")
Back to Journals » Cancer Management and Research » Volume 12
Preventative and Therapeutic Effects of Metformin in Gastric Cancer: A New Contribution of an Old Friend
Authors Zhang J, Wen L, Zhou Q , He K, Teng L
Received 29 May 2020
Accepted for publication 19 August 2020
Published 16 September 2020 Volume 2020:12 Pages 8545—8554
DOI https://doi.org/10.2147/CMAR.S264032
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chien-Feng Li
Jing Zhang, Liping Wen, Quan Zhou, Kuifeng He, Lisong Teng
Department of Surgical Oncology, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China
Correspondence: Lisong Teng
Department of Surgical Oncology, The First Affiliated Hospital, College of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou, Zhejiang 310003, People’s Republic of China
Tel/ Fax +86-571-87236881
Email [email protected]
Abstract: Gastric cancer (GC) is a cancer with high prevalence, and is one of the leading causes of cancer death worldwide. Metformin is a widely used hypoglycemic agent for type-2 diabetes mellitus (T2DM). Recently, metformin has drawn increasing attention in the field of cancer research for its emerging anti-cancer roles. However, the efficacy and underlying molecular mechanisms of metformin in the prevention and treatment for GC remain controversial. This review summarized the present clinical and mechanistic studies that investigated the efficacy of metformin in GC. It was found that the majority of clinical studies affirmed protective roles of metformin in both gastric cancer risk and survival rate. In addition, metformin’s effects in the prevention and treatment for GC involve multiple pathways mainly via AMPK and IGF-1R. It was concluded that metformin presents a unique opportunity for application against GC, but further clinical and mechanistic investigations are required to solidify the roles of metformin in GC.
Keywords: gastric cancer, metformin, AMPK, anti-tumor
Introduction
Gastric cancer (GC) remains as a global issue and is responsible for over 1,000,000 new cases and an estimated of 783,000 deaths in 2018.1 It is the fifth most commonly diagnosed cancer and the third leading cause of cancer-associated death worldwide.1 Even after complete surgical resection and conventional chemotherapy, approximately 50% of GC patients experience recurrence and metastasis,2 and the 5-year relative survival rate among locally advanced GC patients is 36%, according to the MAGIC trial.3 Therefore, the discovery and investigation of new therapeutic options are urgently needed. Repurposing approved drugs for cancer prevention and treatment has been an important strategy of cancer drug discovery. The present study focused on the potential of repurposing metformin for GC prevention and treatment.
Metformin is derived from galegine or isoamylene guanidine, which is the active substance of Galega officinalis, also known as Goat’s Rue, the French lilac or Italian Fitch.4 Metformin has been the most widely used oral hypoglycemic agent in the biguanide class for the treatment of T2DM since its approval in the 1950s in the United Kingdom and in 1995 in the United States.4 Mechanistic studies revealed that metformin activates adenosine monophosphate‑activated protein kinase (AMPK) signaling. Thus, this sensitizes the response of liver and peripheral tissues to insulin signals, and enhances the glucose uptake into muscles and/or increasing fatty acid oxidation in adipose tissues.5,6
Since the early 2000s, several studies have linked the use of metformin with decreased risk of different cancers,7 neurodegeneration8 and cardiac hypertrophy.9 Therefore, it was proposed that the use of metformin could be repurposed as an anti-cancer agent, a neuroprotective agent, and a cardiovascular protective agent.7–9 According to a retrospective study in 2005, metformin decreased the overall cancer incidence by 44% when administered for more than five years.10 Numerous studies have investigated the antineoplastic effect of metformin on liver cancer,11 pancreatic cancer,12,13 colorectal cancer14 and prostate cancer.15 In contrast, limited studies concerning the antitumor effect of metformin on GC has been conducted. However, some meaningful results have been attained in recent years. Therefore, the present study provides an overview of the potential role of metformin in GC prevention and treatment from previous clinical studies. These studies enlightened the mechanism research of metformin. We summarize the latest advances of metformin in perturbing cancer cell metabolism, inhibiting the proliferation, metastasis and stemness, and inducing apoptosis.
The Efficacy of Metformin for Gastric Cancer in Clinical Practice
The effects of metformin on GC risk and survival rate have attracted the attention of clinicians and researchers. Metformin is a widely prescribed first-line drug for T2DM. The large population of metformin users with T2DM provides great opportunity for researchers to investigate metformin’s impact on GC incidence and survival rate. In addition, most clinical studies have investigated the roles of metformin in GC among T2DM patients.
