")
Back to Journals » International Journal of Nanomedicine » Volume 11
Preparation of finasteride capsules-loaded drug nanoparticles: formulation, optimization, in vitro, and pharmacokinetic evaluation
Authors Ahmed T
Received 11 October 2015
Accepted for publication 23 December 2015
Published 2 February 2016 Volume 2016:11 Pages 515—527
DOI https://doi.org/10.2147/IJN.S98080
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Tarek A Ahmed1,2
1Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, King Abdulaziz University, Jeddah, Kingdom of Saudi Arabia; 2Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Al-Azhar University, Cairo, Egypt
Abstract: In this study, optimized freeze-dried finasteride nanoparticles (NPs) were prepared from drug nanosuspension formulation that was developed using the bottom–up technique. The effects of four formulation and processing variables that affect the particle size and solubility enhancement of the NPs were explored using the response surface optimization design. The optimized formulation was morphologically characterized using transmission electron microscopy (TEM). Physicochemical interaction among the studied components was investigated. Crystalline change was investigated using X-ray powder diffraction (XRPD). Crystal growth of the freeze-dried NPs was compared to the corresponding aqueous drug nanosuspension. Freeze-dried NPs formulation was subsequently loaded into hard gelatin capsules that were examined for in vitro dissolution and pharmacokinetic behavior. Results revealed that in most of the studied variables, some of the quadratic and interaction effects had a significant effect on the studied responses. TEM image illustrated homogeneity and shape of the prepared NPs. No interaction among components was noticed. XRPD confirmed crystalline state change in the optimized NPs. An enhancement in the dissolution rate of more than 2.5 times from capsules filled with optimum drug NPs, when compared to capsules filled with pure drug, was obtained. Crystal growth, due to Ostwald ripening phenomenon and positive Gibbs free energy, was reduced following lyophilization of the nanosuspension formulation. Pharmacokinetic parameters from drug NPs were superior to that of pure drug and drug microparticles. In conclusion, freeze-dried NPs based on drug nanosuspension formulation is a successful technique in enhancing stability, solubility, and in vitro dissolution of poorly water-soluble drugs with possible impact on the drug bioavailability.
Keywords: finasteride, nanoparticles, solvent evaporation, optimization, crystal growth, pharmacokinetic
Introduction
Nanosizing is the process that involves the reduction of the drug particle size to the submicron range. This process has found wide applications in the pharmaceutical industry owing to its capacity to enhance the dissolution rate and bioavailability of many poorly water-soluble drugs.1,2 Of the new chemical compounds developed by medicinal drug design programs, more than 40% have limited water solubility.3 Enhancement of aqueous drug solubility affects the process of drug dissolution, and hence both the rate and extent to which the administered drug reaches the systemic circulation are augmented.4 Many techniques have been reported to improve drug aqueous solubility through salt formation, solubilization by cosolvent approach, complexation, micellar formation, and particle size reduction.3 Some other techniques that have illustrated reasonable successes involve enclosing the drug in vesicular nanoparticles (NPs)5 or formulation of the drug in the form of a self-nanoemulsifying drug-delivery system.6–8
A nanosuspension is a submicron colloidal dispersion, approximately between 200 and 600 nm, of poorly water-soluble drug without any matrix that is stabilized by the presence of surfactants and/or polymers that prevent crystal growth or particles aggregation during storage.9 Formulation of the drug in the form of nanosuspension enhances the absorption and bioavailability due to an increase in surface area and dissolution rate, which increases the rate and extent of absorption and hence the bioavailability.9 Nanosuspension is more applicable for drugs that are characterized by high permeability and low solubility such as revaprazan.10 Nanosuspension can be prepared either by the precipitation “bottom–up” or by the disintegration “top–down” technologies. The top–down technologies, which are sometimes also called comminution, include high-pressure homogenization, media milling, and a combination of precipitation and high-pressure/ultrasonic homogenization.11–13 In bottom–up technology, the nanosuspension is prepared from a state of molecular dispersion type and is allowed to associate with subsequent formation of solid particles, which is the same case as of classical precipitation technique. Usually, bottom–up techniques are simple and do not involve the use of sophisticated equipments.
Finasteride is chemically considered a synthetic 4-azasteroid drug. The drug is an effective therapeutic agent in the treatment of benign prostatic hyperplasia.14 Inhibition of the enzyme 5 alpha-reductase is believed to be the mechanism of action of this drug. This enzyme converts testosterone to dihydrotestosterone (DHT), which is a more potent androgenic hormone.15 An increase in the level of DHT in the prostate results in prostate hyperplasia and urinary tract obstruction.16 The drug is practically insoluble in water, with a mean bioavailability of 63%.
The aim of this study was to develop freeze-dried finasteride NPs from aqueous nanosuspension formulation, characterized by enhanced drug solubility and dissolution rate, with subsequent loading into hard gelatin capsules. Preparation of the drug nanosuspension formulation was achieved using the bottom–up technique using the response surface optimization design. Crystal growth and particle agglomeration of the freeze-dried drug NPs were decreased when compared to the corresponding drug nanosuspension formulation. Capsules-loaded lyophilized drug NPs could be considered an alternative to the currently available commercial drug tablet as they exhibited enhanced dissolution rate and better pharmacokinetic behavior.
Materials and methods
Materials
Finasteride was a kind gift from SAJA Pharmaceuticals (Jeddah, Saudi Arabia). Polyvinyl alcohol (PVA; MW: 140,000) was procured from Spectrum Chemicals & Laboratory Products (New Brunswick, NJ, USA). Methanol, lactose, potassium bromide, and tetrahydrofuran were obtained from Sigma-Aldrich (St Louis, MO, USA). Sodium lauryl sulfate were purchased from Fisher Scientific (Pittsburg, PA, USA). Acetonitrile high-performance liquid chromatography (HPLC) grade was obtained from Merck Chemicals Co. (Darmstadt, Germany).
Methods
Preformulation study
Bottom–up technique was implemented to prepare the desired drug NPs formulation. Methanol was selected as a water miscible organic solvent, while PVA was selected as a stabilizer. Both ultrasonic and high-speed homogenization were examined, after evaporation of the drug from its organic solution, to identify the most suitable condition.
A known weight of the drug was dissolved in methanol, and the drug solution was then injected slowly into water containing a known concentration of PVA. The mixture was kept stirring overnight on a magnetic stirrer until complete evaporation of the organic solvent and precipitation of the drug solid particles. The obtained aqueous drug suspension was divided into two parts. The first part was subjected to ultrasonic homogenization, using Sonics Vibra cell, VCX 750 Ultrasonic probe sonicator (Sonics and Materials Inc., Newton, CT, USA), under ice, while the second portion was subjected to high-speed homogenization using Ultra-turrax T-25 1KA (IKA, Staufen, Germany). After characterization of the obtained drug formulations for particle size, high-speed homogenization was selected for the development of finasteride nanosuspension formulation.
