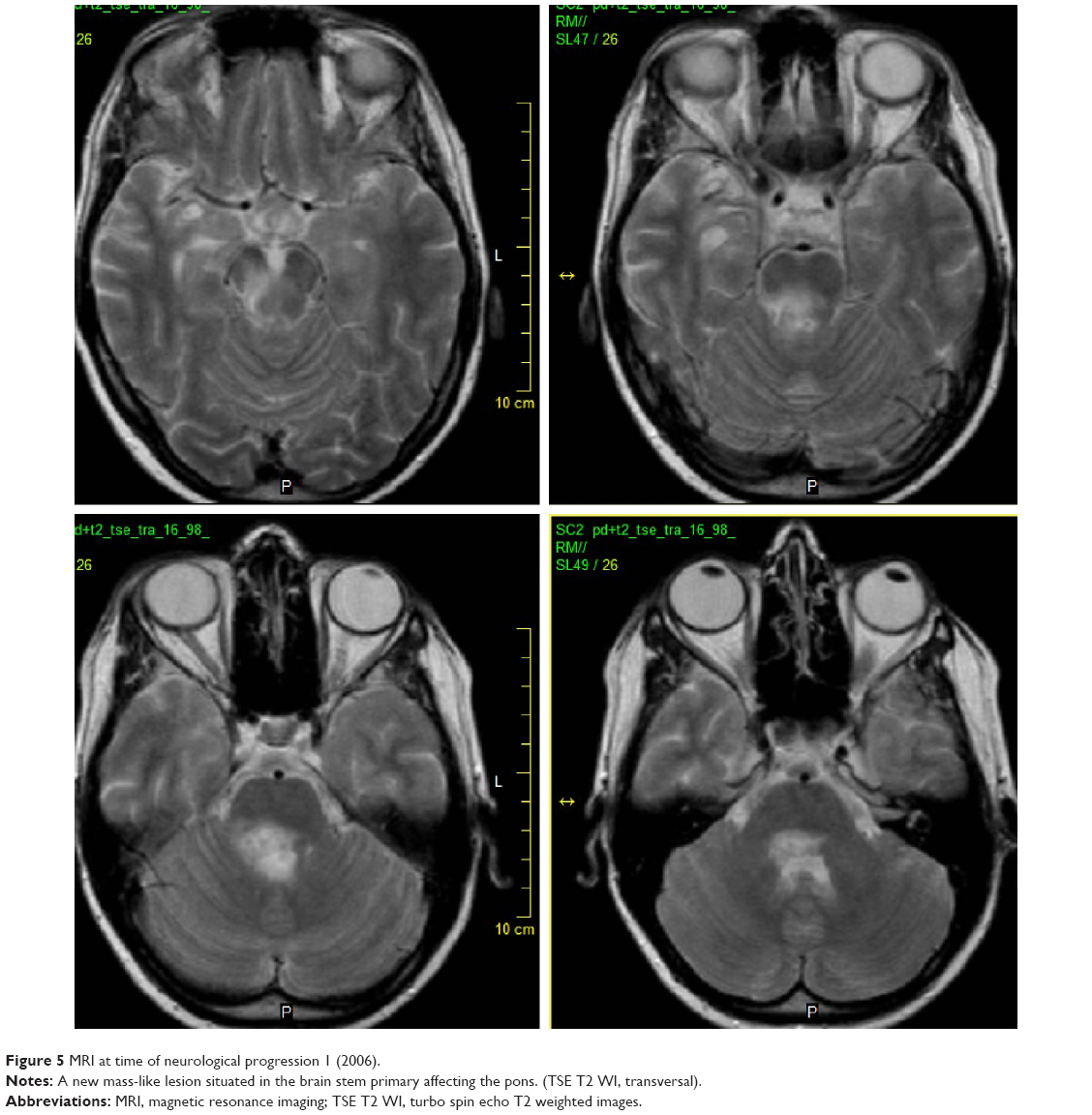 | Figure 5 MRI at time of neurological progression 1 (2006). |
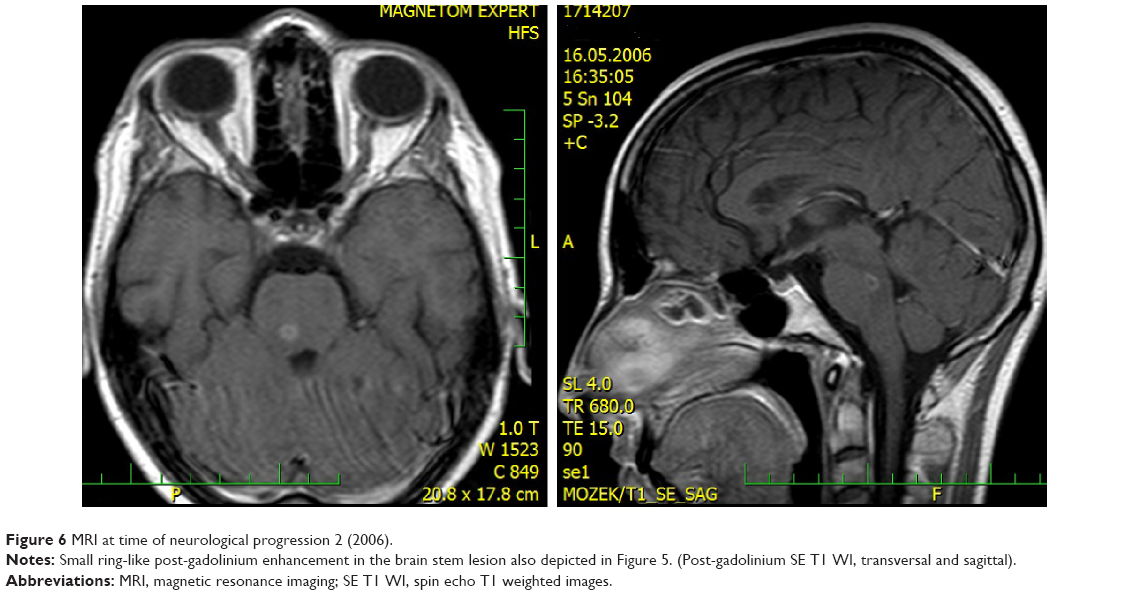 | Figure 6 MRI at time of neurological progression 2 (2006). |
A few months later, the patient stopped visiting our outpatient clinic and we had no contact with her for 8 years. Her next visit took place in October 2014. At that time, she reported that she had stopped taking antibiotics. She denied any worsening of her neurological status in the previous period and was able to attend work as an office clerk. She still suffered from vertical gaze palsy and sixth cranial nerve paresis on the left side. MRI from November 2014 found no new lesions; the previous lesions had partially regressed. Resumption of antibiotic treatment was suggested but was refused by the patient.
In December 2014, the patient was found unconscious by her family. Upon admission to the hospital, she was severely dehydrated with hyperosmolarity, metabolic acidosis, and hypotension. After rehydration and correction of the acid–base dysbalance, she regained consciousness. Right-sided abducens paresis in addition to her previous ocular palsies and severe cognitive impairment was found. She suffered from short- and long-term memory loss and had retrograde amnesia spanning several months prior to the new episode. MRI showed diffuse brain atrophy and severe atrophy of the right hippocampus with hippocampal sclerosis (Figure 7). All enhancing lesions on MRI had regressed, only postinflammatory gliosis in the right temporal lobe and a small periaqueductal pseudocyst were found. Oral antibiotic treatment with trimethoprim–sulfamethoxazole 160/800 mg bid was reestablished. Unfortunately, her neurological status thus far (April 2015) remains the same. The patient signed the informed consent and agreed with publishing medical data in scientific literature in anonymous form. This case report was approved by the ethics committee of Na Homolce Hospital, Prague, Czech Republic.
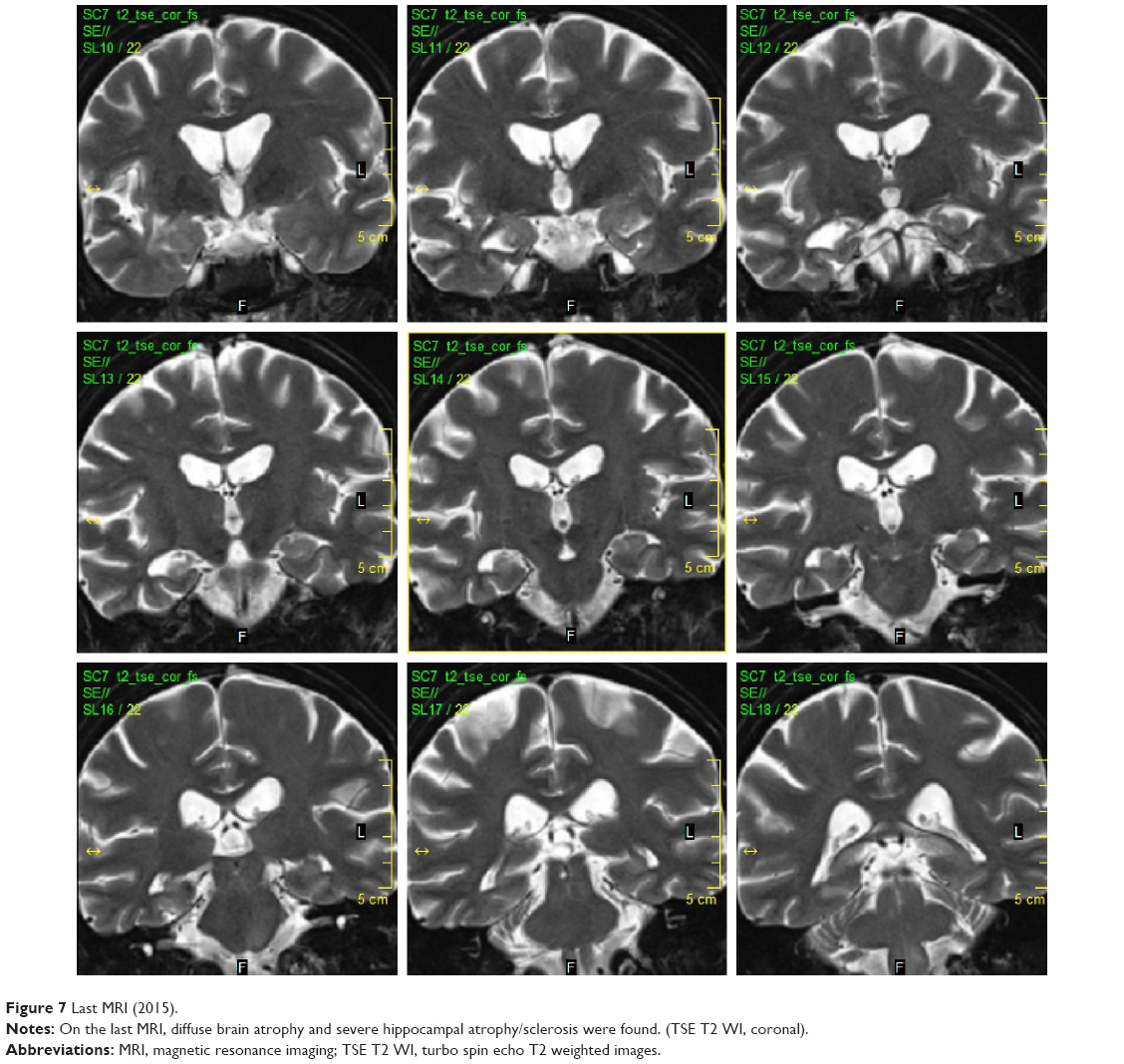 | Figure 7 Last MRI (2015). |
Discussion
Clinical features
We reported a case of primary CNS WD in a patient followed for more than 12 years. Primary CNS WD is an extremely rare form of WD and its incidence is unknown. There are only sporadic case reports in the literature,5 and according to their estimation, only 4%–5% of WD patients present with isolated neurological symptoms.6 Interestingly, our patient did not suffer from any neurological symptoms at the time of diagnosis. The first manifestation of the disease was likely several years before the diagnosis was established, when she suffered from amenorrhea and galactorrhea. These manifestations were likely connected with the hypothalamic lesion apparent on the initial brain MRI. Hypothalamic involvement has been reported in some cases,5,7 typically presenting as polyuria–polydipsia; however, amenorrhea has also been reported.6
The first neurological manifestations in the present case included diplopia and vertical gaze paresis, which developed 2 years after the initial evaluation despite proper antibiotic treatment; cognitive impairment developed more than 10 years after the diagnosis was established. These manifestations appear to be quite frequent according to cases previously reported. Louis et al6 evaluated neurological signs in 84 WD patients with CNS involvement and reported cognitive changes in 71% of patients and supranuclear gaze palsy in 51%. Several studies5,7–9 have reported oculomasticatory myorhythmia, which appears to be a pathognomonic neurologic manifestation of CNS WD. According to the aforementioned survey by Louis et al,6 the frequency of this manifestation is 20%; however, it was not observed in the present case. Others reported manifestations of CNS WD that include cerebellar ataxia, psychiatric signs, myoclonus, seizures, and cranial nerve abnormalities.6,8–10
Diagnostic procedures
In the present case, brain MRI was one of the two essential examinations that led to the diagnosis of CNS WD, and the importance of MRI has been previously stressed.11,12 Panegyres et al5 reported a case of primary CNS WD and compared their results with 19 previous cases. They reported that patients with primary CNS WD can be divided into two groups based on clinical manifestations and brain MRI as: 1) patients with focal neurological manifestations due to solitary mass lesion and 2) those with multiple neurological signs and symptoms and multiple nodular enhancing MRI or CT lesions. The present case shows features of both these groups combined, beginning with a mass lesion in the right temporal lobe with infiltration of hypothalamus and later, despite proper treatment, developing multiple smaller lesions and another mass-like lesion in the brain stem. The last MRI showed cerebral atrophy and marked hippocampal atrophy on the right. Brain atrophy has been previously described.11
The second essential investigation in establishing the diagnosis was brain biopsy, which showed PAS-positive granular, foamy macrophages. Similar histological findings have been described in duodenal biopsies done in systemic WD patients with gastrointestinal symptoms.7,8 As WD is a systemic infection, these histologic findings can be present in various localizations such as the cardiac valves, synovial fluid, or lymph nodes.7 Gerard et al,7 in their report and review article, stress the importance of brain biopsy in primary CNS WD. Other studies support this opinion,6 although the sensitivity of brain tissue biopsy in CNS WD is unknown. In a recently published article, Hagel et al13 comment on the methods used in brain tissue biopsy investigation. They state that PAS staining alone is insufficient for WD diagnosis and may yield false-positive results; further, they stress TW-specific immunohistochemistry and PCR as methods of choice.
PCR of the CSF played an important role in confirming the diagnosis in the present case. Several articles and case reports have mentioned this method and its importance.5,7,13 Furthermore, CSF PCR can be positive not only in primary CNS WD, but also in the classic form of WD with gastrointestinal symptoms,14 which again indicates that WD is a systemic disorder. The aforementioned article by Gerard et al7 also suggests CSF PCR as a method for monitoring the effect of treatment. Our case does not support this recommendation as our patient had negative PCR CSF at the time of unequivocal progression.
Treatment
Our patient was initially treated with IV ceftriaxone for 3 weeks, followed by oral trimethoprim–sulfamethoxazole bid. Several antibiotics have been used in the treatment of WD including chloramphenicol, tetracyclines, penicillin, and streptomycin.15,16 Current treatment recommendations take into consideration the specific features of TW. First is its ability to cross the blood–brain barrier; up to 40% of patients with multisystem WD may have CNS involvement, including positive CSF PCR.3,5 Second is its intracellular persistence.4 Therefore, for several years, treatment recommendations were the following: initial 2-week treatment with IV ceftriaxone 2 g daily, followed by oral trimethoprim–sulfamethoxazole 960 mg bid.7,8 Several articles commented on the efficacy of trimethoprim–sulfamethoxazole and initial ceftriaxone treatment.15,16 However, recent articles mention failures and relapses after trimethoprim–sulfamethoxazole treatment.17,18 This is being attributed to a possible trimethoprim–sulfamethoxazole resistance,17,19 which was demonstrated in vitro.20 Due to these findings, the current recommendations suggest bactericidal treatment with a combination of doxycycline and hydroxychloroquine.17,21 Our patient was treated in 2003–2005, so the previous trimethoprim–sulfamethoxazole guidelines were followed.
To the best of our knowledge, no studies are available on the proper duration of antibiotic treatment.8,17 Our patient was treated with trimethoprim–sulfamethoxazole for 3 years, during which time she underwent two episodes of progression, after the second and third year of treatment. The last episode of disease progression occurred when she was off medication, which she discontinued spontaneously while temporarily lost to clinical follow-up.
Apart from trimethoprim–sulfamethoxazole resistance, another possible cause of relapse in WD patients is the immune reconstitution inflammatory syndrome. This syndrome was first described in HIV-infected patients after initiation of antiretroviral therapy and later was observed in patients with leprosy and tuberculosis. It was demonstrated also in patients with WD.22,23 If this is the case, the patient can profit from corticosteroid therapy; thalidomide efficiency was also described.24 We were not yet aware of the occurrence of immune reconstitution inflammatory syndrome in WD at time our patient relapsed in 2005 and 2006; therefore, steroid therapy was not applied.
Conclusion
We had the unique opportunity to follow a young patient with primary CNS WD for 12 years. During that time, several interesting features presented, such as initial hypothalamic manifestations and lack of proper neurological signs that eventually appeared 2 years after the diagnosis was made. Despite appropriate antibiotic treatment recommended at that time, we observed several episodes of disease progression, both clinically and radiologically, with variable lesions on brain MRI that varied from a tumor-like mass, hypothalamic infiltration, small lesions elsewhere in the brain, and finally brain atrophy and unilateral hippocampal sclerosis combined with the disappearance of all the aforementioned lesions. Thus, we emphasize the importance of early diagnosis, in which neuroimaging as well as brain biopsy may be helpful.
Acknowledgments
The work was supported by Ministry of Health, Czech Republic – conceptual development of research organization (Nemocnice Na Homolce – NNH, 00023884, IG154301). We are indebted to Aaron Rulseh, MD, PhD, for help with English editing.
Disclosure
The authors report no conflicts of interest in this work.
References
Whipple GH. A hitherto undescribed disease characterized anatomically by deposits of fat and fatty acids in the intestinal and mesenteric lymphatic tissues. Bull Johns Hopkins Hosp. 1907;18:382–391. | ||
Dobbins WO 3rd, Ruffin JM. A light and electron microscope study of bacterial invasion in Whipple’s disease. Am J Pathol. 1967;51(2):225–242. | ||
Fenollar F, Puéchal X, Raoult D. Whipple’s disease. N Engl J Med. 2007;356:55–66. | ||
Schnider PJ, Reisinger EC, Gerschlager W, et al. Long term follow-up in cerebral Whipple’s disease. Eur J Gastroenterol Hepatol. 1996;8:899–903. | ||
Panegyres PK, Edis R, Beaman M, Fallon M. Primary Whipple’s disease of the brain: characterization of the clinical syndrome and molecular diagnosis. QJM. 2006;99:609–623. | ||
Louis ED, Lynch T, Kaufmann P, Fahn S, Odel J. Diagnostic guidelines in central nervous system Whipple’s disease. Ann Neurol. 1996;40:561–568. | ||
Gerard A, Sarrot-Reynauld F, Liozon E, et al. Neurologic presentation of Whipple disease report of 12 cases and review of the literature. Medicine. 2002;81(6):443–457. | ||
Marth T, Raoult D. Whipple’s disease. Lancet. 2003;361:239–246. | ||
Mattews BR, Jones LK, Saad DA, Aksamit AJ, Josephs KA. Cerebellar ataxia and central nervous system Whipple disease. Arch Neurol. 2005;62(4):618–620. | ||
Adams M, Rhyner PA, Day J, DeArmond S, Smuckler EA. Whipple’s disease confined to the central nervous system. Ann Neurol. 1987;21:104–108. | ||
Schnider P, Trattnig S, Kollegger H, Auff E. MR of cerebral Whipple disease. AJNR Am J Neuroradiol. 1995;16:1328–1329. | ||
Black DF, Aksamit AJ, Morris JM. MR imaging of central nervous system Whipple disease: a 15-year review. AJNR Am J Neuroradiol. 2010;31:1493–1497. | ||
Hagel S, Epple HJ, Feurle GE, et al. S2k-guideline gastrointestinal infectious diseases and Whipple’s disease. Z Gastroenterol. 2015;53:418–459. | ||
Von Herbay A, Ditton HJ, Schuhmacher F, Maiwald M. Whipple’s disease: staging and monitoring by cytology and polymerase chain reaction analysis of cerebrospinal fluid. Gastroenterology. 1997;113:434–441. | ||
Feurle GE, Junga NS, Marth T. Efficacy of ceftriaxone or meropenem as initial therapies in Whipple’s disease. Gastroenterology. 2010;138:478–486. | ||
Feurle GE, Marth T. An evaluation of antimicrobial treatment for Whipple’s disease tetracycline versus trimethoprim-sulfamethoxazole. Dig Dis Sci. 1994;39(8):1642–1648. | ||
Lagier JC, Fennolar F, Lepidi H, Raoult D. Failure and relapse after treatment with trimetophrim/sulfamethoxazole in classic Whipple’s disease. J Antimicrob Chemother. 2010;65:2005–2012. | ||
Lagier JC, Fennolar F, Lepidi H, Giorgi R. Treatment of classic Whipple’s disease: from in vitro results to clinical outcome. J Antimicrob Chemother. 2014;69:219–227. | ||
Fennolar F, Rolain JM, Alric L, et al. Resistance to trimethoprim/sulfamethoxazole and Tropheryma whipplei. Int J Antimicrob Agents. 2009;34(3):255–259. | ||
Bakkali N, Fennolar F, Biswas S, Rolain JM, Raoult D. Acquired resistance to trimetophrim-sulfamethoxazole during Whipple disease and expression of the causative target gene. J Infect Dis. 2008;198:101–108. | ||
Feurle GE, Moos V, Schneider T, Fennolar F, Raoult D. The combination of chloroquine and minocycline, a therapeutic option in cerebrospinal infection of Whipple’s disease refractory to treatment with ceftriaxone, meropenem and co-trimoxazole. J Antimicrob Chemother. 2012;67(5):1295–1296. | ||
Feurle GE, Moos V, Schinnerling K, et al. The immune reconstitution inflammatory syndrome in Whipple disease: a cohort study. Ann Intern Med. 2010;153(11):710–717. | ||
Moos V, Feurle GE, Schinnerling K, et al. Immunopathology of immune reconstitution inflammatory syndrome in Whipple’s disease. J Immunol. 2013;190:2354–2361. | ||
Le Blay P, Rakotonirainy H, Lagier JC, Raoult D, Puechal X, Pers YM. A severe Whipple disease with an immune reconstitution inflammatory syndrome: an additional case of thalidomide efficiency. Joint Bone Spine. 2014;81(3):260–262. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.