Metformin Use and Gastric Cancer Risk
For the last 10 years, epidemiological studies have shown controversial results for the association between metformin therapy and incidence of GC (Table 1). Six of 11 studies revealed that metformin use in DM patients significantly reduced the risk of developing GC.15,16,19,20,22,24 Kim et al reported that in T2DM patients, metformin significantly reduced GC risk in non-insulin users, but not in insulin users.16 However, further investigations are needed to determine whether exogenous insulin treatment interferes with the protective effect of metformin.16 Several studies have compared metformin with other antidiabetic agents and revealed that lowering GC risk could be a unique feature of metformin. Tseng et al reported that the use of metformin significantly reduced GC risk, especially when the cumulative duration was more than approximately two years.20 Ruiter et al reported that exposure to metformin was generally associated with lower incidence of cancer, specifically gastric cancer, when compared to exposure to sulfonylurea derivatives.15 Similarly, Dulskas et al reported that metformin decreased the risk of GC, while sulfonylureas behaved quite the opposite.24 However, Valent et al concluded that sulfonylurea could also reduce the risk of GC with a similar effect to metformin.19 These studies indicate that the protective roles of metformin against cancer risk might be independent of its hypoglycemic roles. Cheung et al reported that metformin use was associated with lower GC risk among H. pylori-eradicated diabetic patients in a duration- and dose-dependent manner.22
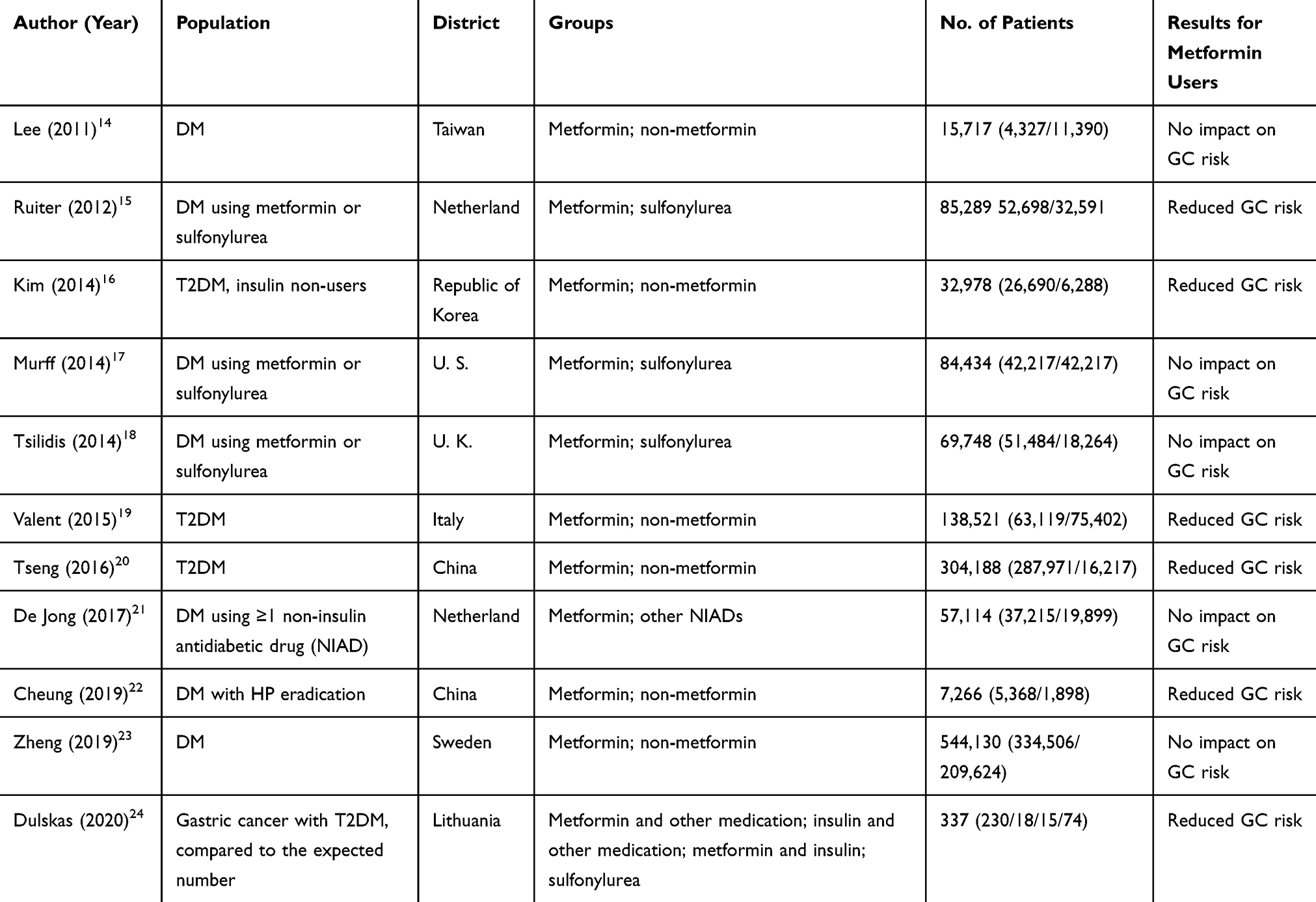 |
Table 1 Characteristics of Studies on Metformin and Gastric Cancer Risk |
On the contrary, a study conducted in the United Kingdom revealed that the use of metformin had a similar incidence for total cancer and gastric cancer, when compared to the use of sulfonylurea.18 Murff et al reported the strong inverse association between metformin and liver cancer, but not gastric cancer.17 Population-based studies in the Netherlands, Sweden and Taiwan also indicated that metformin did not reduce GC risk.14,21,23
Recently, Shuai et al reported a meta-analysis that the use of metformin reduced 21% of GC incidence (HR: 0.790, 95% CI 0.624–1.001). The subgroup analysis revealed that compared to Western populations, Asian patients with T2DM are more likely to benefit from metformin against the development of GC.25 Randomized controlled trials of metformin for the prevention of GC would be useful to verify these observations.
Metformin Use and Gastric Cancer Survival
Accumulating studies have determined whether metformin could improve survival in tumor patients.26,27 Studies on GC are presently limited, and there is only one ongoing prospective clinical trial registered at clinicaltrials.gov.
To date, only four retrospective studies have investigated the association between metformin use and gastric cancer survival (Table 2). Among these studies, three of four reported that use of metformin improved GC patient survival. The study led by Baglia et al did not present the significant impact of metformin, and this might be deviated by the relatively smaller cohort (130 patients only) in the study.31 Lee et al and Seo et al both reported that increasing the cumulative duration of metformin use could significantly improve the survival and reduce the recurrence rate in GC patients with diabetes who underwent gastrectomy.28,30 It has been shown that in general, DM patients have a poorer prognosis, when compared to non-DM patients with cancers. Interestingly, gastrectomy per se has been shown to relieve T2DM, which made the problem more complicated.32,33 Furthermore, Seo et al reported that the DM+metformin group even had significantly a better overall survival, when compared to the non-DM group.30 The study based on a Belgian population indicated that metformin improved the overall survival, but not cancer-specific survival.29 It remains unclear whether metformin would have similar beneficial effects in patients with gastric cancer without DM. Hence, further prospective studies to evaluate metformin’s role as adjuvant therapy in non-DM GC patients are needed.34
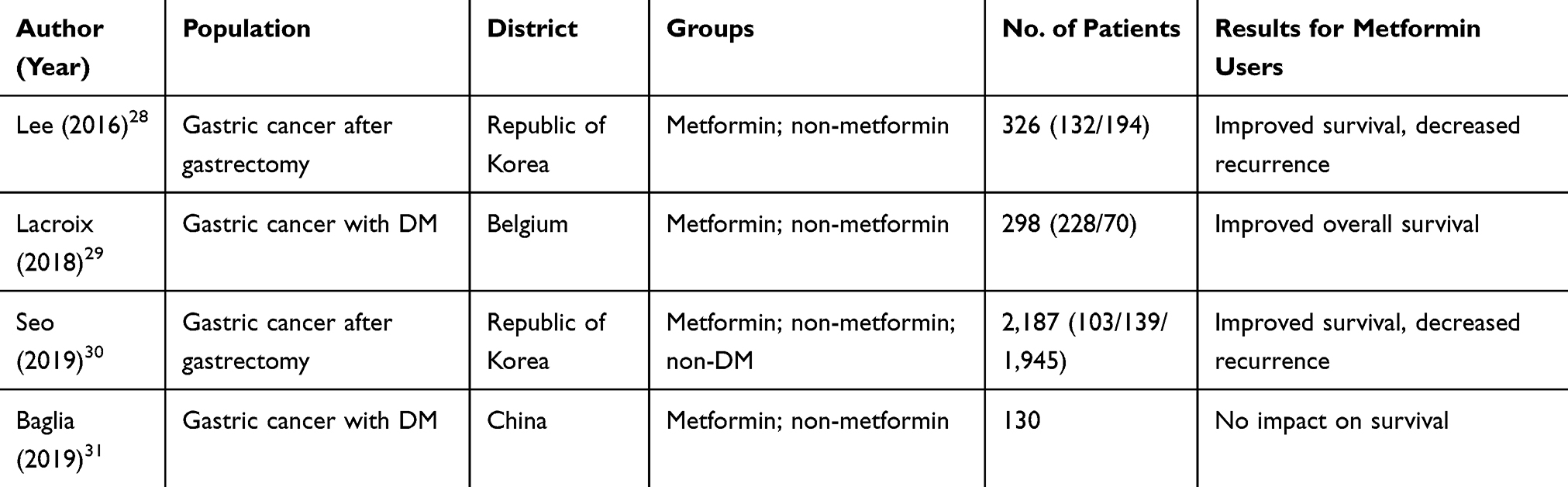 |
Table 2 Characteristics of Studies on Metformin and Gastric Cancer Survival |
In summary, these present limited studies suggest that the use of metformin might significantly improve the survival of GC patients. However, these studies are not able to address whether metformin has a direct anti-cancer effect. Therefore, the direct impact of metformin on GC cells and its potential molecular mechanisms need to be further explored, which will be the focus of the following sections of the present study.
Metformin Perturbs Cancer Cell Metabolism
Altered metabolism is one of the 10 emerging hallmarks of cancer.35 Back in 1924, Otto Warburg proposed a connection between cellular metabolism and malignancy, where tumor tissue or cells preferentially utilize glycolysis, rather than oxidative phosphorylation (OXPHOS), even when oxygen is sufficient.36 Mitochondrial Complex I is a crucial enzyme in the electron transfer chain (ECT) in the mitochondrial in OXPHOS. Metformin has a very polar guanidine structure and a highly hydrophilic base, which exists in a positive charged form under physiological conditions. The uptake of metformin involves transmembrane transporters, such as organic cation transporters (OCTs), multidrug and toxic extrusion transporters (MATE), and plasma membrane monoamine transporter (PMAT).37 Following its uptake, metformin accumulates within the mitochondria inner membrane driven by the mitochondria transmembrane electrochemical potential, and directly targets complex I. The inhibition of complex I by metformin decreases the proton gradient and mitochondrial oxygen consumption rate (OCR), which slows down tricarboxylic acid (TCA) cycle and decreases cellular ATP level.38,39 As a compensatory response, cells can increase the glucose uptake and upregulate glycolysis39 to partially refuel the TCA cycle and provide intermediates for biosynthesis. If the compensatory upregulation of glycolysis fails to meet the cellular ATP demand, the cellular AMP/ATP ratio would increase, and AMP would bind to the AMPK complex, and promote its phosphorylation and activation.40 The activation of AMPK would result in the potentiation of catabolic metabolism and downregulation of anabolic reactions.39 If the AMPK cascade continues to be active, and cell proliferation would be ultimately inhibited.41 Overall, the end result of cells exposed to metformin would be cellular energetic stress. In addition, metformin can act as a cytotoxic agent when cancer cells fail to compensate for this energetic stress, or a cytostatic agent when the cells are able to compensate.42
The protein complex AMPK belongs to a family of serine-threonine protein kinase complexes, and this serves as a major cellular energy sensor that maintains cellular energy homeostasis via a variety of pathways. AMPK is a heterotrimer that comprises a catalytic subunit (AMPKα) and two regulatory subunits (AMPKβ and AMPKγ).40 The complex can be activated via the phosphorylation of Thr172 in the α-subunit mainly by three kinases, liver kinase B1 (LKB1),43,44 calmodulin-mediated kinase kinase b (CaMKKb),45,46 and transforming growth factor-b activated kinase 1 (TAK1).47,48 Metformin can activate the catalytic AMPKα, inducing cell cycle arrest, autophagy, apoptosis and migration.41
Apart from its direct effect on cancer cells, metformin can also exert an indirect effect on cancer cells by reducing serum glucose, and thereby, the insulin levels.49 Metformin inhibited hepatic gluconeogenesis by opposing glucagon-mediated signaling in the liver, resulting in the decrease of glucose.50,51 On the other hand, metformin improved the affinity of insulin receptor for glucose uptake in skeletal muscle, leading to the reduction in insulin resistance52 and the decline in hyperinsulinemia.49 Hyperinsulinemia is responsible for the increased level of insulin-like growth factors (IGFs), leading to the increase in free or bioactive IGF-1 levels and the activation of insulin-like growth factor receptor (IGF-1R).53 The activation of IGF-1R resulted in cell growth survival through the MAPK and PI3K signaling pathways.54 However, metformin blocks the GC cell cycle in G0-G1 in vitro and in vivo through the reduction of IGF-1R activity.55
Metformin’s Anti-Tumor Effects on Gastric Cancer
As mentioned in the last section, metformin can act as either a cytostatic or cytotoxic agent in cancer cells via the activation of AMPK and the inhibition of IGF-1R. This section reviews the roles of a variety of gene products that are responsible for metformin’s anti-cancer effects downstream of the AMPK activation. These genes can be categorized based on their involvement in different biological processes, namely, the proliferation, apoptosis, metastasis and maintenance of stemness, which are summarized in Figure 1.
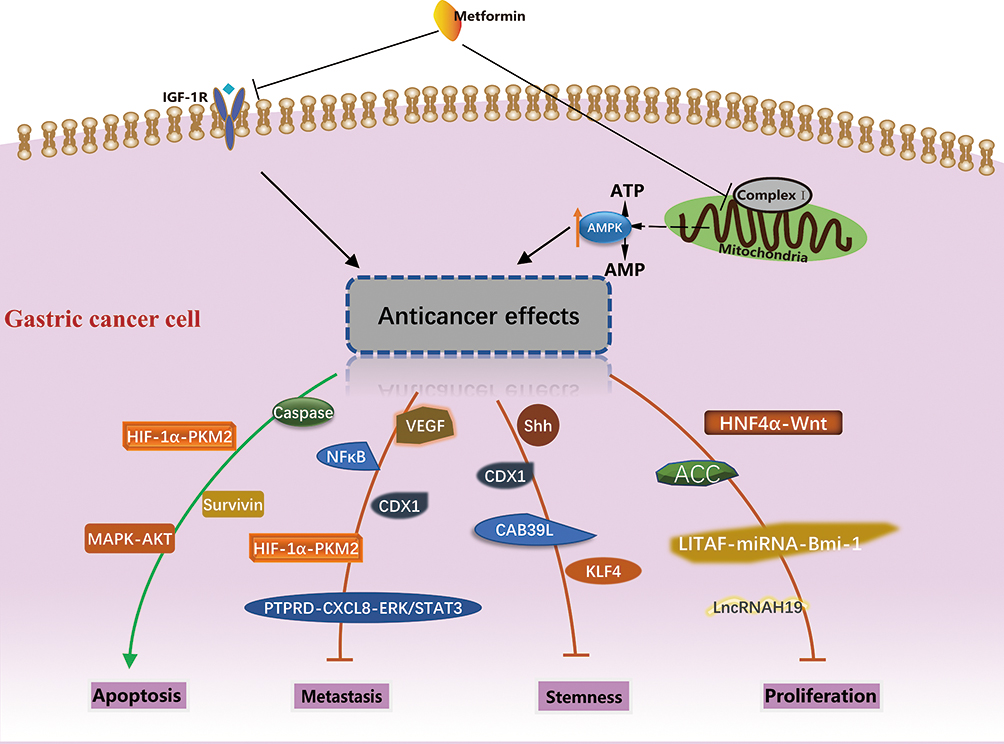 |
Figure 1 Potential molecular mechanism of anti-cancer activity of metformin in gastric cancer. The different biological processes and molecules involved were summarized. |
Metformin Inhibits Gastric Cancer Cell Proliferation
The phosphorylation and activation of AMPK lead to the phosphorylation and inactivation of Acetyl-CoA carboxylase (ACC), the rate-limiting enzymes in de novo fatty acid synthesis, thereby causing a reduction in lipogenesis.56 Fang et al reported that in GC patients, lower expression of pACC was significantly correlated with poorer differentiation, higher lymph node and overall TNM stage, and worse overall survival.57 A further mechanistic study conducted by the same group revealed that metformin inhibits GC cell proliferation and colony formation by upregulating pACC through the activation of AMPK.57
Hepatocyte nuclear factor-4α (HNF4α), a transcription factor, is overexpressed in many types of cancer, and is involved in cell proliferation.58 Chang et al reported that the upregulation of HNF4α is a key event in the development of GC in both Asian and Caucasian populations.59 Furthermore, they found that HNF4α can be downregulated by AMPKα signaling and AMPK agonist metformin in vivo and in vitro.59 In addition, they reported WNT5A as a direct target gene of HNF in GC.60 Further animal models confirmed the AMPK-HNF4α-WNT signaling cascade involved in cyclin downregulation, cell cycle arrest, and tumor growth inhibition.59
A previous study identified lipopolysaccharide-induced TNFα factor (LITAF) as a downstream effector of AMPK in the regulation of tumor cell growth in prostate cancer.61 Huang et al first depicted the regulatory axis sequentially tethering AMPK-LITAF-miRNAs-Bmi-1 in GC cells and other cancer cells.62 B-lymphoma Moloney murine leukemia virus insertion region-1 (Bmi-1) is a transcriptional regulator that promotes tumor cell self-renewal, and epithelial to mesenchymal transition and its upregulation is associated with tumor progression.63 AMPK activation increased the abundance of LITAFs and concurrently reduced the expression of Bmi-1, while the knockdown of LITAF upregulated Bmi-1 and promoted the aggressive behaviors of GC cells.62 Metformin upregulated expression of miR-15a, miR-128, miR-192 and miR-194, which can be abolished by the knockdown of LITAF.62 Similarly, Kato et al reported that metformin inhibits human GC cell proliferation and tumor growth, possibly by suppressing the cell-cycle-related molecules via the alteration of miRNAs.55 They found that the expression of has-mir-638 in metformin-treated cells was 3.3 times higher than that in untreated cells, suggesting that has-mir-638 might be a new therapeutic target in GC.55
Apart from miRNAs, long noncoding RNA (lncRNA) H19 was found to be overexpressed in GC tissues, when compared to normal tissue. In addition, the increased expression of H19 was correlated with more advanced pathological tumor stage.64 Metformin could not further activate AMPK in H19 knocked down GC cells, indicating that H19 is a key component in the AMPK signaling pathway of metformin for suppressing GC cell proliferation and invasion.64
Metformin treatment inhibited GC cell growth in vitro and in vivo by blocking the cell cycle with the decreased expression of Cyclin D1.55 Yu et al further demonstrated that cell growth and colony formation were inhibited with metformin treatment in a dose-dependent manner, and that this is correlated with the decreased expression of Cyclin D1 and CDK4.65
Metformin Induces Gastric Cancer Cell Apoptosis
Han et al reported that metformin selectively induces apoptosis in GC, but not noncancerous gastric cells, and they suggested that the apoptotic effect of metformin appears to be associated with poorer differentiation of GC.66 The study conducted by Lu demonstrated that metformin‑induced apoptosis was accompanied by the upregulation of AMPK Thr172 phosphorylation, and downregulation of AKT (Ser473), mTOR (Ser2448) and p70S6K (Ser424) phosphorylation.67 The data also demonstrated that the attenuation of AMPK signaling using an AMPK inhibitor (Compound C) abrogated the effects of metformin on the viability of gastric adenocarcinoma cells (AGS).67 AMPK/mTOR-mediated inhibition of survivin expression contributes, at least in part, to the metformin-induced apoptosis of GC cells.66 In addition, metformin promotes the caspase-dependent mitochondria-derived apoptosis in AGS cells.67
The increase in hypoxia-induced factor 1α (HIF-1α) level is associated with increased risk of mortality in multiple human cancers, including gastric cancer.68 Chen et al reported that the expression of HIF1α and pyruvate kinase M2 (PKM2) were higher in GC tissues than in gastritis tissues, and found that metformin significantly induced apoptosis, inhibited cell invasion and migration of GC cells.69 In addition, their data support a model where metformin exhibits its anti-tumor effect for GC by inducing intrinsic apoptosis via the inhibition of HIF1α/PKM2 signaling pathway.69 In addition, HIF1α plays a critical role in the regulation of tumor angiogenesis in response to hypoxia.68
Limited studies have provided little available information on the effect of metformin combined with chemotherapy. The combination of rapamycin and metformin appears to be more favorable in anti-gastric cancer.65 However, the effect of metformin combined with cisplatin remains controversial. Yu et al reported that on MKN45 xenografts and BGC823 cells, the combined administration with metformin enhanced the chemo-induced reduction of tumor growth in GC.65 On the contrary, Lesan et al reported that on the human gastric MKN-45 cell line, metformin diminishes the anti-proliferative effects of cisplatin when used in combination with metformin.70 The antagonistic effect of metformin on cisplatin could be through the survivin and mTOR/AKT signaling pathways in the MKN-45 cell line.70 The discrepancy may lie in the difference in cancer type and cell type.
Metformin Inhibits Cancer Metastasis
One of the key features of malignancy in cancer cells is their ability to reduce cell-cell adhesion and invade surrounding tissues.71 Metformin could inhibit the metastasis of GC, but its specific mechanism remains on debate. Cadherins are a group of cell membrane proteins critical in the formation of cell-cell adherence junctions and can be classified by their distinct immunological and tissue specificities, such as E- for epithelial and N- for neural. Loss of E-cadherin can lead to initiation of epithelial–mesenchymal transition (EMT), which is characterized by reduced cell polarity, and increased migratory and invasive growth properties.72 Different from the general metastatic phenotype alternation, Valaee et al indicated that EMT inhibition by metformin in the GC cell line was in concordant with E-cadherin enhancement.73 On the other hand, Jun et al reported that brain metastasis in advanced GC was significantly correlated with the expression of vascular endothelial growth factor (VEGF), but not E- or N-cadherin.74 Metformin treatment affects the metastatic capacity of GC by reducing VEGF expression and blocking EMT.74
E-cadherin can be regulated by Protein tyrosine phosphatase receptor delta (PTPRD) to influence cell migration.75 PTPRD is frequently inactivated in GC. The loss of PTPRD induces CXCL8, via both ERK and STAT3 signaling, promoting angiogenesis, and consequently, metastasis.76 Researchers have further proposed that metformin could exert its anti-cancer effect by reversing the downregulated PTPRD expression.76
In addition, loss of E-cadherin could enhance the inflammatory signals, particularly NF-κB induced EMT.77,78 More specifically, Sekino et al reported that metformin inhibited the GC peritoneal metastasis on a NF-κB-, but not AMPK-, dependent manner.79
Metformin Inhibits Cancer Cell Stemness
Cancer stem cells (CSC), or tumor-initiating cells, are a subset of cancer cells that are considered to have indefinite potential of self-renewal, and could lead to tumorigenesis.80,81 Courtois et al reported that metformin can target GC stem cells, and decrease their self-renewal ability both in vitro and in vivo.82 Previous evidence has demonstrated that in GC cells, the Sonic hedgehog (Shh) pathway are abnormally activated, and the overexpression of Shh protein could contribute to tumorogenesis.83 Glioma associated oncogene (Gli), a downstream target gene of the hedgehog signal pathway, is represented by Gli-1, Gli-2 and Gli-3 in mammalians.84 In the study conducted by Song, the Shh pathway maintained the GC stem-like cells (CSLCs) have characteristics, including chemoresistance and tumorigenic capacity.85 A follow-up study led by the same group showed that both protein and mRNA levels of Shh and Gli 1, Gli2, Gli3 were decreased by metformin in two GC cell lines in a dose- and time-dependent manner.86
Caudal type homeobox 1 (CDX1), as a transcription factor, act a pivotal part in human intestinal development and maintenance.87 The aberrant expression of CDX1 can be triggered by H. pylori virulence factor CagA.88 Patients with H. pylori tended to have more CDX1 expression than non-infected and enhanced GC tumorigenesis and progression.88 Human tissue samples with higher CDX1 levels revealed the prominent stem cell markers CD44/SOX2 expression.88 Likewise, in a murine xenograft model, combining metformin or shCDX1 with cisplatin-reduced tumor growth, increased caspase-3 cleavage, and reduced the expression of the stem cell markers CD44 and MMP-9 to a greater degree, when compared to cisplatin alone.88
In addition, Li et al identified the aberrant silencing of Calcium Binding Protein 39-Like (CAB39L) in GC by promoter hypermethylation, and represented an epigenetic mechanism that contributes to metabolic dysregulation and GC development.89 CAB39L elicited an anti-Warburg effect via a LKB1-AMPK-PGC1α axis to inhibit gastric tumorigenesis, and CAB39L-silenced GC cells exhibited an increased sensitivity to metformin.89
Recently, Miao et al demonstrated that AMPK activates Krϋppel-like factor 4 (KLF4) in progenitors to govern gastric epithelial progenitor differentiation, reduce self-renewal, and promote parietal cell (PC) fate, while the PC-specific activation of AMPK-PGC1α promotes maturation.90 This may provide a potential suggestion for why metformin increases acid-secretion and reduces GC risk.90
Conclusion and Future Perspective
Clinical studies have suggested that the regular use of metformin may decrease the risk and mortality of multiple cancers in T2DM patients. In terms of GC, several clinical studies have revealed that metformin can reduce GC risk and improve overall survival. Furthermore, studies have shown that metformin is an anti-cancer agent, and can inhibit proliferation, metastasis and stemness, promote tumor apoptosis, and increase the chemosensitivity of GC cells. Mechanistic studies have revealed a wide range of gene products and signaling pathways involved in metformin’s anti-cancer effect.
Compared with liver cancer, pancreatic cancer and colon cancer, both clinical studies and mechanism researches in GC are still relatively rare. In clinical practice, more prospective clinical trials are needed to confirm its effect, and the synergistic action between metformin use and chemotherapy or immunotherapy is also an attractive aspect for GC. For the mechanism research, uptake is an important process for metformin to exert its activity. It was found that promoter variants in transporter MATE and the heterogeneity of transporter OCT1 expression might affect the pharmacokinetics and pharmacodynamics of metformin.91,92 Further investigations are needed to elucidate the uptake of metformin in different types of GC cells and the results it caused. On the other hand, accumulating evidence has indicated the potential roles of microbiome on the anti-cancer activity of metformin in pancreatic and colorectal cancer,93,94 the interaction between metformin and gut microbiota is worth studying in gastric cancer prevention and treatment.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 81301809), the Clinical Research Fund of Zhejiang Medical Association (Grant No.2017ZYC-A12), and the Zhejiang Provincial Medical and Health Major Science and Technology Plan Project (Grant No. 2017209495).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
2. Al-Batran SE, Homann N, Pauligk C, et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): a randomised, Phase 2/3 trial. Lancet. 2019;393(10184):1948–1957.
3. Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med. 2006;355(1):11–20.
4. Witters LA. The blooming of the French lilac. J Clin Invest. 2001;108(8):1105–1107.
5. Coughlan KA, Valentine RJ, Ruderman NB, Saha AK. AMPK activation: a therapeutic target for type 2 diabetes? Diabetes Metab Syndr Obes. 2014;7:241–253.
6. Jalving M, Gietema JA, Lefrandt JD, et al. Metformin: taking away the candy for cancer? Eur J Cancer. 2010;46(13):2369–2380.
7. Quinn BJ, Kitagawa H, Memmott RM, Gills JJ, Dennis PA. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab. 2013;24(9):469–480.
8. Gupta A, Bisht B, Dey CS. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and alzheimer’s-like changes. Neuropharmacology. 2011;60(6):910–920.
9. Ahmed FW, Rider R, Glanville M, Narayanan K, Razvi S, Weaver JU. Metformin improves circulating endothelial cells and endothelial progenitor cells in type 1 diabetes: MERIT study. Cardiovasc Diabetol. 2016;15(1):116.
10. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–1305.
11. Zhang ZJ, Zheng ZJ, Shi R, Su Q, Jiang Q, Kip KE. Metformin for liver cancer prevention in patients with type 2 diabetes: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2012;97(7):2347–2353.
12. Bhaw-Luximon A, Jhurry D. Metformin in pancreatic cancer treatment: from clinical trials through basic research to biomarker quantification. J Cancer Res Clin Oncol. 2016;142(10):2159–2171.
13. Gong J, Robbins LA, Lugea A, Waldron RT, Jeon CY, Pandol SJ. Diabetes, pancreatic cancer, and metformin therapy. Front Physiol. 2014;5:426.
14. Lee MS, Hsu CC, Wahlqvist ML, Tsai HN, Chang YH, Huang YC. Type 2 diabetes increases and metformin reduces total, colorectal, liver and pancreatic cancer incidences in Taiwanese: a representative population prospective cohort study of 800,000 individuals. BMC Cancer. 2011;11:20.
15. Ruiter R, Visser LE, van Herk-sukel MP, et al. Lower risk of cancer in patients on metformin in comparison with those on sulfonylurea derivatives: results from a large population-based follow-up study. Diabetes Care. 2012;35(1):119–124.
16. Kim YI, Kim SY, Cho SJ, et al. Long-term metformin use reduces gastric cancer risk in type 2 diabetics without insulin treatment: a nationwide cohort study. Aliment Pharmacol Ther. 2014;39(8):854–863.
17. Murff HJ, Roumie CL, Greevy RA, et al. Metformin use and incidence cancer risk: evidence for a selective protective effect against liver cancer. Cancer Causes Control. 2018;29(9):823–832.
18. Tsilidis KK, Capothanassi D, Allen NE, et al. Metformin does not affect cancer risk: a cohort study in the U.K. clinical practice research datalink analyzed like an intention-to-treat trial. Diabetes Care. 2014;37(9):2522–2532.
19. Valent F. Diabetes mellitus and cancer of the digestive organs: an Italian population-based cohort study. J Diabetes Complications. 2015;29(8):1056–1061.
20. Tseng CH. Metformin reduces gastric cancer risk in patients with type 2 diabetes mellitus. Aging (Albany NY). 2016;8(8):1636–1649.
21. de Jong RG, Burden AM, de Kort S, et al. No decreased risk of gastrointestinal cancers in users of metformin in the Netherlands; a time-varying analysis of metformin exposure. Cancer Preve Res. 2017;10(5):290–297.
22. Cheung KS, Chan EW, Wong AYS, et al. Metformin use and gastric cancer risk in diabetic patients after helicobacter pylori eradication. J Natl Cancer Inst. 2019;111(5):484–489.
23. Zheng J, Xie SH, Santoni G, Lagergren J. Metformin use and risk of gastric adenocarcinoma in a Swedish population-based cohort study. Br J Cancer. 2019;121(10):877–882.
24. Dulskas A, Patasius A, Kaceniene A, et al. Study of antihyperglycemic medication exposure and gastric cancer risk. J Clin Med. 2020;9(2):435.
25. Shuai Y, Li C, Zhou X. The effect of metformin on gastric cancer in patients with type 2 diabetes: a systematic review and meta-analysis. Clin Transl Oncol. 2020:1–11.
26. Coyle C, Cafferty FH, Vale C, Langley RE. Metformin as an adjuvant treatment for cancer: a systematic review and meta-analysis. Ann Oncol. 2016;27(12):2184–2195.
27. Gandini S, Puntoni M, Heckman-Stoddard BM, et al. Metformin and cancer risk and mortality: a systematic review and meta-analysis taking into account biases and confounders. Cancer Preve Res. 2014;7(9):867–885.
28. Lee CK, Jung M, Jung I, et al. Cumulative metformin use and its impact on survival in gastric cancer patients after gastrectomy. Ann Surg. 2016;263(1):96–102.
29. Lacroix O, Couttenier A, Vaes E, Cardwell CR, De Schutter H, Robert A. Impact of metformin on gastric adenocarcinoma survival: a Belgian population based study. Cancer Epidemiol. 2018;53:149–155.
30. Seo HS, Jung YJ, Kim JH, Lee HH, Park CH. The effect of metformin on prognosis in patients with locally advanced gastric cancer associated with type 2 diabetes mellitus. Am J Clin Oncol. 2019;42(12):909–917.
31. Baglia ML, Cui Y, Zheng T, et al. Diabetes Medication use in association with survival among patients of breast, colorectal, lung, or gastric cancer. Cancer Res Treat. 2019;51(2):538–546.
32. Kwon Y, Abdemur A, Lo ME, Park S, Szomstein S, Rosenthal RJ. The foregut theory as a possible mechanism of action for the remission of type 2 diabetes in low body mass index patients undergoing subtotal gastrectomy for gastric cancer. Surg Obes Relat Dis. 2014;10(2):235–242.
33. An JY, Kim YM, Yun MA, Jeon BH, Noh SH. Improvement of type 2 diabetes mellitus after gastric cancer surgery: short-term outcome analysis after gastrectomy. World J Gastroenterol. 2013;19(48):9410–9417.
34. Greenhill C. Gastric cancer. Metformin improves survival and recurrence rate in patients with diabetes and gastric cancer. Nat Rev Gastroenterol Hepatol. 2015;12(3):124.
35. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47.
36. Foretz M, Viollet B. New promises for metformin: advances in the understanding of its mechanisms of action. Med Sci (Paris). 2014;30(1):82–92.
37. Markowicz-Piasecka M, Komeil I, Huttunen J, Sikora J, Huttunen KM. Effective cellular transport of ortho-halogenated sulfonamide derivatives of metformin is related to improved antiproliferative activity and apoptosis induction in MCF-7 cells. Int J Mol Sci. 2020;21:7.
38. Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014;2:12.
39. Griss T, Vincent EE, Egnatchik R, et al. Metformin antagonizes cancer cell proliferation by suppressing mitochondrial-dependent biosynthesis. PLoS Biol. 2015;13(12):e1002309.
40. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8(10):774–785.
41. Daugan M, Dufay Wojcicki A, d’Hayer B, Boudy V. Metformin: an anti-diabetic drug to fight cancer. Pharmacol Res. 2016;113(Pt A):675–685.
42. Andrzejewski S, Siegel PM, St-Pierre J. Metabolic profiles associated with metformin efficacy in cancer. Front Endocrinol (Lausanne). 2018;9:372.
43. Woods A, Johnstone SR, Dickerson K, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13(22):2004–2008.
44. Hawley SA, Boudeau J, Reid JL, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2(4):28.
45. Woods A, Dickerson K, Heath R, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2(1):21–33.
46. Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2(1):9–19.
47. Neumann D. Is TAK1 a direct upstream kinase of AMPK? Int J Mol Sci. 2018;19(8):2412.
48. Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281(35):25336–25343.
49. Del Barco S, Vazquez-Martin A, Cufi S, et al. Metformin: multi-faceted protection against cancer. Oncotarget. 2011;2(12):896–917.
50. Pernicova I, Korbonits M. Metformin–mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10(3):143–156.
51. Minassian C, Tarpin S, Mithieux G. Role of glucose-6 phosphatase, glucokinase, and glucose-6 phosphate in liver insulin resistance and its correction by metformin. Biochem Pharmacol. 1998;55(8):1213–1219.
52. Pollak M. Overcoming drug development bottlenecks with repurposing: repurposing biguanides to target energy metabolism for cancer treatment. Nat Med. 2014;20(6):591–593.
53. Kourelis TV, Siegel RD. Metformin and cancer: new applications for an old drug. Med Oncol. 2012;29(2):1314–1327.
54. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11(1):9–15.
55. Kato K, Gong J, Iwama H, et al. The antidiabetic drug metformin inhibits gastric cancer cell proliferation in vitro and in vivo. Mol Cancer Ther. 2012;11(3):549–560.
56. Loubiere C, Goiran T, Laurent K, Djabari Z, Tanti JF, Bost F. Metformin-induced energy deficiency leads to the inhibition of lipogenesis in prostate cancer cells. Oncotarget. 2015;6(17):15652–15661.
57. Fang W, Cui H, Yu D, Chen Y, Wang J, Yu G. Increased expression of phospho-acetyl-CoA carboxylase protein is an independent prognostic factor for human gastric cancer without lymph node metastasis. Med Oncol. 2014;31(7):15.
58. Courtois S, Lehours P, Bessede E. The therapeutic potential of metformin in gastric cancer. Gastric Cancer. 2019;22(4):653–662.
59. Chang HR, Nam S, Kook MC, et al. HNF4alpha is a therapeutic target that links AMPK to WNT signalling in early-stage gastric cancer. Gut. 2016;65(1):19–32.
60. Nam S, Chang HR, Kim KT, et al. PATHOME: an algorithm for accurately detecting differentially expressed subpathways. Oncogene. 2014;33(41):4941–4951.
61. Zhou J, Yang Z, Tsuji T, et al. LITAF and TNFSF15, two downstream targets of AMPK, exert inhibitory effects on tumor growth. Oncogene. 2011;30(16):1892–1900.
62. Huang D, He X, Zou J, et al. Negative regulation of Bmi-1 by AMPK and implication in cancer progression. Oncotarget. 2016;7(5):6188–6200.
63. Park IK, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. 2004;113(2):175–179.
64. De Luise M, Guarnieri V, Ceccarelli C, et al. DNA mutation associates with dysfunction of HIF1alpha in a von hippel-lindau renal oncocytoma. J Diabetes Res. 2019;2019:8069583.
65. Yu G, Fang W, Xia T, et al. Metformin potentiates rapamycin and cisplatin in gastric cancer in mice. Oncotarget. 2015;6(14):12748–12762.
66. Han G, Gong H, Wang Y, Guo S, Liu K. AMPK/mTOR-mediated inhibition of survivin partly contributes to metformin-induced apoptosis in human gastric cancer cell. Cancer Biol Ther. 2015;16(1):77–87.
67. Lu CC, Chiang JH, Tsai FJ, et al. Metformin triggers the intrinsic apoptotic response in human AGS gastric adenocarcinoma cells by activating AMPK and suppressing mTOR/AKT signaling. Int J Oncol. 2019;54(4):1271–1281.
68. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123(9):3664–3671.
69. Chen G, Feng W, Zhang S, et al. Metformin inhibits gastric cancer via the inhibition of HIF1alpha/PKM2 signaling. Am J Cancer Res. 2015;5(4):1423–1434.
70. Lesan V, Ghaffari SH, Salaramoli J, et al. Evaluation of antagonistic effects of metformin with cisplatin in gastric cancer cells. Int J Hematol Oncol Stem Cell Res. 2014;8(3):12–19.
71. Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392(6672):190–193.
72. Garcia de Herreros A, Baulida J. Cooperation, amplification, and feed-back in epithelial-mesenchymal transition. Biochim Biophys Acta. 2012;1825(2):223–228.
73. Valaee S, Yaghoobi MM, Shamsara M. Metformin inhibits gastric cancer cells metastatic traits through suppression of epithelial-mesenchymal transition in a glucose-independent manner. PLoS One. 2017;12(3):e0174486.
74. Jun KH, Lee JE, Kim SH, et al. Clinicopathological significance of N-cadherin and VEGF in advanced gastric cancer brain metastasis and the effects of metformin in preclinical models. Oncol Rep. 2015;34(4):2047–2053.
75. Funato K, Yamazumi Y, Oda T, Akiyama T. Tyrosine phosphatase PTPRD suppresses colon cancer cell migration in coordination with CD44. Exp Ther Med. 2011;2(3):457–463.
76. Bae WJ, Ahn JM, Byeon HE, Kim S, Lee D. PTPRD-inactivation-induced CXCL8 promotes angiogenesis and metastasis in gastric cancer and is inhibited by metformin. J Exp Clin Cancer Res. 2019;38(1):484.
77. Kuphal S, Poser I, Jobin C, Hellerbrand C, Bosserhoff AK. Loss of E-cadherin leads to upregulation of NFkappaB activity in malignant melanoma. Oncogene. 2004;23(52):8509–8519.
78. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196.
79. Sekino N, Kano M, Matsumoto Y, et al. The antitumor effects of metformin on gastric cancer in vitro and on peritoneal metastasis. Anticancer Res. 2018;38(11):6263–6269.
80. Lei Y, Yi Y, Liu Y, et al. Metformin targets multiple signaling pathways in cancer. Chin J Cancer. 2017;36(1):17.
81. Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–284.
82. Courtois S, Duran RV, Giraud J, et al. Metformin targets gastric cancer stem cells. Eur J Cancer. 2017;84:193–201.
83. Pasca di Magliano M, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer. 2003;3(12):903–911.
84. Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15(6):801–812.
85. Song Z, Yue W, Wei B, et al. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS One. 2011;6(3):e17687.
86. Song Z, Wei B, Lu C, Huang X, Li P, Chen L. Metformin suppresses the expression of Sonic hedgehog in gastric cancer cells. Mol Med Rep. 2017;15(4):1909–1915.
87. Grainger S, Hryniuk A, Lohnes D. Cdx1 and Cdx2 exhibit transcriptional specificity in the intestine. PLoS One. 2013;8(1):e54757.
88. Choi SI, Yoon C, Park MR, et al. CDX1 expression induced by CagA-expressing helicobacter pylori promotes gastric tumorigenesis. Mol Cancer Res. 2019;17(11):2169–2183.
89. Li W, Wong CC, Zhang X, et al. CAB39L elicited an anti-warburg effect via a LKB1-AMPK-PGC1alpha axis to inhibit gastric tumorigenesis. Oncogene. 2018;37(50):6383–6398.
90. Miao ZF, Adkins-Threats M, Burclaff JR, et al. A metformin-responsive metabolic pathway controls distinct steps in gastric progenitor fate decisions and maturation. Cell Stem Cell. 2020.
91. Stocker SL, Morrissey KM, Yee SW, et al. The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther. 2013;93(2):186–194.
92. Segal ED, Yasmeen A, Beauchamp MC, Rosenblatt J, Pollak M, Gotlieb WH. Relevance of the OCT1 transporter to the antineoplastic effect of biguanides. Biochem Biophys Res Commun. 2011;414(4):694–699.
93. Dong TS, Chang HH, Hauer M, et al. Metformin alters the duodenal microbiome and decreases the incidence of pancreatic ductal adenocarcinoma promoted by diet-induced obesity. Am J Physiol Gastrointest Liver Physiol. 2019;317(6):G763–G772.
94. Jones GR, Molloy MP. Metformin, Microbiome and protection against colorectal cancer. Dig Dis Sci. 2020:1–6.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.