Draper–Lin small composite design
Statgraphics® plus software, version 4 (Manugistics Inc., Rockville, MD, USA) was used in the design of the drug nanosuspension formulation. A 43 Draper–Lin small composite design was applied; the studied variables, their level, and the responses are listed in Table 1. The stabilizer concentration (X1), miscible solvent ratio (X2), homogenization speed (X3), and homogenization time (X4) were selected as independent variables, while the particle size (Y1) of the obtained drug nanosuspensions and the solubility enhancement of the freeze-dried drug NPs (Y2) were studied as dependent responses. A total of 18 runs were obtained as listed in Table 2.
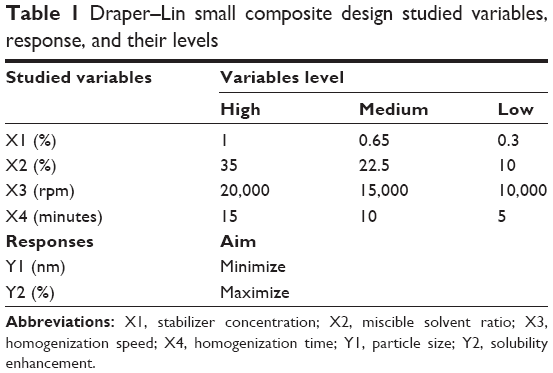 | Table 1 Draper–Lin small composite design studied variables, response, and their levels |
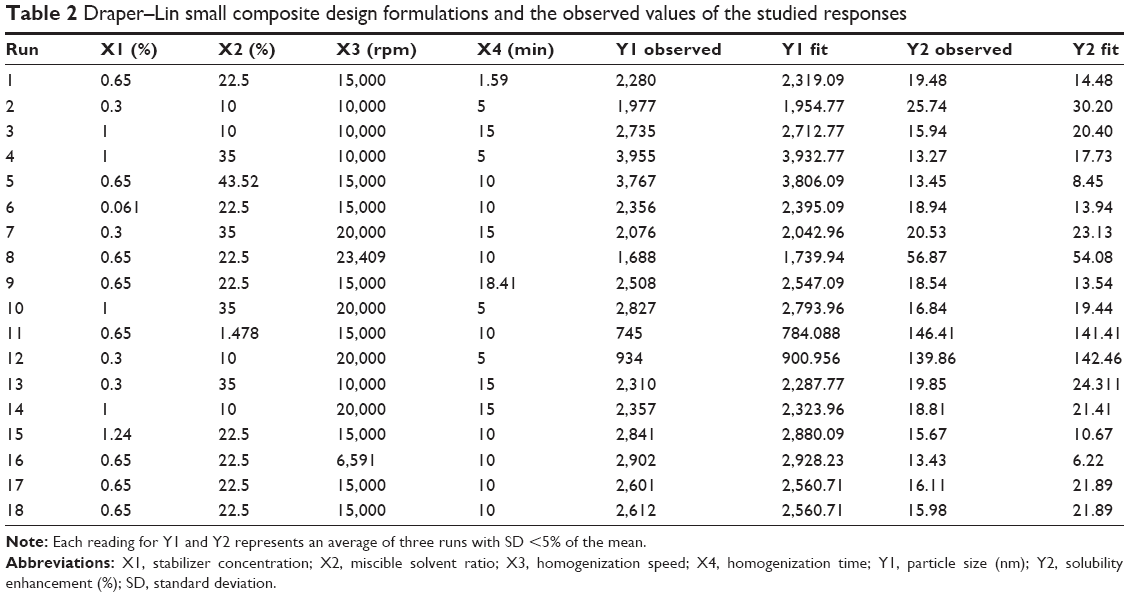 | Table 2 Draper–Lin small composite design formulations and the observed values of the studied responses |
Preparation of the formulation
Finasteride nanosuspension formulations listed in Table 2 were prepared by the solvent evaporation high-speed homogenization technique. Briefly, a known weight of the drug was dissolved in methanol to obtain a drug concentration of 100 mg/mL. The drug solution was then added dropwise into 50 mL water containing known concentration of PVA (0.3%–1%), and the mixture was kept stirring overnight at 1,000 rpm on a magnetic stirrer until complete evaporation of the organic solvent and precipitation of the drug solid particles. The obtained suspension was homogenized at the specified speed for the predetermined time using UltraTurax, IKA® T18 basic Homogenizer (IKA, Campinas, Brazil).
Characterization of the formulation
Particle size and zeta potential measurement
Dynamic light scattering is the technique that was used in the measurement of the particle size and zeta potential of the obtained 18 formulations, Zetatrac of Microtrac Inc., (Montgomeryville, PA, USA) was the instrument used. Characterization of both properties was done in triplicate.
Solubility study
The prepared 18 formulations were subjected to freeze drying using Christ Alpha 1-2 LD Plus Lyophilizer (Martin Christ GmbH, Ostrode am Harz, Germany). Before freeze drying, mannitol was added as a cryoprotectant (5 wt%).17 Solubility study for the freeze-dried formulations was compared to that of the pure drug by placing excess amount of each sample in screw cap vials containing 3 mL of distilled water; the vials were kept shaking for 72 hours at room temperature in a thermostatically controlled shaking water bath (Model 1031; GFL Corporation, Burgwedel, Germany). Aliquots of each vial were taken every day, filtered using Acrodisc® syringe filter of 0.45 μm, and analyzed by HPLC, utilizing the condition that will be fully described in the Pharmacokinetic study section, until equilibrium solubility was reached. The enhancement in the aqueous solubility of the prepared drug formulation was calculated using the following equation:
|
|
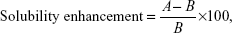
where A is the aqueous solubility of the NPs formulations and B is the aqueous solubility of the pure drug.
Draper–Lin small composite design statistical analysis
The results obtained for the particle size and solubility enhancement were introduced into the response columns in the Statgraphics® plus software and the model was run. Statistical analysis of the obtained data was considered to be significant at P<0.05.
Preparation and characterization of the optimum formulation
After identifying the optimum level for each independent variable that achieves our goal, an optimized formulation was suggested. This formulation was prepared, characterized for the particle size, zeta potential, and solubility enhancement as previously mentioned, and the obtained results were compared to the predicted responses.
Transmission electron microscopy
Transmission electron microscope (TEM) (Model JEM-1230, JOEL, Tokyo, Japan) was used to study the morphological characteristics of the prepared optimized drug nanosuspension. A few drops of the sample were mounted on a carbon-coated grid, left for 2 minutes to allow better adsorption on the carbon film, and a filter paper was used to remove the excess liquid. A drop of phosphotungstic acid (1%) was added and the sample was examined by TEM.
Physicochemical characterization
The thermal behavior of the drug, PVA, their physical mixture, and the optimized freeze-dried drug NPs formulation was investigated by differential scanning calorimetry (DSC) using Shimadzu DSC TA-50 ESI DSC apparatus (Shimadzu, Tokyo, Japan).
All samples used in the DSC study were also subjected to characterization using Fourier-transformed infrared (FTIR) spectroscopy characterized using Perkin Elmer Spectrum One, Model 16 PC (Germany). Each component was mixed with KBr and compressed to a transparent disc, and then tested in the range 4,000–400 cm−1.
The crystalline state of the prepared finasteride NPs compared to that of the pure drug powder was studied using powder X-ray diffractometer (D/max 2500, Rigaku, Tokyo, Japan). The diffraction patterns of both samples were recorded at a scan speed of 0.5000 degree/min.
Stability study
The optimized finasteride aqueous suspension formulation was stored at room temperature in hermetically sealed glass vials in which the particle size was determined, using dynamic light scattering as previously described, every 2 months for 6 months. The optimized freeze-dried drug NPs formulation was also subjected to particle size analysis for the same period.
Preparation of finasteride capsules
Hard gelatin capsules size 3 filled with either the optimized freeze-dried finasteride NPs or drug alone were prepared. Freeze-dried NPs or pure drug was mixed with lactose in a clean jar. Proper mixing was achieved by following the geometric dilution method. Each jar, for the drug or the NPs, was tumbled after each addition of the diluents. Finally, the capsules were filled manually, in which 5 mg of finasteride was loaded into each unit.
In vitro dissolution test
The dissolution profile of the prepared capsules was studied using USP dissolution apparatus Type II (DT 700; Erweka GmbH, Heusenstamm, Germany). A volume of 500 mL distilled water containing 0.05% sodium lauryl sulfate, to provide sink condition,18 was used as a dissolution medium; the temperature was adjusted at 37°C±0.5°C, and the stirring speed was set at 50 rpm. Samples of 2 mL, followed by instantaneous substitution, were withdrawn from the dissolution medium after 5, 10, 15, 30, 45, 60, 90, and 120 minutes. The withdrawn samples were analyzed by HPLC, that will be described in the “Pharmacokinetic study” section. Six samples were studied for each formulation.
Pharmacokinetic study
Study design
The pharmacokinetics of finasteride from capsules loaded with the optimized freeze-dried drug NPs was compared with capsules loaded with either freeze-dried drug microparticles or pure drug. Male New Zealand white rabbits weighing from 2 to 2.5 kg were used; the animals were divided into four groups (n=4): Group I: administered 0.5% CMC solution (negative control group), Group II: administered pure drug powder suspension in 0.5% CMC (positive control), Group III: administered an optimized freeze-dried finasteride NPs formulation that was suspended in 0.5% CMC, and Group IV: administered finasteride microparticles of 0.9 μm particle size that was suspended in 0.5% CMC. Animal care, use, and handling in this study was approved by the local Institutional Review Board for Preclinical and Clinical Research, which ensured the care and use of animals conformed to the EU Directive 2010/63/EU on the protection of animals used for scientific purposes and Guiding Principle in Care and Use of Animals (DHEW publication NIH 80-23) and followed the Principles of Laboratory Animal Care (NIH publication #85-23, revised in 1985). During the experiment, rabbits were allowed full access to normal standard diet and tap water ad libitum. The animals were housed; two per cage in standard rabbit cages maintained at 22°C±3°C under a 12-hour light and 12-hour dark cycles and were acclimatized at least 1 week before the beginning of the experiment. Rabbits were kept fasted for at least 24 hours before the experiments and administered a single oral dose of finasteride (0.4 mg/kg) using a single-dose one-period parallel design. The dose for each group was administered to the animals in the form of oral suspension using gastric tube.
Sample collection
Blood samples of 1 mL were withdrawn from the marginal ear vein at predetermined time intervals: 0, 0.5, 1, 1.5, 2, 4, 6, 8, 12, and 24 hours. Since finasteride has a mean terminal half-life that is 4.7–7.1 hours,15 the extent of the sampling points were determined as per USFDA guidance, which stated that:
The sampling times should extend to at least three multiples of the drug’s apparent terminal elimination half-life, beyond the time when maximum blood concentrations are achieved.19
The collected blood samples were then centrifuged at 6,000 rpm for 10 minutes, and the clear plasma (200 μL) was collected, deproteinized using acetonitrile, and stored at −20°C till analysis using HPLC.
HPLC analysis of finasteride in rabbit plasma
Determination of finasteride, in the form of drug microplates, NPs or pure drug, in the collected plasma samples was achieved using Waters HPLC system (Waters, MA, USA) equipped with Agilent C18 100 mm ×3.3 mm, 4.5 μm particle size column (Agilant, Littelfall, CA, USA). An isocratic mobile phase consists of acetonitrile: 15 mM phosphate buffer:tetrahydrofuran (42:55:3) v/v was delivered at a flow rate of 0.4 mL/min, and the UV detector was set at 220 nm wave length. The method was selected and reproduced with slight modification to that previously published by Anutra et al.20
Pharmacokinetic data analysis
Different pharmacokinetic parameters of finasteride following oral administration of the optimized NPs, pure drug, and drug microplates were estimated from the obtained drug plasma concentration values using WinNonlin®, version 1.5 (Scientific Consulting, Inc., Cary, NC, USA). Maximum plasma concentration (Cmax), time to reach the maximum plasma concentration (tmax), elimination rate constant (ke), and mean residence time (MRT) after oral administration were determined. The measured drug plasma concentrations were also used to calculate the area under the plasma concentration–time curve from time zero to the last concentration time point (AUC0−t) and the area under the plasma concentration–time curve from time zero to infinity (AUC0–∞). AUC0−t was calculated according to the trapezoidal rule, while AUC0–∞ was calculated by the sum of AUC0−t and the last measured concentration divided by the elimination constant (Ct/ke). The obtained data were expressed as mean ± standard deviation and were statistically analyzed using GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA) to verify the differences between the tested groups. Two-way analysis of variance (ANOVA) followed by Tukey’s multicomparison test was used to assess the difference and P<0.05 was considered to be statistically significant. In addition, the 95% confidence level is considered for testing.
Results and discussion
Production of nanosuspension by bottom–up technique may be achieved by employment of precipitation, followed by a second step of high-energy process such as homogenization to produce drug NPs in a liquid medium.13 The obtained nanosuspension is a thermodynamically unstable colloid dispersion system that has an inherent property to aggregate owing to the Ostwald ripening phenomenon and to a positive Gibbs free energy change.21 Hence, the formulation must be permanently stabilized with the appropriate stabilizer to prevent aggregation of the NPs during the preparation process or storage. The stabilizers used should be adsorbed onto the drug nanosuspension surfaces and produce a steric or electrostatic stabilization effect. Usually, in addition to steric repulsion to establish effective stabilization, strong and fast adsorption at full coverage of the drug particles and a long desorption time are all required. PVA is a water-soluble synthetic polymer that was selected as a stabilizer since it acts as a protective colloid that is adsorbed on the drug NPs during the size- reduction process. Previous studies illustrated the effective role of PVA in producing small size and stable NPs during development of drug nanosuspension such as felodipen when compared to hydroxypropyl methyl cellulose-based drug nanosuspension.22 A similar finding has also been reported for albendazole nanosuspension, in which the authors have mentioned that poloxamer 188 produced drug NPs that were significantly larger in particle size than PVA.23 Some other studies reported that other stabilizers such as hydroxypropyl methyl cellulose, NaCMC, polysorbate 80, PVP, pluronic F 68, F188, and F127 are also effective, but with other techniques such wet-milling,24 sonoprecipitation,25 and emulsion–diffusion process.26 Methanol was selected as the water miscible organic solvent as finasteride is freely soluble in this solvent. In addition, on the basis of our preliminary investigation, high-speed homogenization was selected as it produced particles in the nanosize range, while ultrasonic homogenization produced drug microparticles. These drug microparticles were used in the pharmacokinetic part of this study for comparative purpose. A brief description for the ultrasonic homogenization technique will be submitted as a separate work.
Optimization of the formulation and processing variables is critical during nanosuspension development. For example, inadequate stabilizer concentration will not provide full coverage of particle, which is necessary for repulsion between the particles, while higher concentration of the stabilizer will form micelles that play a critical role in nanosuspension thermal instability.21 Other variables such as the homogenization speed and time have been previously reported to affect the process.21 Draper–Lin small experimental design is one of the response surface methodology that has been used in this study to investigate the influence of four variables on the particle size and solubility enhancement of finasteride NPs. The design helps investigate the main, interaction, and quadratic effects of the studied variables on the selected responses. Eighteen formulations have been proposed by the software. Each formulation has been prepared, characterized for the particle size and solubility enhancement, and the results are listed in Table 2. Zeta potential, which is an important characteristic that reflects the surface charges on the particles and affects the stability of the prepared NPs,27 was in the range 0.94–2.40 mv. This value is relatively low and needs to be improved to maintain a constant particle size. Previous reports mentioned similar finding of low zeta potential value for nitrendipine nanosuspensions prepared using PVA.28 The authors also mentioned improvement in this value and production of constant particle size following surface modification of the particles after incorporation of cationic polymer such as chitosan. Improvement of the obtained zeta potential value will be studied in our upcoming work by optimizing the concentration of different cationic and anionic polymer. Formulation F15 and F17 showed the higher zeta potential values among the studied formulation, an effect that could be attributed to the low percent of PVA in these formulations. Adsorption of PVA on the surface of the particles leads to a decrease in zeta potential values, which could be attributed to the presence of acetate group in the PVA polymer chain and shift of the slipping plane.
Effect of the studied parameters on the selected responses
Statistical analysis for the effect of the studied variables on the particle size and solubility enhancement was carried out by multiple regression analysis and two-way ANOVA using statgraphics software. Table 3 lists the estimated effects of the selected factors, the F-ratios, and the associated P-values for the two responses resulting from ANOVA. A positive sign for the estimated effect values indicates synergistic effect of the studied independent variables on the selected response, while negative sign indicates antagonistic effect. F-ratio compares between the actual and expected variation of variable averages; an F-ratio greater than 1 indicates a location effect and hence the P-value reports the significance level. A factor is considered to influence the studied response if the P-value differs from 0 and is less than 0.05.
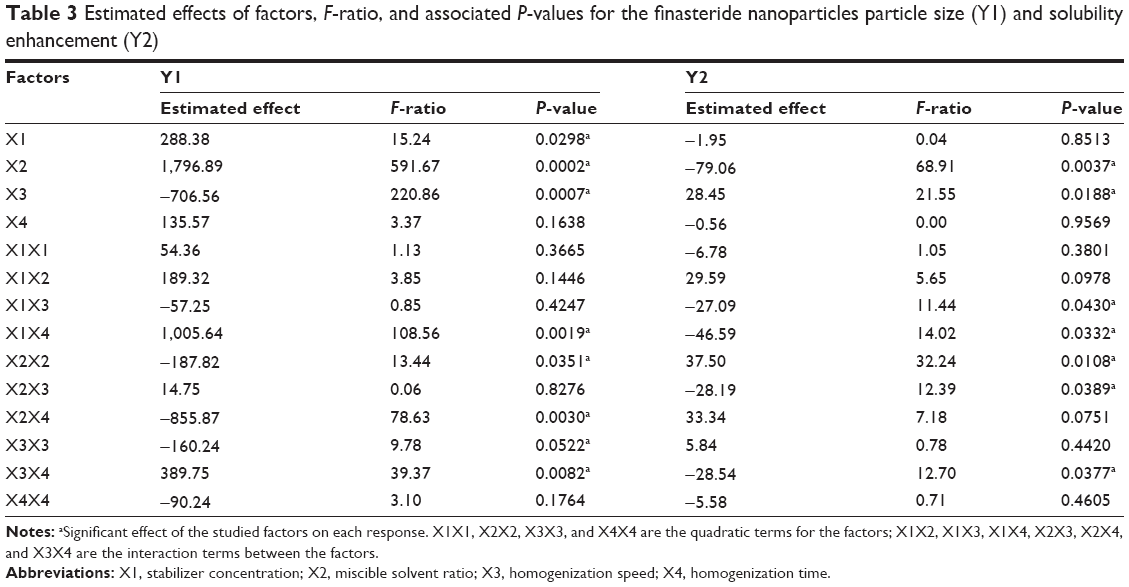 | Table 3 Estimated effects of factors, F-ratio, and associated P-values for the finasteride nanoparticles particle size (Y1) and solubility enhancement (Y2) |
The solvent ratio (X2), homogenization speed (X3), and stabilizer concentration (X1) had a significant effect on the particle size of the prepared nanosuspension as illustrated in the standardized Pareto chart (Figure 1). X2 and X1 were of positive (synergistic) effect on Y1 due to the effect of increasing the drug load and to formation of PVA micelles molecules at high PVA concentration, respectively. Increase in the homogenization speed will increase the impact force on the drug particles that results in decrease in particle size. The interaction effect of X1X4, X2X4 and X3X4, and the quadratic effect of X2 and X3 were also significantly affecting the particle size. This was obvious in the figure as it includes a vertical line at the critical P-value of 0.05. An effect that exceeds the vertical line is considered to be statistically significant.
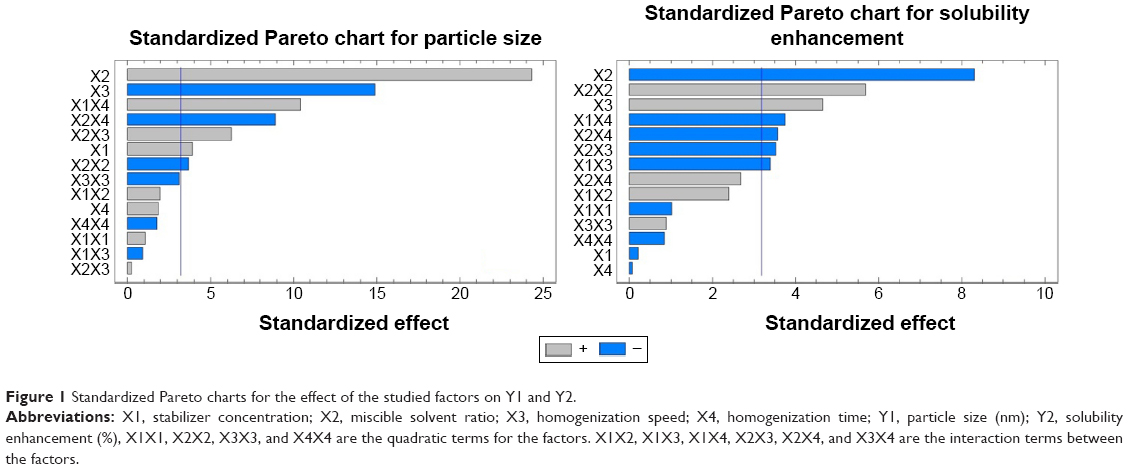 | Figure 1 Standardized Pareto charts for the effect of the studied factors on Y1 and Y2. |
The R-squared statistic indicates that the model as fit explains 99.7948% of the variability in particle size. The adjusted R-squared statistic, which is more suitable for comparing models with different numbers of independent variables, is 98.8372%.
The main effect plot for the studied variables on the particle size indicated the significance of X2, X3, and X1. To illustrate the effect of X1 and X2 on the particle size when X3 and X4 were kept at their intermediate levels, estimated response surface and contour plots were constructed (Figure 2). The equation of the fit model is as follows:
|
|
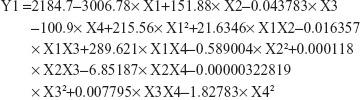
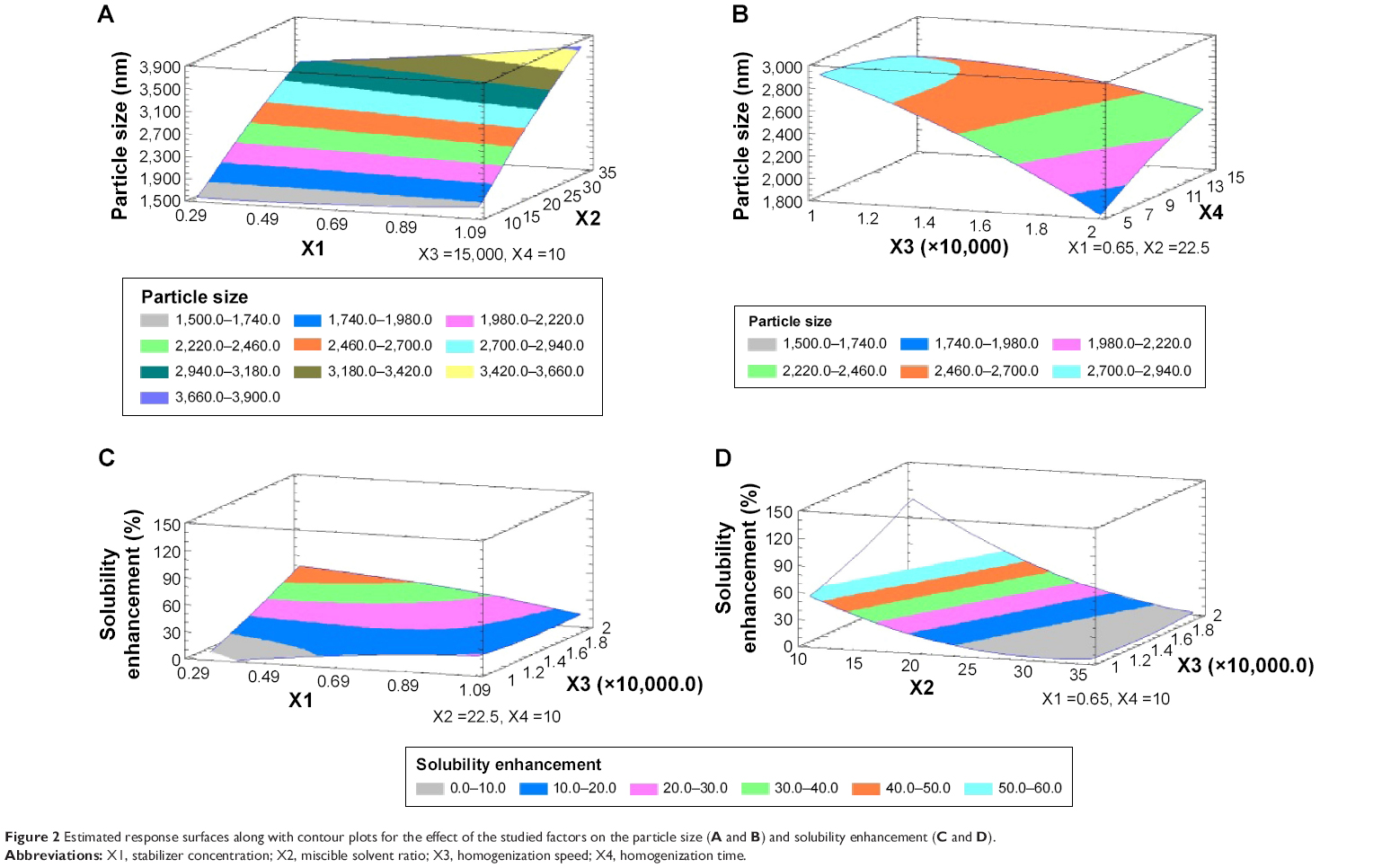 | Figure 2 Estimated response surfaces along with contour plots for the effect of the studied factors on the particle size (A and B) and solubility enhancement (C and D). |
Regarding the effect of the studied variables on the solubility enhancement, seven effects have P<0.05, indicating that they are significantly different from 0 at the 95.0% confidence level. X2 and X3, the interaction effect of X1X4, X3X4, X2X3, and X1X3, and the quadratic effect of X2 were significantly affecting Y2 as illustrated in Figure 1. The effect of X2 and X3 on Y2 was directly related to their effect on the particle size. Small particles showed enhanced aqueous drug solubility and vice versa. The R-squared statistic indicates that the model as fit explains 98.4841% of the variability in solubility enhancement. The adjusted R-squared statistic, which is more suitable for comparing models with different numbers of independent variables, is 91.4101%. The main effect plot for the studied variables on Y2 indicated the significance of X2 and X3. Estimated response surface and contour plots (Figure 2) were constructed to illustrate the effect of X1 and X2 on the solubility enhancement when X3 and X4 were kept at their intermediate levels. The equation of the fit model is as follows:
|
|
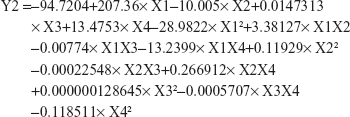
Equations 1 and 2 imitate the quantitative influence of the formulation variables on Y1 and Y2 responses.
Optimum desirability
To prepare an optimized finasteride nanosuspension formulation characterized by smaller particle size and higher solubility enhancement values of 677.214 nm and 155.383%, respectively, the optimize desirability was identified and the composition of the optimized formulation was suggested to contain 0.66867, 1.54226, 17,660, and 10.5844 of X1, X2, X3, and X4, respectively. This optimized formulation was prepared and characterized for the particle size and solubility enhancement, in which the observed values were 645 nm and 159.24%, respectively, indicating the validity of the model.
Transmission electron microscopy
Figure 3 illustrates the TEM image of the optimized finasteride nanosuspension formulation. The obtained image indicates that the prepared drug nanosuspension particles were homogenous, almost spherical in shape, and no aggregation was noticed. This finding is in good agreement with the work done by Thakkar et al29 for Olmesartan medoxomil nanosuspension. The size of the particles obtained by the TEM image seems to be smaller than that of the dynamic light scattering due to dilution of the sample and proper dispersion of the particles before the sample has been mounted on a carbon-coated grid, the effect that prevents NP aggregation. Both techniques are quite different; TEM characterizes dehydrated and immobilized particles on a solid support that leads to structural distortions compared to characterization of the same particles in the solvent swollen state in dynamic light scattering.30
 | Figure 3 TEM image for the optimized finasteride nanoparticles formulation. |
Physicochemical characterization
DSC describes the material thermal events and appears as deviations from the baseline, in either an exothermic or endothermic direction, depending upon the quantity of energy, whether more or less, that has to be supplied to the sample relative to the reference material. The DSC thermogram shows the direction of the exothermic event (exo down). The thermal events that are shown in the thermogram represent the material phase transformations. The intercept between the baseline and the tangent of the melting peak onset provides the melting point.31
DSC thermogram of pure drug showed a characteristic peak at 250.81 corresponding to the drug-melting endotherm. Mixing the drug with PVA as a physical mixture or the optimum finasteride formulation did not affect the characteristic drug-melting endotherm as illustrated in Figure 4.
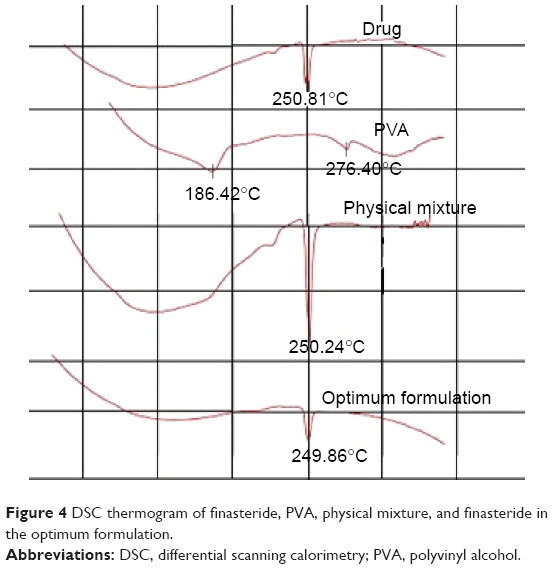 | Figure 4 DSC thermogram of finasteride, PVA, physical mixture, and finasteride in the optimum formulation. |
FTIR for the pure drug, its physical mixture with PVA, and the drug in the optimized formulation, is depicted in Figure 5. Finasteride IR spectra showed characteristic peaks at 1,666 and 1,598 cm−1 corresponding to the two amide groups. Other peaks at 1,395–1,385/1,365 cm−1 corresponding to tert-butyl group. Nagalaxmi et al32 demonstrated three functional groups: amide, ketone, and alkyl groups after finasteride FTIR scanning. Finasteride physical mixture with PVP and the optimum drug micronized formulation did not interfere with the characteristic drug peaks as shown in Figure 5, which confirms the compatibility.
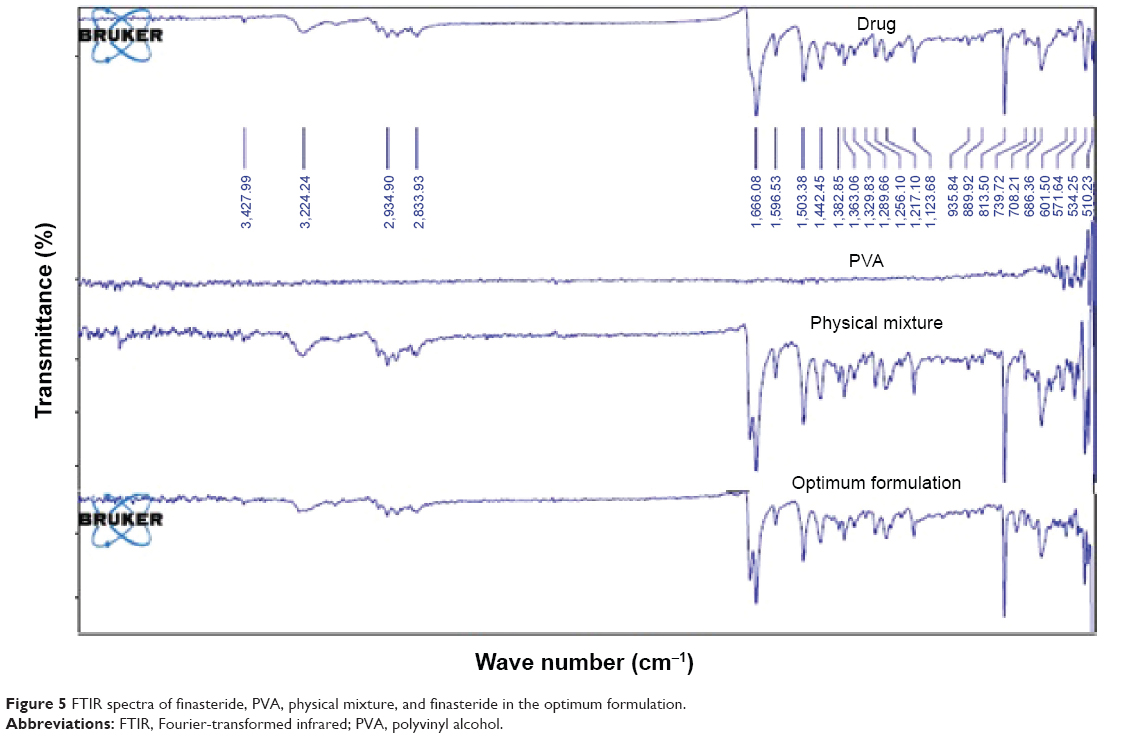 | Figure 5 FTIR spectra of finasteride, PVA, physical mixture, and finasteride in the optimum formulation. |
X-ray powder diffraction (XRPD) was performed to investigate the physical state of finasteride in the pure drug state and also in the optimized freeze-dried NPs formulation. Pure finasteride powder showed intense characteristic peaks of the crystalline drug structure, while the X-ray diffraction pattern of the optimized freeze-dried NPs was different from that of the pure drug, which is an indication of the crystalline state transformation into the amorphous form (Figure 6). A previous study reported the same crystalline state transformation for atorvastatin following atorvastatin nanocrystals development.33 This transformation accounts for the enhanced solubility and dissolution rate and is expected to improve the bioavailability from the prepared NPs.
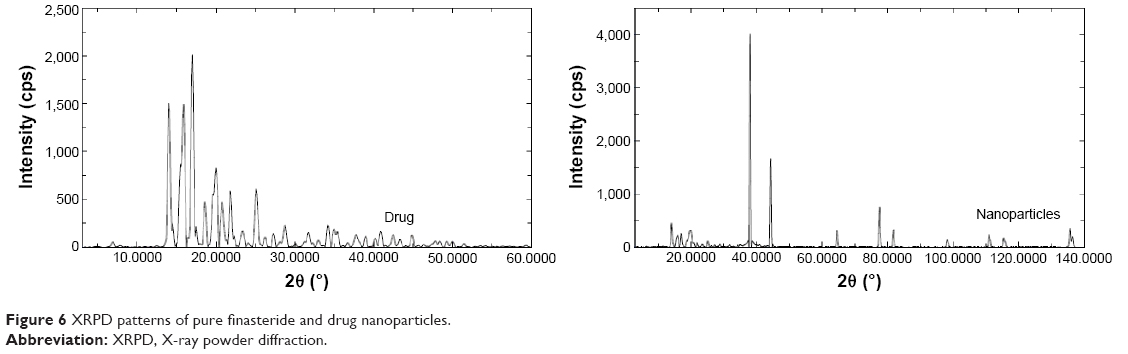 | Figure 6 XRPD patterns of pure finasteride and drug nanoparticles. |
Stability study
It has been previously stated that the solidified state is generally preferred when compared to the corresponding aqueous nanosuspension formulation owing to the significant decrease in particle aggregation and other instability problems such as hydrolysis.34 Our results for particle size analysis for both freeze-dried drug NPs and aqueous nanosuspension formulation confirmed this finding. After 6 months storage, there was a significant increase in particle size (>24.8%) in the aqueous nanosuspension drug formulation when compared to freeze-dried NPs (<7.4%). Although the steric and/or ionic stabilization effect of the stabilizer molecules counterbalances the positive surface energy of the particles in the aqueous condition, the effect that is highly decreased in the solid state since the stabilizer chain solidify,35 yet incorporation of mannitol as a cryoprotectant agent prevents crystal growth due to the formation of new strong layer around the particles during the freeze-drying sublimation step.21 Crystal growth in the freeze-dried NPs was also prevented or decreased due to the protective coating of the NPs by the PVA molecules.
In vitro dissolution of the prepared capsules
The dissolution profile of the prepared capsules filled with either the optimized freeze-dried finasteride NPs or drug alone was studied. Results revealed enhancement in the rate of drug dissolution by more than 2.5 times from the capsules filled with the optimized drug NPs when compared to capsules filled with the pure drug (data not shown). This enhancement is attributed to improvement in the drug aqueous solubility owing to a decrease in the drug particle size with subsequent increase in the drug surface area, an effect that results in increase of contact between the solvent and the drug surface with possible increase in the solubility and dissolution. A previous study mentioned enhancement in the dissolution of felodipine NPs prepared as nanosuspension when compared to the pure drug.36 It has been previously stated that the aqueous solubility of finasteride is almost constant between pH 1 and 13, and so water was used as dissolution medium instead of HCl/phosphate buffer since water is less expensive, easily available, and environmentally friendly.37 In vitro release of drugs with a release rate independent of pH value of the medium has been reported, in which similar results to those obtained in HCl or phosphate buffer were observed.37,38
Pharmacokinetic study
Study of the pharmacokinetics of a new drug formulation is of prime importance to ensure satisfactory drug plasma level, acceptable bioavailability, and effectiveness of this formulation.39 The study requires administration of this formulation, followed by blood sampling during which the animal physiological status must not be disturbed.40
It has been reported that enhancing the dissolution and absorption of water-insoluble drugs, such miconazole, improves the drug pharmacokinetic parameters, and hence the drug bioavailability.39
On the basis of our preliminary study, finasteride microparticles of 1.9 μm were prepared and administered to Group IV animals, and the results were compared with the pure drug and the optimized freeze-dried NPs formulation to study the effect of decreasing the particle size on drug bioavailability.
As shown in Figure 7, the plasma level–time curve for Group III, administered the drug NPs formulation, showed enhanced bioavailability when compared with the other two groups as indicated by higher Cmax and area under the curve. It is noteworthy to mention that a good in vitro–in vivo correlation was observed after considering the drug pharmacokinetic profile. As previously illustrated in the in vitro dissolution, when the drug particle size was decreased, the surface area was increased and, therefore, the solubility and dissolution rate were enhanced. Drug NPs that showed a solubility enhancement of 155.38% significantly affected the observed pharmacokinetic profile. The studied pharmacokinetic parameters are listed in Table 4. Significant differences in Cmax and tmax among the studied groups were noticed, which is an indication for the effect of drug particle size reduction. Values for the area under the curves were also different in the three groups and confirmed superiority of NPs over microplates and pure drug in enhancing the pharmacokinetic and, thus, drug bioavailability.
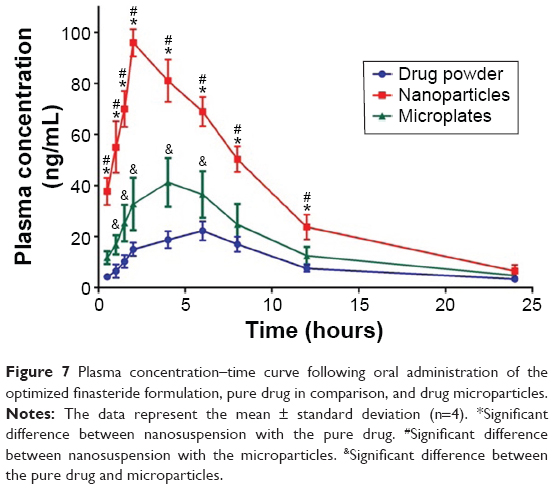 | Figure 7 Plasma concentration–time curve following oral administration of the optimized finasteride formulation, pure drug in comparison, and drug microparticles. |
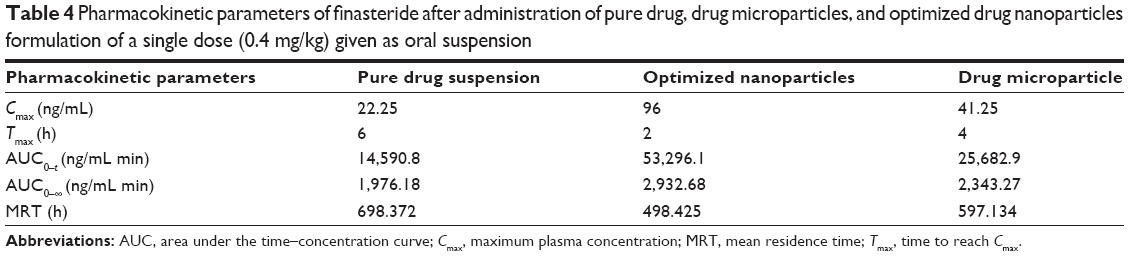 | Table 4 Pharmacokinetic parameters of finasteride after administration of pure drug, drug microparticles, and optimized drug nanoparticles formulation of a single dose (0.4 mg/kg) given as oral suspension |
Conclusion
It can be concluded that the experimental design has been successfully implemented to develop finasteride NPs with uniform particle size and solubility enhancement. Stability of the freeze-dried NPs was superior to that of the corresponding aqueous drug nanosuspension. Reduction in the particle size enhanced the bioavailability of finasteride, the effect that could be attributed to enhancing the drug solubility, dissolution rate, and oral absorption from the prepared freeze-dried NPs. Hence, finasteride NPs formulation loaded into hard gelatin capsules could be used as an efficient oral medication.
Acknowledgments
This work was supported by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under the grant number 166-592-D 1435. The author gratefully acknowledges the DSR technical and financial support.
Disclosure
The author reports no conflicts of interest in this work.
References
Khadka P, Ro J, Kim H, et al. Pharmaceutical particle technologies: an approach to improve drug solubility, dissolution and bioavailability. Asian J Pharm Sci. 2014;9(6):304–316. doi:10.1016/j.ajps.2014.05.005. | ||
Badr-Eldin SM, Ahmed TA, Ismail HR. Aripiprazole-cyclodextrin binary systems for dissolution enhancement: effect of preparation technique, cyclodextrin type and molar ratio. Iran J Basic Med Sci. 2013;16(12):1223–1231. | ||
Shid RL, Dhole SN, Kulkarni N, Shid SL. Nanosuspension: a review. Int J Pharm Sci Rev Res. 2013;22(1):98–106. | ||
Kumar GP, Krishna KG. Nanosuspensions: the solution to deliver hydrophobic drugs. Int J Drug Deliv. 2011;3(4):546–557. doi:10.5138/385. | ||
Ahmed TA. Preparation of transfersomes encapsulating sildenafil aimed for transdermal drug delivery: Plackett–Burman design and characterization. J Liposome Res. 2015;25(1):1–10. doi:10.3109/08982104.2014.950276. | ||
El-Say KM, Ahmed TA, Badr-Eldin SM, Fahmy U, Aldawsari H, Ahmed OAA. Enhanced permeation parameters of optimized nanostructured simvastatin transdermal films: ex vivo and in vivo evaluation. Pharm Dev Technol. 2015;20(8):919–926. doi:10.3109/10837450.2014.938859. | ||
Ahmed OAA, Badr-Eldin SM, Tawfik MK, Ahmed TA, El-Say KM, Badr JM. Design and optimization of self-nanoemulsifying delivery system to enhance quercetin hepatoprotective activity in paracetamol-induced hepatotoxicity. J Pharm Sci. 2014;103(2):602–612. doi:10.1002/jps.23834. | ||
Al-Subaie MM, Hosny KM, Mohamed El-Say K, Ahmed TA, Aljaeid BM. Utilization of nanotechnology to enhance percutaneous absorption of acyclovir in the treatment of herpes simplex viral infections. Int J Nanomedicine. 2015;10:3973–3985. doi:10.2147/IJN.S83962. | ||
Sutradhar KB, Khatun S, Luna IP. Increasing possibilities of nanosuspension. J Nanotechnol. 2013;2013:1–12. doi:10.1155/2013/346581. | ||
Li W, Yang Y, Tian Y, et al. Preparation and in vitro/in vivo evaluation of revaprazan hydrochloride nanosuspension. Int J Pharm. 2011;408(1–2):157–162. doi:10.1016/j.ijpharm.2011.01.059. | ||
Jacobs C, Kayser O, Müller RH. Production and characterisation of mucoadhesive nanosuspensions for the formulation of bupravaquone. Int J Pharm. 2001;214(1–2):3–7. | ||
Patravale VB, Date AA, Kulkarni RM. Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol. 2004;56(7):827–840. doi:10.1211/0022357023691. | ||
Keck CM, Müller RH. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur J Pharm Biopharm. 2006;62(1):3–16. doi:10.1016/j.ejpb.2005.05.009. | ||
Faller B, Farley D, Nick H. Finasteride: a slow-binding 5 alpha-reductase inhibitor. Biochemistry. 1993;32(21):5705–5710. | ||
Steiner JF. Clinical pharmacokinetics and pharmacodynamics of finasteride. Clin Pharmacokinet. 1996;30(1):16–27. | ||
Prahalada SR, Keenan KP, Hertzog PR, et al. Qualitative and quantitative evaluation of prostatic histomorphology in rats following chronic treatment with finasteride, a 5-alpha reductase inhibitor. Urology. 1994;43(5):680–685. | ||
Wang Y, Liu Z, Zhang D, et al. Development and in vitro evaluation of deacety mycoepoxydiene nanosuspension. Colloids Surf B Biointerfaces. 2011;83(2):189–197. doi:10.1016/j.colsurfb.2010.10.029. | ||
Ibrahim HM, Ahmed TA, Hussain MD, et al. Development of meloxicam in situ implant formulation by quality by design principle. Drug Dev Ind Pharm. 2013;9045:1–8. doi:10.3109/03639045.2012.746360. | ||
US Food and Drug Administration. Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA. US Department of Health and Human Services: Rockville, MD, USA; 2013. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm377465.pdf | ||
Anutra K, Kumsorn B, Rojanasthien N. Bioequivalence study of generic finasteride in healthy male volunteers. Chiang Mai Med Bull. 2003;42(4):131–137. | ||
Wang Y, Zheng Y, Zhang L, Wang Q, Zhang D. Stability of nanosuspensions in drug delivery. J Control Release. 2013;172(3):1126–1141. doi:10.1016/j.jconrel.2013.08.006. | ||
Sahu BP, Das MK. Optimization of felodipine nanosuspensions using full factorial design. Int J PharmTech Res. 2013;5(2):553–561. | ||
El-Badry M, Fetih G, Salem-Bekhit MM, Shakeel F. Formulation and evaluation of nanosuspension of albendazole for dissolution enhancement. Nanosci Nanotechnol Lett. 2013;5(9):1024–1029. doi:10.1166/nnl.2013.1645. | ||
Pandya VM, Patel JK, Patel DJ. Formulation and optimization of nanosuspensions for enhancing simvastatin dissolution using central composite design. Dissolut Technol. 2011;18:40–45. | ||
Esfandi E, Ramezani V, Vatanara A, Rouholamini Najafabadi A, Hadipour Moghaddam SP. Clarithromycin dissolution enhancement by preparation of aqueous nanosuspensions using sonoprecipitation technique. Iran J Pharm Res. 2014;13(3):809–818. | ||
Mishra B, Arya N, Tiwari S. Investigation of formulation variables affecting the properties of lamotrigine nanosuspension using fractional factorial design. Daru. 2010;18(1):1–8. | ||
Wiśniewska M. The temperature effect on electrokinetic properties of the silica-polyvinyl alcohol (PVA) system. Colloid Polym Sci. 2011;289(3):341–344. doi:10.1007/s00396-010-2341-4. | ||
Quan P, Shi K, Piao H, et al. A novel surface modified nitrendipine nanocrystals with enhancement of bioavailability and stability. Int J Pharm. 2012;430(1–2):366–371. doi:10.1016/j.ijpharm.2012.04.025. | ||
Thakkar HP, Patel BV, Thakkar SP. Development and characterization of nanosuspensions of olmesartan medoxomil for bioavailability enhancement. J Pharm Bioallied Sci. 2011;3(3):426–434. doi:10.4103/0975-7406.84459. | ||
Ito T, Sun L, Bevan MA, Crooks RM. Comparison of nanoparticle size and electrophoretic mobility measurements using a carbon-nanotube-based coulter counter, dynamic light scattering, transmission electron microscopy, and phase analysis light scattering. Langmuir. 2004;20(16):6940–6945. doi:10.1021/la049524t. | ||
Brown ME. Introduction to Thermal Analysis: Techniques and Applications. Berlin, Germany: Springer Science & