")
Back to Journals » OncoTargets and Therapy » Volume 12
Prevention Of Skeletal Related Events In Multiple Myeloma: Focus On The RANK-L Pathway In The Treatment Of Multiple Myeloma
Authors Parrondo RD, Sher T
Received 6 May 2019
Accepted for publication 20 September 2019
Published 14 October 2019 Volume 2019:12 Pages 8467—8478
DOI https://doi.org/10.2147/OTT.S192490
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yao Dai
Ricardo D Parrondo, Taimur Sher
Department of Medicine, Hematology-Oncology, Mayo Clinic Florida, Jacksonville, FL, USA
Correspondence: Taimur Sher
Department of Medicine, Hematology-Oncology, Mayo Clinic Florida, 4500 San Pablo Road S, Jacksonville, FL 32224, USA
Email [email protected]
Abstract: More than 90% of patients with multiple myeloma (MM) have osteolytic bone lesions which increase the risk of skeletal-related events (SRE). The cytokine milieu in the bone marrow microenvironment (BMME) of MM plays a key role in myeloma bone disease by impairing the balance between osteoclastogenesis and osteoblastogenesis. This is orchestrated by the malignant plasma cell (MPC) with the ultimate outcome of MPC proliferation and survival at the expense of excess osteoclast activation resulting in osteolytic bone lesions. Prevention of SRE is currently accomplished by the inhibition of osteoclasts. Bisphosphonates (BPs) are pyrophosphate analogues that cause apoptosis of osteoclasts and have been proven to prevent and delay SRE. Denosumab, a fully humanized monoclonal antibody that binds and inhibits receptor activator of nuclear factor-ĸB ligand (RANKL), a key molecule in the BMME crucial for osteoclastogenesis, is also approved for the prevention of SRE in MM. The addition of BPs and denosumab to standard MM treatment affords a survival benefit for patients with MM. Specifically, the addition of denosumab to standard MM treatments results in superior PFS compared to BPs, highlighting the key role of the RANKL pathway in MM. This review focuses on the pathophysiology of myeloma bone disease as well as on the importance of targeting the RANK-L pathway for the treatment of MM and prevention of SRE.
Keywords: multiple myeloma, denosumab, RANKL, bisphosphonates, skeletal-related events
Introduction
Multiple myeloma (MM) is a plasma cell neoplasm that accounts for 13% of all hematologic cancers. With an age-adjusted incidence of nearly 6 per 100,000 persons per year, MM is the second most common hematological malignancy.1 MM is characterized by malignant proliferation of monoclonal plasma cells in the bone marrow with resultant elevation in monoclonal paraprotein, hypercalcemia, renal dysfunction, anemia and osteolytic lesions.2,3 Diffuse osteopenia, pathologic fractures, focal lytic lesions and bony pain are common clinical manifestations in patients with MM. Using Positron Emission Tomography and Computed Tomography (PET-CT) and Magnetic Resonance Imaging (MRI), bone involvement can be found in 91 and 95% of MM patients respectively.4 These osteolytic bone lesions result in an increased risk of skeletal-related events (SRE). SRE are defined as pathological fractures, radiation or surgery to bone, spinal cord compression and hypercalcemia that often lead to diminished quality of life and increased morbidity and mortality.5,6 Approximately 60% of MM patients will develop a fracture during their disease course.7
Via interactions with the bone marrow microenvironment (BMME), malignant plasma cells (MPC) are able to orchestrate the production of osteoclast-activating factors and osteoblast-inhibitory factors which leads to asynchronous bone turnover, net bone loss and osteolytic lesions.8 MPC and stromal cells secrete factors such as RANKL, macrophage inflammatory protein 1 alpha (MIP-1α), interleukin 3 (IL-3), and interleukin 6 (IL-6), which increase osteoclast activity and additional factors such as dickkopf-1 (Dkk-1) and secreted frizzled-related protein 2 (sFRP-2) which inhibit osteoblast function.9–12 Signaling between MPC and osteocytes induce osteocyte apoptosis which leads to increased RANKL and sclerosin secretion. The former recruits and activates osteocytes while the later inhibits osteoblast function.13 RANKL in particular is a key mediator of osteoclast formation, activation and survival. In MM, production of RANKL is significantly increased by osteocytes, bone marrow stromal cells and MPC resulting in increased osteoclast activity and increased bone resorption.13–15 MPC ability to upregulate RANKL secretion in the BMME results in a vicious cycle of osteoclast activation, increased bone resorption and MPC proliferation and survival.14,16–18
Bisphosphonates (BPs) were the first class of drugs approved for treating MM bone disease. By causing osteoclast apoptosis, BPs such as zoledronic acid (ZA) and pamidronate led to a decrease and a delay in the development of SRE in MM.19–22 Furthermore, BPs have been found to reduce mortality and improve overall survival in MM which suggests that BPs have an anti-MM effect due to their disruption of the feedback loop between MPC and osteoclasts.23 Denosumab, a fully human monoclonal antibody that binds RANKL and inhibits the RANK pathway has proven to be non-inferior to ZA in delaying SRE in a phase III trial and resulted in superior progression free survival.24,25 This review focuses on the role of the RANKL pathway in the prevention of SRE and in the treatment of MM.
Pathophysiology Of Myeloma Bone Disease: The Key Role Of RANKL In The Myeloma Bone Marrow Microenvironment
Under normal physiologic conditions, osteoblasts and osteoclasts work in unison to remodel bone by bone formation and bone resorption, respectively.26,27 Immature osteoblasts secrete cytokines such as IL-6 to upregulate osteoclasts and mature osteoblasts secrete osteoprotegerin (OPG) to inhibit the activation of osteoclasts. As new bone is formed, osteoblasts become trapped and differentiate into osteocytes which contribute factors to both osteoclastogenesis and osteoblastogenesis.8 MPC cause the dysregulation and uncoupling of this bone remodeling process by interacting with the BMME to induce osteoclast-activating factors to promote osteoclastogenesis while simultaneously secreting osteoblast inhibitory factors to inhibit osteoblastogenesis (Figure 1).12 In the initial stages of the disease, both osteoblasts and osteoclasts are recruited to initiate bone resorption. MPC produce IL-1 and tumor necrosis factor (TNF) which stimulate osteoblast progenitor cells to differentiate into osteoblasts. Osteoblasts in turn secrete IL-6 which acts as MPC growth factor and promoter of osteoclastogenesis.11,28 Once myeloma bone disease (MBD) is established, osteoblasts decrease in number. MPC and bone marrow stromal cells secrete Dkk-1 while osteocytes secrete sclerosin, both of which inhibit the canonical Wnt pathway and result in a decrease in osteoblastogenesis.29,30 Dkk-1 additionally inhibits mesenchymal stromal cells from differentiating into osteoblasts which enables the maximum amount of IL-6 to be secreted thus promoting MPC growth.31 sFRP-2, a Wnt antagonist secreted by MPC, further inhibits osteoblastogenesis.27,32
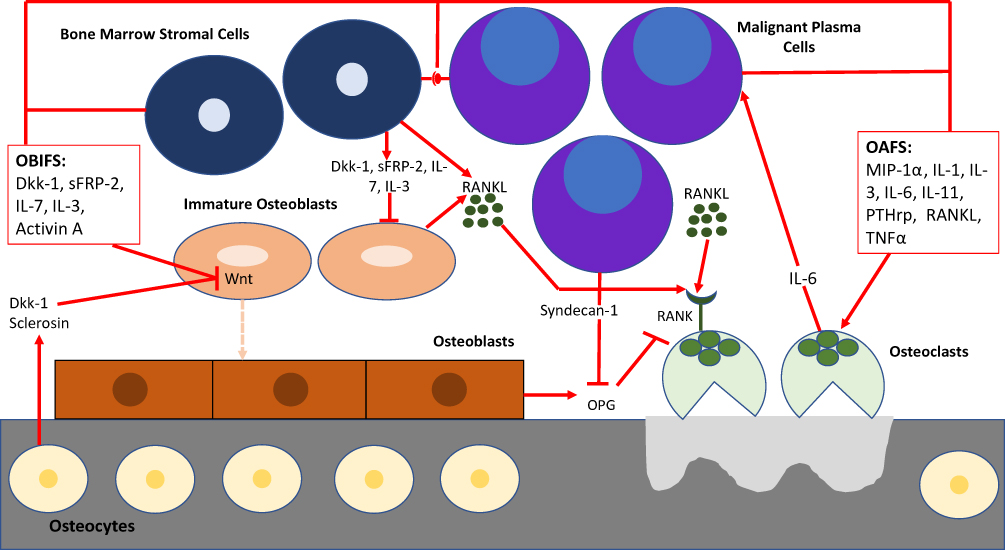 |
Figure 1 BMME in myeloma bone disease. MPC cause the dysregulation and uncoupling of bone remodeling by interacting with the BMME to induce osteoclast-activating factors (OAFS) to promote osteoclastogenesis while simultaneously secreting osteoblast inhibitory factors (OBIFS) to inhibit osteoblastogenesis.Abbreviations: Dkk-I, dickkopf-1; sFRP2, secreted frizzled-related protein 2; IL-1, interleukin-1; IL-3, interleukin-3; IL-6, interleukin-6; IL-7, interleukin-7; IL-11, interleukin-11; PTHrp, parathyroid hormone related peptide; MIP-1 α, macrophage inflammatory protein-1 alpha; RANKL, receptor activator of nuclear factor kappa B, TNFα, tumor necrosis factor alpha; OPG, osteoprotegerin. |
The balance between osteoblasts and osteoclasts is maintained by the ratio of OPG:RANKL.33 The interaction between RANK and RANKL activate downstream nuclear factor kappa B (NF-kB) which in turn activates osteoclast precursors and causes their differentiation to mature osteoclasts and decreases osteoclast apoptosis.9 OPG is a soluble decoy receptor for RANKL that inhibits the RANK-RANKL interaction via molecular mimicry in order to increase osteoblast activity and promote bone formation.27,34 MPC interact with the BMME and activate molecular cascades that ultimately result in increased RANKL and decreased OPG expression.35,36 MPC secrete soluble RANKL as well as PTHrP, IL-1, IL-6, IL-11 and other cytokines which in turn stimulate RANKL expression by osteoblasts and bone marrow stromal cells.8,37,38 In addition, MPC express syndecan-1 which binds to OPG resulting in subsequent endocytosis and degradation of OPG by MPC.39
Greater serum RANKL/OPG ratios are associated with shorter survival. At 60 months, the survival probability for patients with soluble RANKL/OPG <1 was 89% and for patients with a ratio of 1–3 was 32%. The level of soluble RANKL also correlated with the extent of bone disease as examined by radiographic imaging.40 MPC are able to tip the balance of RANKL/OPG in favor of greater levels of RANKL with subsequent suppression of osteoblastogenesis, hyperactivation of osteoclasts and the propagation of osteolytic lesions throughout the entire bone marrow.8
Anti-Myeloma Therapies And Their Effect On The RANKL Pathway And Bone Remodeling
Given the key role of the RANKL pathway and osteoclastogenesis in MPC survival, anti-myeloma therapies that simultaneously target MPC and osteoclast differentiation have the potential to cause deep clinical responses as well as prevent SRE.
Proteasome inhibitors (PIs) such as bortezomib, carfilzomib and ixazomib have been reported to affect bone remodeling via their ability to modulate the RANK/RANKL pathway. One of the main cytotoxic effects of proteasome inhibitors is attributed to inhibition of NF-kB activity.41 Given that binding of RANKL to RANK on the surface of osteoclast precursors activates NF-kB which promotes osteoclast maturation and bone resorption, proteasome-dependent inhibition of NF-kB by PIs lead to a reduction in RANKL-mediated osteoclast differentiation.42,43 In patients with MM, bortezomib was associated with an increase in the levels of biomarkers associated with bone formation and decreased serum levels of RANKL and markers of bone resorption.44 Carfilzomib has been shown to directly inhibit osteoclast formation and bone resorption in vitro, while enhancing osteogenic differentiation and matrix mineralization. Carfilzomib increased trabecular bone volume, decreased bone resorption and enhanced bone formation in mouse models of MM.45 Ixazomib has demonstrated the ability to inhibit in vitro osteoclastogenesis and resorption and these effects on osteoclasts were partially mediated by inhibition of RANKL-induced NF-κB signaling. Ixazomib also stimulates osteogenic differentiation of mesenchymal cells in vitro and promotes osteoblast function and matrix mineralization.46
Immunomodulatory drugs such as thalidomide, lenalidomide and pomalidomide possess anti-myeloma properties including immune-modulation, anti-angiogenic, anti-inflammatory and anti-proliferative effects. Lenalidomide has been shown to inhibit osteoclast formation and activation through inhibition of key factors during osteoclastogenesis in vitro. The combination of lenalidomide and bortezomib blocked osteoclast-derived secretion of growth and survival factors and RANKL secretion from bone marrow stromal cells. Furthermore, lenalidomide treatment decreased serum bone-remodeling markers in patients with relapsed and refractory MM.47 In patients with relapsed and refractory MM, intermediate doses of thalidomide (200mg/day) with dexamethasone led to significant reduction of the soluble RANKL/OPG ratio and markers of bone remodeling.48 Pomalidomide has been shown to inhibit osteoclastogenesis by downregulating transcription factor PU.1 and by significantly blunting RANKL upregulation normalizing the RANKL/OPG ratio in human osteoprogenitor cells when co-cultured with MM cells.49,50
Monoclonal antibodies against CD 38 are the newest group of drugs that have revolutionized anti-MM therapy. Daratumumab, an anti-CD38 monoclonal antibody, has shown in-vitro inhibition of osteoclastogenesis and bone resorption activity in bone marrow cells of MM patients by blocking the interaction of CD 38 expressing monocytes and early osteoclast progenitors.51 Furthermore, the inhibition of T-cell proliferation caused by osteoclasts is partially overcome by another anti-CD38 monoclonal antibody, isatuximab, via inhibition of multiple immune checkpoint molecules expressed on osteoclasts which in turn decrease the immune-evasive properties of MPC.52
In a study of 51 MM patients, patients who received high dose chemotherapy followed by autologous stem cell transplant (ASCT) had a significant reduction of sRANKL/OPG ratio, with a concomitant decrease in markers of bone resorption starting the second month post-ASCT. Bone formation markers started to increase after the 9th month post-ASCT while the increase of OPG preceded this. Thus, it is postulated that high dose chemotherapy followed by ASCT normalizes the abnormal bone resorption in MM patients through the decrease of the RANKL/OPG ratio.53
Histone deacetylase inhibitors (HDACs) inhibit HDAC enzymes, curtailing the aberrant HDAC enzyme activity in MPC.54 Vorinostat has been shown to inhibit RANKL-induced osteoclast formation by suppressing the induction of the osteoclastogenic transcription factor c-Fos.55 Panobinostat has also been shown to inhibit RANKL-mediated osteoclast formation in vitro and in a mouse model of MM.56
By targeting the RANKL pathway, the most active myeloma therapies not only cause apoptosis of the MPC but also inhibit osteoclastogenesis and other key signaling events that underlie SRE.
Bisphosphonates For The Prevention Of Skeletal-Related Events And Treatment Of Multiple Myeloma
BPs are pyrophosphate analogues characterized by two phosphate groups linked to a P-C-P core.57 They inhibit osteoclast activity by inhibiting farnesyl pyrophosphate synthase and accumulate in the mineral phase of the bone.58 There are two groups of BPs, one that contains nitrogen and one that does not. Ibandronate, pamidronate, and ZA contain nitrogen; etidronate and clodronate do not. Nitrogen-containing BPs inhibit farnesyl pyrophosphate synthase, which is essential for osteoclast survival and activity while non-nitrogen containing BPs are metabolized to cytotoxic adenosine triphosphate
analogues that induce osteoclast apoptosis.59 BPs have also been shown to stimulate the innate anti-cancer immune response by upregulating γδT-cells.60 A direct anti-MPC activity has also been described for N-BPs in vitro.61 Nitrogen-containing BPs have also been shown to inhibit RANKL-induced osteoclast formation in vitro.62 Nitrogen-containing BPs have potencies that are 100 to 10,000 times higher than BPs without nitrogen.63 Multiple clinical trials have shown the efficacy of BPs in preventing SRE in patients with MM (Table 1).
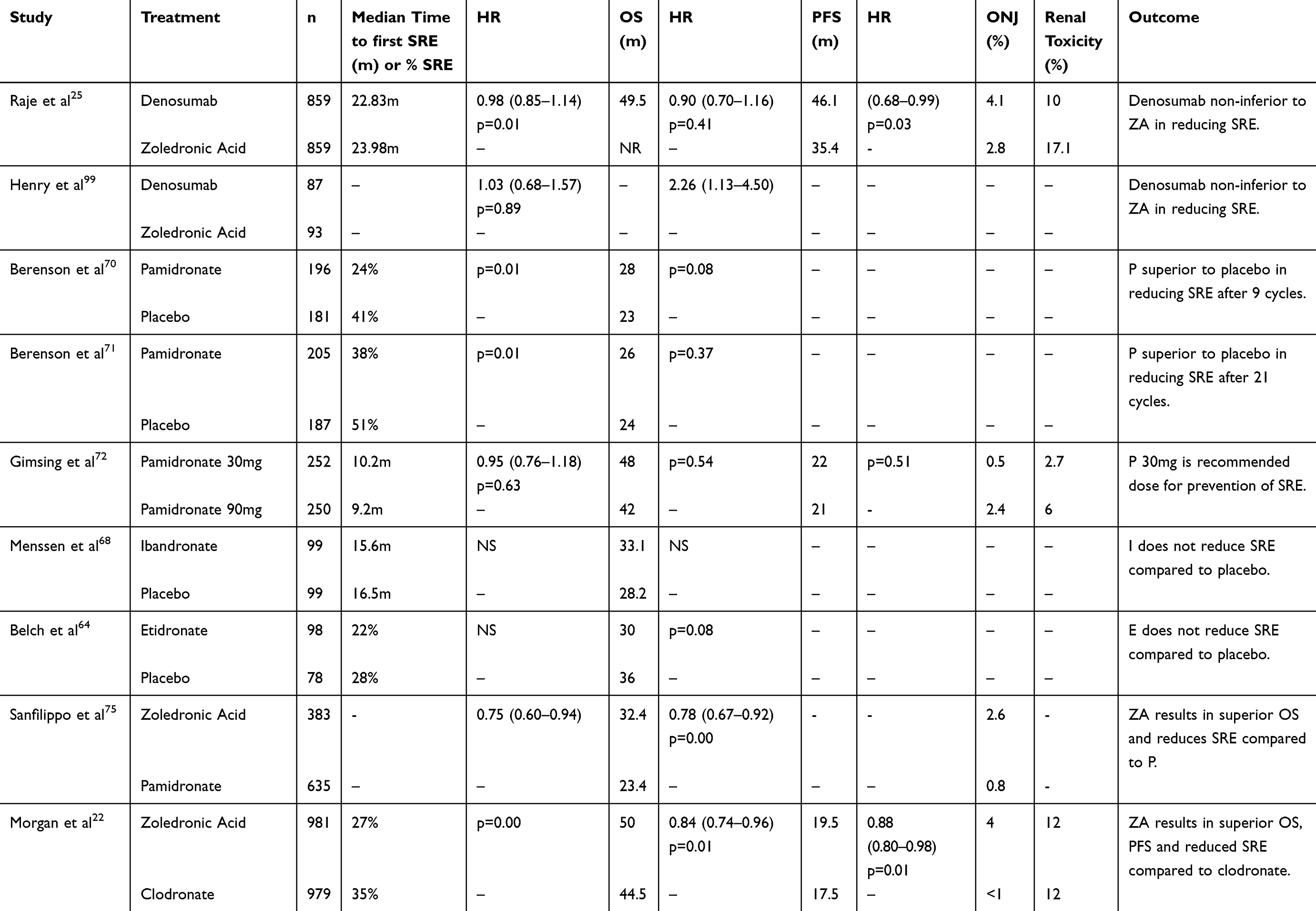 |
Table 1 Randomized Studies Of Bisphosphonates And Denosumab For The Prevention Of Skeletal-Related Events In Multiple Myeloma |
A randomized double-blind study of patients with newly diagnosed MM which compared the effect of daily etidronate vs placebo on the osteolysis of MM found that etidronate therapy did not have a clinically significant effect in MM.64 The results of three randomized trials comparing oral clodronate to placebo showed variable clinical results. A Finnish trial with 350 MM patients found that the proportion of patients with progression of osteolytic bone lesions was twice as high in the placebo group than in the clodronate group respectively (24 vs 12%, p = 0.026).65 Another Finnish trial reported no significant differences in the reduction of vertebral or nonvertebral fractures.66 The Medical Research Council reported a significant reduction in nonvertebral fractures (6.8% vs 13.2%, p=0.04) and vertebral fractures (38% vs 55%, p=0.01) in the clodronate arm compared to the placebo arm respectively.67
A phase III double-blind trial comparing ibandronate vs placebo in addition to anti-myeloma therapy found that ibandronate did not show significant benefits in reducing SRE in MM patients with lytic bone disease.68 Oral pamidronate (300mg/d) was evaluated in a double-blind randomized trial in patients with newly diagnosed MM. After a median duration of 18 months, no significant reduction was apparent in SRE, hypercalcemic episodes or survival between treatment arms.69 A large, randomized, double-blind study conducted to determine the effect of monthly 90mg infusions of pamidronate in patients with MM revealed that after nine cycles of therapy, 24% of pamidronate-treated patients developed a SRE compared to 41% of patients who received placebo.70 Patients who received pamidronate also had a significant decrease in bone pain, no deterioration in performance status and no increase in analgesic use at the end of 9 months. The proportion of patients developing an SRE remained significantly lower in the pamidronate group after an additional 12 cycles of treatment.71 Overall survival was increased in the subset of patients with MM receiving second-line antimyeloma therapy (21 months vs 14 months; p=0.041) compared with placebo. A double-blind randomized phase III trial comparing monthly 90mg vs 30mg of IV pamidronate found that the median time to a SRE was 9.2 months in the 90 mg group and 10.2 months in the 30 mg group (p=0.63).72 Given that a higher proportion of patients in the pamidronate 90 mg group developed osteonecrosis of the jaw (ONJ) and renal toxicity compared to patients in the 30 mg group, monthly infusion of pamidronate 30 mg was the recommended dose for prevention of bone disease in patients with MM.
A phase III trial evaluated two doses of ZA (4 and 8mg) compared with pamidronate (90mg) infused every 3 to 4 weeks for the treatment of patients with MM or breast cancer with metastatic bone disease. The results of the study showed that the proportion of patents developing SRE did not differ between the ZA (4mg) and pamidronate-treated patients.73 After 25 months of follow-up, the overall proportion of patients developing an SRE remained similar between the ZA (4mg) and pamidronate group.74 However, an additional multiple-events analysis showed that patients treated with ZA had a 16% reduction in the risk of developing an SRE compared with patients who received pamidronate.74 In a study which evaluated ZA vs pamidronate in 1,018 United States Veterans with newly diagnosed MM, patients receiving ZA had a 25% reduction in SRE as well as a 22% reduction in risk of death compared to pamidronate.75 A randomized phase III trial comparing 4 mg IV ZA every 3–4 weeks or 1600 mg oral clodronic acid (CA) daily amongst 1970 patients found that ZA reduced mortality by 16% compared to CA (p=0.0118). ZA also extended median overall survival by 5.5 months (50.0 months vs 44.5 months; p=0.04), significantly improved progression-free (PFS) survival by 12% (p=0.0179), and increased median PFS by 2.0 months (19.5 months vs 17.5 months; p=0.07) compared to CA.23 While ZA has been administered every 4 weeks in the aforementioned trials, a randomized phase III trial has shown that ZA administered every 12 weeks is noninferior to ZA administered every 4 weeks.76
BPs not only prevent SRE in MM but also provide a survival benefit possibly due to their anti-MPC properties. In vitro studies suggest that pamidronate may possess anti-MPC properties based on its ability to induce apoptosis of MPC, suppression of IL-6 production and antiangiogenesis.64,77,78 ZA has also been shown to possess antiangiogenic properties in vitro.79 In a long-term follow-up (8.6 years) of a placebo-controlled trial, the subset of CA-treated patients who did not have vertebral fractures at baseline had significantly longer OS vs patients who received placebo (median OS, 59 months vs 37 months, respectively; p=0.006).80 In a retrospective analysis of a phase III trial comparing ZA (4 mg) with pamidronate (90 mg), patients with high baseline bone-specific alkaline phosphatase levels had significantly better 25-month survival with zoledronic acid than with pamidronate (82 vs 53%, respectively; p=0.041).81 In a clinical trial in which 94 patients were randomized to receive either ZA (4mg) or not, after 49.6 months median follow-up, the ZA-treated group had superior 5-year event-free survival (80% vs 52%, p=<0.01) and 5 year OS (80% vs 46%, p=<0.01) compared to the control group.82 As aforementioned, several other trials have shown that BPs prolong survival in patients with MM.23,75 Taken together, these studies conclusively established the role of the nitrogenous BPs ZA, and to a slightly lesser extent pamidronate, for management of MBD.
By interfering with the crosstalk between MPC and osteoclasts, BPs are able to reduce SRE and prolong survival in MM patients via apoptosis of osteoclasts, immunemodulation and direct anti-MPC activity.
Side Effects And Toxicity Of Bisphosphonates
Despite being effective agents for the prevention of SRE in MM patients, the long term use of BPs has come under scrutiny due to their side effects. Notable and well characterized toxicities of BPs include flu-like symptoms, renal toxicity requiring dose reduction in patients with renal insufficiency, ONJ, gastrointestinal upset, atrial fibrillation and atypical femoral fracture.8
Approximately 40% of patients will experience a flu-like syndrome with the first administration of an IV nitrogen-containing BP. Symptoms include fever, fatigue, malaise, myalgia, arthralgia and bone pain that are caused by release of cytokines from γδT cells and macrophages.83 Patients treated with alendronate for osteoporosis were noted to develop low-energy fractures associated with minor trauma, most commonly in the subtrochanteric region of the femur.84 These fractures have usually been associated with extended BP treatment duration of 4–10 years.85 In MM patients treated with IV BPs, cases of atypical fractures resembling those seen in patients on alendronate have been reported.86,87
Renal injury is a major limiting factor in BP use, with acute tubular necrosis as the main pathology.88 Nephrotoxicity is related to the dose, infusion time and maximum plasma concentration that affects the intracellular concentration of BPs.89 There is the potential for BPs with prolonged renal tissue half-life, such as ZA, to accumulate in renal tissue and cause damage. Pamidronate has been associated with collapsing focal segmental glomerulosclerosis.90 With ZA, the risk of kidney injury (rise in creatinine of 0.5 mg/dl) was observed in the initial phase III trials comparing ZA to pamidronate, prompting a dose reduction from 8mg to 4mg and increase in the duration of the infusion from 5 to 15 min.73 To minimize toxicity, the 2007 American Society of Clinical Oncology guidelines suggested dose adjustments of ZA in patients with creatinine clearance ranging from 30–60 mL/min. ZA is not recommended for patients with creatinine clearance <30 mL/min.91 For patients with severe renal impairment, pamidronate 90mg administered over 4–6 hrs is the preferred BP. This nephrotoxicity is not unique to nitrogenous BPs as in a randomized phase III trial comparing 4 mg IV ZA every 3–4 weeks or 1600 mg oral CA daily amongst 1970 patients with MM, both groups had the same rate of renal toxicity (12%).22 Dose dependence of renal toxicity was demonstrated in a phase III trial comparing monthly 90mg vs 30mg of IV pamidronate which found a greater incidence of renal toxicity in the 90mg group compared to the 30mg group (6% vs 2.7%; p=0.072).72
ONJ is defined as a lesion of exposed bone in the maxilla or mandible that persists for 8 weeks in patients treated with BPs who are not receiving radiotherapy to the craniofacial area.92 Clinical signs and symptoms of ONJ include pain, swelling and/or ulceration of the oral mucosa, loose teeth or a nonhealing socket after tooth extraction. The severity of ONJ can vary from asymptomatic forms to severe lesions complicated by the appearance of fistula or fracture.93 The incidence of ONJ in patients with MM treated with BPs has been noted to be as high as 8.5%. Ibandronate and pamidronate appear to have a better safety profile compared to ZA.94 In a study comparing ZA vs pamidronate in 1,018 United States Veterans with newly diagnosed MM, the patients who received ZA had a higher incidence of ONJ compared to patients who received pamidronate (2.6% vs 0.8%).75 The phase III trial comparing ZA to CA in 1970 patients found a higher risk of developing ONJ in the ZA group compared to the CA group (4% vs <1%).22 The phase III trial comparing monthly 90mg vs 30mg of IV pamidronate found a greater amount of ONJ in the 90mg group (2.4%) vs the 30mg group (0.5%).72 The complete removal of necrotic bone, smoothing of sharp bony
edges and careful wound closure, accompanied by perioperative antibiotic treatment is generally considered to be the most suitable approach to achieve ONJ healing.95 With surgical management, resolution with complete healing of ONJ has been noted greater than 80% of cases.96
Denosumab: Combining Inhibition Of Osteoclastogenesis With Anti-Myeloma Activity And An Improved Side-Effect Profile
Denosumab is a fully human monoclonal antibody that binds and inhibits RANKL and is administered subcutaneously. Unlike BPs, denosumab does not accumulate or persist in bone and is cleared through the reticuloendothelial system hence not dependent on renal clearance. Denosumab has a half-life of approximately 26 days.97 The positive effect of denosumab on bone remodeling was initially demonstrated in the treatment of osteoporosis. A phase III trial of 7886 women with osteoporosis demonstrated that denosumab (60mg given subcutaneously every 6 months) was superior to placebo in reducing the risk of vertebral, nonvertebral and hip fractures.98
Two clinical trials have evaluated the efficacy of denosumab for the prevention of SRE in MM (Table 1).
The first double-blind study which evaluated the efficacy of denosumab in MM was the 244 study which compared denosumab 120mg subcutaneously vs ZA 4mg IV every 4 weeks.99 This study excluded breast and prostate cancer patients and the largest proportion of patients had lung cancer (40%) and MM (10%). The median time to first SRE was longer with denosumab compared to ZA (20.6 vs 16.3 months, p=0.06). Denosumab improved quality of life and also reduced the need for radiation and preventing worsening of pain.100 When all patients in the 244 study were analyzed together, there was no difference in overall survival. However, in the MM cohort (n=180), patients treated with denosumab had a worse overall survival (HR=2.26). Due to the small number of MM patients, the 244 study had many limitations and confounding factors that favored the ZA arm; the denosumab arm had more patients with renal dysfunction (which confers a worse prognosis) and the patients in the ZA arm received more intensive treatment with newer agents and high dose melphalan and ASCT. In addition, there was more censoring from early withdrawal in the ZA arm.
To address the limitations of the 244 study, a large phase III study was carried out randomizing 1718 patients with newly diagnosed MM with at least one bone lesion to either denosumab 120mg subcutaneously vs ZA 4mg IV every 4 weeks.25 The primary end point of the study was time to SRE, defined in the trial as pathologic fracture, need for radiation therapy or bone surgery or spinal cord compression. Denosumab was non-inferior to ZA in time to SRE (22.83 vs 23.98 months; HR=0.98 [0.84–1.14]; p=0.01 for non-inferiority) and OS was similar in both arms, (49.5 months vs not reached; HR=0.90 [0.70–1.16]; p=0.41). PFS survival was longer in the denosumab arm compared with the ZA arm (46 vs 35.4 months; HR=0.82 [0.68–0.99], p=0.036).
ONJ was reported in 4.1% of patients in the denosumab arm and in 2.8% of patients in the ZA arm, though the difference was not statistically significant (p=0.147). Renal toxicity was significantly lower in the denosumab arm compared to the ZA arm, 10 vs 17.1% (p=<0.001) respectively. This difference in renal toxicity was highlighted in patients with renal insufficiency at baseline (creatinine clearance ≤ 60ml/min) where renal toxicity was reduced by half with denosumab compared with ZA; 12.9 vs 26.4% respectively. Fewer acute phase reactions were noted in the denosumab group (5.4%) compared to the ZA group (8.7%). Hypocalcemia occurred in 16.9% of patients in the denosumab arm compared to 12.4% in the ZA arm. Grade 3–4 hypocalcemia was uncommon, occurring in 0.9% of patients in the denosumab arm and 0.2% of patients in the ZA arm. On the basis of these findings, the FDA and the European Medicines Agency approved denosumab for the prevention of SRE in patients with MM.101,102
In a large series of 1027 patients with newly diagnosed MM, half of the patients were found to have an elevated creatinine and 20% had a serum creatinine >2 mg/dl.103 Given these findings and the fact that renal dysfunction often presents a major barrier to effective and continued use of osteoclast-targeted therapy with BPs, denosumab is an ideal agent for preventing SRE in patients with MM and renal disease as its dosing does not depend on creatinine clearance. The improved PFS noted in the denosumab group compared to the ZA group warrants further investigation. Given the key role of RANKL in osteoclastogenesis and the importance of osteoclast cytokine signaling crucial for MPC survival, it is possible that denosumab’s anti-myeloma effects arises from its interference of the crosstalk between osteoclasts and MPC via RANKL.18 The first therapeutic study on RANKL blockade in an animal model of myeloma bone disease revealed that RANKL inhibition markedly reduced tumor burden assessed histologically and by serum paraprotein in the SCID-hu-MM mice.24 In a phase II study of single-agent denosumab in the treatment of 93 relapsed or plateau-phase multiple myeloma patients, treatment with denosumab did not result in reduction of serum monoclonal protein level in the range of complete response, partial response or minimal response.104 However, eleven subjects (21%) with myeloma who entered the study with progressive disease maintained stable disease for a maximum of 16.5 months (median duration: 2.6 months) and 19 subjects (46%) with plateau-phase myeloma maintained stable disease for a maximum of 18.3 months (median duration: 10.2 months). It is important to consider that this study was dealing with a relatively-drug resistant population which may have made it difficult to detect an anti-myeloma effect with single agent denosumab. The stabilization of disease observed in some subjects raises the possibility that cytostatic effects through alteration of the BMME could influence the growth of MPC. Further research into the anti-neoplastic role of RANKL inhibition and its clinical benefit is merited, especially in the context of recent therapeutic advances in the treatment of MM.
As noted above, the toxicity profile of denosumab is fairly established. Hypocalcemia and ONJ are important acute and long term toxicities, respectively. One of the major concerns of denosumab treatment is the increased risk of vertebral fractures when the drug is discontinued. Multiple case reports of vertebral fractures, including multiple vertebral fractures, soon after discontinuation of denosumab in osteoporosis patients have been published.105,106 A post hoc analysis of postmenopausal women with osteoporosis discontinuing denosumab in the FREEDOM trial revealed evidence of increased vertebral fractures.107 The risk of multiple vertebral fractures was 3.4% after stopping denosumab and 2.2% after stopping placebo (p = 0.049), with the risk being 3.9 (95% CI 2.1–7.2) times higher in those with a prior vertebral fracture before or during treatment compared with those having no prior vertebral fracture. Treating with BPs after stopping denosumab can prevent rebound fractures. Limited evidence suggests that ZA given 7–8 months after the last dose of denosumab may be the preferred clinical strategy.108 Further evaluation of denosumab discontinuation and fracture risk in MM patients is warranted.
Conclusion
SRE are a hallmark of MM and lead to increased morbidity and mortality. Via interactions with the BMME, MPC stimulate osteoclastogenesis which in turn leads to MPC survival and osteolytic bone lesions. Osteoclast inhibition with BPs, particularly ZA and pamidronate, is the standard of care in preventing and delaying SRE in MM as well as in prolonging OS due to their anti-myeloma properties. However, administration of BPs is challenging in MM patients due to their renal clearance, potential to cause nephrotoxicity and the inherent renal dysfunction associated with MM. RANKL is a key molecule in the BMME involved in osteoclastogenesis. Anti-myeloma therapies including PIs, immunomodulatory agents, high dose chemotherapy followed by ASCT and HDACs have been shown to inhibit osteoclastogenesis via inhibition of RANKL. Denosumab, a fully human monoclonal antibody against RANKL has proven to be noninferior to ZA in preventing and delaying SRE in MM. Denosumab has also shown to prolong PFS in MM patients compared to ZA. Favorable renal tolerance makes denosumab an attractive candidate for use in MM patients with renal disease. Vertebral fractures upon discontinuation of therapy represent an important toxicity and need to be monitored for carefully. Given the direct anti-MM effect observed in several studies, well planned clinical trials combining denosumab with novel immunotherapeutic approaches are desirable to expand the therapeutic armamentarium for MM.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Becker N. Epidemiology of multiple myeloma. In: Moehler T, Goldschmidt H, editors. Multiple Myeloma. Vol. 183, Recent Results in Cancer Research. Berlin: Springer; 2011:25–35.
2. Dimopoulos MA, Kastritis E, Anagnostopoulos A, et al. Osteonecrosis of the jaw in patients with multiple myeloma treated with bisphosphonates: evidence of increased risk after treatment with zoledronic acid. Haematologica. 2006;91:968–971.
3. Qian Y, Arellano J, Bhowmik D, et al. Healthcare resource use and costs associated with renal impairment in US patients with bone metastases from solid tumors. J Oncol Pharm Pract. 2017;23:195–202. doi:10.1177/1078155216629826
4. Moreau P, Attal M, Caillot D, et al. Prospective evaluation of magnetic resonance imaging and [18F] fluorodeoxyglucose positron emission tomography-computed at diagnosis and before maintenance therapy in symptomatic patients with multiple myeloma included in the IFM/DFCI 2009 trial: results of the IMAJEM study. J Clin Oncol. 2017;35(25):2911–2918. doi:10.1200/JCO.2017.72.2975
5. Tosi P. Diagnosis and treatment of bone disease in multiple myeloma: spotlight on spinal involvement. Scientifica (Cairo). 2013;2013:104546.
6. Zagouri F, Kastritis E, Zomas A, et al. Hypercalcemia remains an adverse prognostic factor for newly diagnosed multiple myeloma patients in the era of novel antimyeloma therapies. Eur J Haematol. 2017;99:409–414. doi:10.1111/ejh.12923
7. Kyle RA. Multiple myeloma: review of 869 cases. Mayo Clin Proc. 1975;50:29–40.
8. Ring ES, Lawson MA, Snowden JA, Jolley I, Chantry AD. New agents in the treatment of myeloma bone disease. Calcif Tissue Int. 2018;102:196–209. doi:10.1007/s00223-017-0351-7
9. Silbermann R, Roodman GD. Current controversies in the management of myeloma bone disease. J Cell Physiol. 2016;231:2374–2379. doi:10.1002/jcp.25173
10. Mundy GR, Raisz LG, Cooper RA, Schechter GP, Salmon SE. Evidence for the secretion of an osteoclast stimulating factor in myeloma. N Engl J Med. 1974;291:1041–1046. doi:10.1056/NEJM197411142912001
11. Bataille R, Chappard D, Marcelli C, et al. Recruitment of new osteoblasts and osteo- clasts is the earliest critical event in the pathogenesis of human multiple myeloma. J Clin Invest. 1991;88:62–66. doi:10.1172/JCI115305
12. Abildgaard N, Glerup H, Rungby J, et al. Biochemical markers of bone metabolism reflect osteoclastic and osteoblastic activity in multiple myeloma. Eur J Haematol. 2000;64:121–129. doi:10.1034/j.1600-0609.2000.90074.x
13. Delgado-Calle J, Bellido T, Roodman GD. Role of osteocytes in multiple myeloma bone disease. Curr Opin Support Palliat Care. 2014;8(4):407–413. doi:10.1097/SPC.0000000000000090
14. Todoerti K, Lisignoli G, Storti P, et al. Distinct transcriptional profiles characterize bone microenvironment mesenchymal cells rather than osteoblasts in relationship with multiple myeloma bone disease. Exp Hematol. 2010;38:141–153. doi:10.1016/j.exphem.2009.11.009
15. Moreaux J, Hose D, Kassambara A, et al. Osteoclast-gene expression profiling reveals osteoclast-derived CCR2 chemokines promoting myeloma cell migration. Blood. 2011;117:1280–1290. doi:10.1182/blood-2010-04-279760
16. Lai FP, Cole-Sinclair M, Cheng WJ, et al. Myeloma cells can directly contribute to the pool of RANKL in bone bypassing the classic stromal and osteoblast pathway of osteoclast stimulation. Br J Haematol. 2004;126:192–201. doi:10.1111/j.1365-2141.2004.05018.x
17. Abe M, Hiura K, Wilde J, et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: a vicious cycle between bone destruction and myeloma expansion. Blood. 2004;104:2484–2491. doi:10.1182/blood-2003-11-3979
18. Yaccoby S, Wezeman MJ, Henderson A, et al. Cancer and the microenvironment: myeloma-osteoclast interactions as a model. Cancer Res. 2004;64:2016–2023. doi:10.1158/0008-5472.CAN-03-1131
19. Rosen LS, Gordon D, Kaminski M, et al. Long-term efficacy and safety of zoledronic acid in the treatment of skeletal metastases in patients with nonsmall cell lung carcinoma and other solid tumors: a randomized, Phase III, double-blind, placebo-controlled trial. Cancer. 2004;100(12):2613–2621. doi:10.1002/cncr.20308
20. Rosen LS, Gordon D, Tchekmedyian S, et al. Zoledronic acid versus placebo in the treatment of skeletal metastases in patients with lung cancer and other solid tumors: a phase III, double-blind, randomized trial–the Zoledronic Acid Lung Cancer and Other Solid Tumors Study Group. J Clin Oncol. 2003;21(16):3150–3157. doi:10.1200/JCO.2003.04.105
21. Saad F, Gleason DM, Murray R, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. 2002;94(19):1458–1468. doi:10.1093/jnci/94.19.1458
22. Morgan GJ, Davies FE, Gregory WM, et al. First-line treatment with zoledronic acid as compared with clodronic acid in multiple myeloma (MRC Myeloma IX): a randomised controlled trial. Lancet. 2010;376:1989–1999. doi:10.1016/S0140-6736(10)62051-X
23. Wong MH, Stockler MR, Pavlakis N. Bisphosphonates and other bone agents for breast cancer. Cochrane Database Syst Rev. 2012;2:CD003474.
24. Pearse RN, Sordillo EM, Yacoby S, et al. Multiple Myeloma disrupts the TRANCE/osteoprotegerin cytokine axis to trigger bone destruction and promote tumor progression. Proc Natl Acad Sci. 2001;98(20):11581–11586. doi:10.1073/pnas.201394498
25. Raje N, Terpos E, Willenbacher W, et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: an international, double-blind, double- dummy, randomised, controlled, phase 3 study. Lancet Oncol. 2018;19:370–381. doi:10.1016/S1470-2045(18)30144-X
26. Hameed A, Brady JJ, Dowling P, Clynes M, Gorman P. Bone disease in multiple myeloma: pathophysiology and management. Cancer Growth Metastasis. 2014;7:33–42. doi:10.4137/CGM.S16817
27. Walker RE, Lawson MA, Buckle CH, Snowden JA, Chantry AD. Myeloma bone disease: pathogenesis, current treatments and future targets. Br Med Bull. 2014;111:117–138. doi:10.1093/bmb/ldu016
28. Bataille R, Harousseau J-L. Multiple myeloma. N Engl J Med. 1997;336:1657–1664.
29. Tian E, Zhan F, Walker R, et al. The role of the Wnt-Signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–2494. doi:10.1056/NEJMoa030969
30. McDonald MM, Reagan MR, Youlten SE, et al. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple myeloma. Blood. 2017;129:3452–3464. doi:10.1182/blood-2017-03-773341
31. Gunn WG, Conley A, Deininger L, Olson SD, Prockop DJ, Gregory CA. A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK1 and interleukin-6: a potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells. 2006;24:986–991. doi:10.1634/stemcells.2005-0220
32. Oshima T, Abe M, Asano J, et al. Myeloma cells suppress bone formation by secreting a soluble Wnt inhibitor, sFRP-2. Blood. 2005;106(9):3160–3165. doi:10.1182/blood-2004-12-4940
33. Giuliani N, Bataille R, Mancini C, Lazzaretti M, Barille ́ S. Myeloma cells induce imbalance in the osteoprotegerin/osteoprotegerin ligand system in the human bone marrow environment. Blood. 2001;98:3527–3533. doi:10.1182/blood.v98.2.351
34. Croucher PI, Shipman CM, Lippitt J, et al. Osteoprotegerin inhibits the development of osteolytic bone disease in multiple myeloma. Blood. 2001;98:3534–3540. doi:10.1182/blood.v98.2.351
35. Delgado-Calle J, Anderson J, Cregor MD, et al. Bidirectional notch signaling and osteocyte-derived factors in the bone marrow microenvironment promote tumor cell proliferation and bone destruction in multiple myeloma. Cancer Res. 2016;76:1089–1100. doi:10.1158/0008-5472.CAN-16-0584
36. Toscani D, Palumbo C, Dalla Palma B, et al. The proteasome inhibitor bortezomib maintains osteocyte viability in multiple myeloma patients by reducing both apoptosis and autophagy: a new function for proteasome inhibitors. J Bone Miner Res. 2016;31:815–827. doi:10.1002/jbmr.2741
37. Buckle CH, De Leenheer E, Lawson MA, et al. Soluble rank ligand produced by myeloma cells causes generalised bone loss in multiple myeloma. PLoS One. 2012;7(8):e41127. doi:10.1371/journal.pone.0041127
38. Cafforio P, Savonarola A, Stucci S, et al. PTHrP produced by myeloma plasma cells regulates their survival and pro-osteoclast activity for bone disease progression. J Bone Miner Res. 2014;29:55–66. doi:10.1002/jbmr.2022
39. Standal T, Seidel C, Hejertner O, et al. Osteoprotegerin is bound, internalized, and degraded by multiple myeloma cells. Blood. 2002;100:3002–3007. doi:10.1182/blood-2002-03-0706
40. Terpos E, Szydlo R, Apperley JF, et al. Soluble receptor activator of nuclear factor kappaB ligand-osteoprotegerin ratio predicts survival in multiple myeloma: proposal for a novel prognostic index. Blood. 2003;102:1064–1069. doi:10.1182/blood-2003-02-0380
41. Hideshima T, Chauhan D, Richardson P, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277(19):16639–16647. doi:10.1074/jbc.M200360200
42. Zavrski I, Krebbel H, Wildemann B, et al. Proteasome inhibitors abrogate osteoclast differentiation and osteoclast function. Biochem Biophys Res Commun. 2005;333(1):200–205. doi:10.1016/j.bbrc.2005.05.197
43. vonMetzler I, Krebbel H, Hecht M, et al. Bortezomib inhibits human osteoclastogenesis. Leukemia. 2007;21(9):2025–2034. doi:10.1038/sj.leu.2404806
44. Terpos E, Heath DJ, Rahemtulla A, et al. Bortezomib reduces serum dickkopf-1 and receptor activator of nuclear factor-kappaB ligand concentrations and normalises indices of bone remodeling in patients with relapsed multiple myeloma. Br J Haematol. 2006;135:688–692. doi:10.1111/j.1365-2141.2006.06356.x
45. Hurchla MA, Garcia-Gomez A, Hornick MC, et al. The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects. Leukemia. 2013;27:430–440. doi:10.1038/leu.2012.183
46. Garcia-Gomez A, Quwaider D, Canavese M, et al. Pre-clinical activity of the oral proteasome inhibitor MLN9708 in myeloma bone disease. Clin Cancer Res. 2014;20:1542–1554. doi:10.1158/1078-0432.CCR-13-3045
47. Breitkreutz I, Raab MS, Vallet S, et al. Lenalidomide inhibits osteoclastogenesis, survival factors and bone-remodeling markers in multiple myeloma. Leukemia. 2008;22:1925–1932. doi:10.1038/sj.leu.2404889
48. Terpos E, Mihou D, Szydlo R, et al. The combination of intermediate doses of thalidomide with dexamethasone is an effective treatment for patients with refractory/relapsed multiple myeloma and normalizes abnormal bone remodeling, through the reduction of sRANKL/osteoprotegerin ratio. Leukemia. 2005;19:1969–1976. doi:10.1038/sj.leu.2403890
49. Anderson G, Gries M, Kurihara N, et al. Thalidomide derivative CC-4047 inhibits osteoclast formation by down-regulation of PU.1. Blood. 2006;107:3098–3105. doi:10.1182/blood-2005-08-3450
50. Bolzoni M, Storti P, Bonomini S, et al. Immunomodulatory drugs lenalidomide and pomalidomide inhibit multiple myeloma-induced osteoclast formation and the RANKL/OPG ratio in the myeloma microenvironment targeting the expression of adhesion molecules. Exp Hematol. 2013;41:387–397. doi:10.1016/j.exphem.2012.11.005
51. Costa F, Toscani D, Chillemi A, et al. Expression of CD38 in myeloma bone niche: A rational basis for the use of anti-CD38 immunotherapy to inhibit osteoclast formation. Oncotarget. 2017;8(34):56598–56611. doi:10.18632/oncotarget.17896
52. An G, Acharya C, Feng X, et al. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: therapeutic implication. Blood. 2016;128(12):1590–1603. doi:10.1182/blood-2016-06-724161
53. Terpos E, Politou M, Szydlo R, et al. Autologous stem cell transplantation normalizes abnormal bone remodeling and sRANKL/osteoprotegerin ratio in patients with multiple myeloma. Leukemia. 2004;18:1420–1426. doi:10.1038/sj.leu.2403423
54. Petrella A, Fontanella B, Carratu A, Bizzarro V, Rodriquez M, Parente L. Histone deacetylase inhibitors in the treatment of hematological malignancies. Mini Rev Med Chem. 2011;11:519–527.
55. Kim HN, Ha A, Lee JH, et al. Trichostatin A inhibits osteoclastogenesis and bone resorption by suppressing the induction of c-Fos by RANKL. Eur J Pharmacol. 2009;623:22–29. doi:10.1016/j.ejphar.2009.09.025
56. Imai Y, Ohta E, Takeda S, et al. Histone deacetylase inhibitor panobinostat induced calcineurin degradation in multiple myeloma. JCI Insight. 2016;1(5):e85061. doi:10.1172/jci.insight.85061
57. Fleisch H. Development of bisphosphonates. Breast Cancer Res. 2002;4:30–34.
58. Favus MJ. Bisphosphonates for osteoporosis. N Engl J Med. 2010;363(21):2027–2035. doi:10.1056/NEJMct1004903
59. D’Oronzo S, Coleman R, Brown J, Silvestris F. Metastatic bone disease: pathogenesis and therapeutic options: up-date on bone metastasis management. J Bone Oncol. 2018;15:100205. doi:10.1016/j.jbo.2018.10.004
60. Naoe M, Ogawa Y, Takeshita K, et al. Zoledronate stimulates gamma delta T cells in prostate cancer patient. Oncol Res. 2010;18(10):493–501. doi:10.3727/096504010X12671222663638
61. Baulch-Brown C, Molloy TJ, Yeh SL, Ma D, Spencer A. Inhibitors of the mevalonate pathway as potential therapeutic agents in multiple myeloma. Leuk Res. 2007;31:341–352. doi:10.1016/j.leukres.2006.07.018
62. Tsubaki M, Komai M, Itoh T, et al. Nitrogen-containing bisphosphonates inhibit RANKL- and M-CSF-induced osteoclast formation through the inhibition of ERK1/2 and Akt activation. J Biomed Sci. 2014;21(1):10. doi:10.1186/1423-0127-21-10
63. Mahindra A, Pozzi S, Noopur R. Clinical trials of bisphosphonates in multiple myeloma. Clin Adv Hematol Oncol. 2012;10(9):582–587.
64. Belch AR, Bergsagel DE, Wilson K, et al. Effect of daily etidronate on the osteolysis of multiple myeloma. J Clin Oncol. 1991;9:1397–1402. doi:10.1200/JCO.1991.9.8.1397
65. Lahtinen R, Laakso M, Palva I, Virkkunen P, Elomaa I. Randomised, placebo-controlled multicentre trial of clodronate in multiple myeloma. Lancet. 1992;340:1049–1052. doi:10.1016/0140-6736(92)93075-x
66. Heim ME, Clemens MR, Queisser W, et al. Prospective randomized trial of dicholoromethylene bisphosphonate (clodronate) in patients with multiple myeloma requiring treatment: a multicenter study. Onkologie. 1995;18:439–448.
67. McCloskey EV, MacLennan CM, Drayson MT, et al. A randomized trial of the effect of clodronate on skeletal morbidity in multiple myeloma. Br J Haematol. 1998;101:317–325. doi:10.1046/j.1365-2141.1998.00567.x
68. Menssen HD, Saklova A, Fontana A, et al. Effects of long-term intravenous ibandronate therapy on skeletal-related events, survival, and bone resorption markers in patients with advanced multiple myeloma. J Clin Oncol. 2002;20:2353–2359. doi:10.1200/JCO.2002.02.032
69. Brincker H, Westin J, Abildgaard N, et al. Failure of oral pamidronate to reduce skeletal morbidity in multiple myeloma: a double-blind placebo-controlled trial. Br J Haematol. 1998;101:280–286. doi:10.1046/j.1365-2141.1998.00695.x
70. Berenson JR, Lichtenstein A, Porter L, et al. Efficacy of pamidronate in reducing the skeletal events in patients with advanced multiple myeloma. N Engl J Med. 1996;334:488–493. doi:10.1056/NEJM199602223340802
71. Berenson J, Lichtenstein A, Porter L, et al. Long- term pamidronate treatment of advanced multiple myeloma patients reduces skeletal events. J Clin Oncol. 1998;16:593–602. doi:10.1200/JCO.1998.16.2.593
72. Gimsing P, Carlson K, Turesson I, et al. Effect of pamidronate 30 mg versus 90 mg on physical function in patients with newly diagnosed multiple myeloma (Nordic Myeloma Study Group): a double-blind, randomised controlled trial. Lancet Oncol. 2010;11(10):973–982. doi:10.1016/S1470-2045(10)70198-4
73. Rosen LS, Gordon D, Antonio BS, et al. Zoledronic acid versus pamidronate in the treatment of skeletal metastases in patients with breast cancer or osteolytic lesions of multiple myeloma: a phase III, double-blind, comparative trial. Cancer J. 2001;7:377–387.
74. Rosen LS, Gordon D, Kaminski M, et al. Long-term efficacy and safety of zoledronic acid compared with pamidronate disodium in the treatment of skeletal complications in patients with advanced multiple myeloma or breast carcinoma: a randomized, double- blind, multicenter, comparative trial. Cancer. 2003;98:1735–1744. doi:10.1002/cncr.11701
75. Sanfilippo KM, Gage B, Luo S, et al. Comparative effectiveness on survival of zoledronic acid versus pamidronate in multiple myeloma. Leuk Lymphoma. 2015;56(3):615–621. doi:10.3109/10428194.2014.924117
76. Himelstein AL, Foster JC, Khatcheressian JL, et al. Effect of longer-interval vs standard dosing of zoledronic acid on skeletal events in patients with bone metastases. JAMA. 2017;317(1):48–58. doi:10.1001/jama.2016.19425
77. Aparicio A, Gardner A, Tu Y, Savage A, Berenson J, Lichtenstein A. In vitro cytoreductive effects on multiple myeloma cells induced by bisphosphonates. Leukemia. 1998;12:220–229. doi:10.1038/sj.leu.2400892
78. Savage AD, Belson DJ, Vescio RA, et al. Pamidronate reduces IL-6 production by bone marrow stroma from multiple myeloma patients. Blood. 1996;88:105a.
79. Wood J, Bonjean K, Ruetsz S, et al. Novel antiangiogenic effects of the bisphosphonate compound zoledronic acid. J Pharmacol Exp Ther. 2002;302:1055–1061. doi:10.1124/jpet.102.035295
80. McCloskey EV, Dunn JA, Kanis JA, MacLennan IC, Drayson MT. Long-term follow-up of a prospective, double-blind, placebo- controlled randomized trial of clodronate in multiple myeloma. Br J Haematol. 2001;113:1035–1043. doi:10.1046/j.1365-2141.2001.02851.x
81. Berenson JR, Dimopoulos MA, Chen YM. Zoledronic acid may improve survival compared to pamidronate in patients with MM and high BALP levels: univariate and multivariate models of hazard ratios. Blood. 2006;108 Abstract 3589.
82. Aviles A, Nambo MJ, Neri N, Castaneda C, Cleto S, Huerta-Guzman J. Antitumor effect of zoledronic acid in previously untreated patients with multiple myeloma. Med Oncol. 2007;24:227–230. doi:10.1007/BF02698044
83. Olson K, Van Poznak C. Significance and impact of bisphosphonate- induced acute phase responses. J Oncol Pharm Pract. 2007;13:223–229. doi:10.1177/1078155207080806
84. Lenart BA, Lorich DG, Lane JM. Atypical fractures of the femoral diaphysis in postmenopausal women taking alendronate. N Engl J Med. 2008;358:1304–1306. doi:10.1056/NEJMc0707493
85. Odvina CV, Levy S, Rao S, Zerwekh JE, Rao DS. Unusual mid-shaft fractures during long-term bisphosphonate therapy. Clin Endocrinol (Oxf). 2010;72:161–168. doi:10.1111/j.1365-2265.2009.03581.x
86. Wernecke G, Namdari S, DiCarlo EF, Schneider R, Lane J. Case report of spontaneous, nonspinal fractures in a multiple myeloma patient on long-term pamidronate and zoledronic acid. HSS J. 2008;4:123–127. doi:10.1007/s11420-008-9077-4
87. Grasko JM, Herrmann RP, Vasikaran SD. Recurrent low-energy femoral shaft fractures and osteonecrosis of the jaw in a case of multiple myeloma treated with bisphosphonates. J Oral Maxillofac Surg. 2009;67:645–649. doi:10.1016/j.joms.2008.11.005
88. Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int. 2008;74(11):1385–1393. doi:10.1038/ki.2008.356
89. Body JJ, Pfister T, Bauss F. Preclinical perspectives on bisphosphonate renal safety. Oncologist. 2005;10:3–7. doi:10.1634/theoncologist.10-90001-3
90. Kunin M, Kopolovic J, Avigdor A, Holtzman EJ. Collapsing glomerulopathy induced by long-term treatment with standard-dose pamidronate in a myeloma patient. Nephrol Dial Transplant. 2004;19:723–726. doi:10.1093/ndt/gfg567
91. Kyle RA, Yee GC, Somerfield MR, et al. American Society of Clinical Oncology 2007 clinical practice guideline update on the role of bisphosphonates in multiple myeloma. J Clin Oncol. 2007;25:2464–2472. doi:10.1200/JCO.2007.12.1269
92. Ruggiero SL, Fantasia J, Carlson E. Bisphosphonate-related osteonecrosis of the jaw: background and guidelines for diagnosis, staging and management. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;102:433–441. doi:10.1016/j.tripleo.2006.06.004
93. Ruggiero SL, Dodson TB, Assael LA, Landesberg R, Marx RE, Mehrotra B. American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaws-2009 update. J Oral Maxillofac Surg. 2009;67:2–12. doi:10.1016/j.joms.2009.01.009
94. Vahtsevanos K, Kyrgidis A, Verrou E, et al. Longitudinal cohort study of risk factors in cancer patients of bisphosphonate-related osteonecrosis of the jaw. J Clin Oncol. 2009;27:5356–5362. doi:10.1200/JCO.2009.21.9584
95. Ristow O, Otto S, Troeltzsch M, Hohlweg-Majert B, Pautke C. Treatment perspectives for medication-related osteonecrosis of the jaw (MRONJ). J Craniomaxillofac Surg. 2015;43:290–293. doi:10.1016/j.jcms.2014.11.014
96. Schiodt M, Otesen C, Dalsten H, Oturay P, Kofod T. Surgical Treatment Outcome of 141 Consecutive Patients with Medication-related Osteonecrosis of the Jaws (MRONJ) from the Copenhagen ONJ Cohort, 13–16 September 2016. London (UK): European Association for Cranio Maxillo-Facial Surgery (EACMFS). p. Oral Presentation Session 4.
97. Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone. 2011;48(4):677–692. doi:10.1016/j.bone.2010.11.020
98. Cummings SR, San Martin J, Mcclung MR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361(8):756–765. doi:10.1056/NEJMoa0809493
99. Henry DH, Costa L, Goldwasser F, et al. Randomized double-blind study of denosumab versus zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. J Clin Oncol. 2011;29(9):1125–1132. doi:10.1200/JCO.2010.31.3304
100. Vadhan-Raj S, Von Moos R, Fallowfield LJ, et al. Clinical benefit in patients with metastatic bone disease: results of a phase III study of denosumab versus zolendronic acid. Ann Oncol. 2012;23(12):3045–3051. doi:10.1093/annonc/mds175
101. OncLive. FDA approves denosumab for multiple myeloma. 2018. Available from: http://www.onclive.com/web-exclusives/fda-approves-denosumab-for-multiple-myeloma.
102. European Medicines Agency. Xgeva (denosumab). Summary of product characteristics. 2018. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002173/WC500110381.pdf.
103. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clinic Proc. 2003;78(1):21–33. doi:10.4065/78.1.21
104. Vij R, Horvath N, Spencer A, et al. An open-label, phase 2 trial of denosumab in the treatment of relapsed or plateau-phase multiple myeloma. Am J Hematol. 2009;84(10):650–656. doi:10.1002/ajh.21509
105. Anastasilakis AD, Polyzos SA, Makras P, Aubry-Rozier B, Kaouri S, Lamy O. Clinical features of 24 patients with rebound- associated vertebral fractures after denosumab discontinuation: systematic review and additional cases. J Bone Miner Res. 2017;32:1291–1296. doi:10.1002/jbmr.3110
106. Lamy O, Gonzalez-Rodriguez E, Stoll D, Hans D, Aubry-Rozier B. Severe rebound-associated vertebral fractures after denosumab discontinuation: 9 clinical cases report. J Clin Endocrinol Metab. 2017;102:354–358. doi:10.1210/jc.2016-3170
107. Cummings SR, Ferrari S, Eastell R, et al. Vertebral fractures after discontinuation of denosumab: a post hoc analysis of the randomized placebo- controlled FREEDOM trial and its extension. J Bone Miner Res. 2018;33:190–198. doi:10.1002/jbmr.3337
108. Horne AM, Mihov B, Reid IR. Bone loss after romosozumab/denosumab: effects of bisphosphonates. Calcif Tissue Int. 2018;103:55–61. doi:10.1007/s00223-018-0404-6
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.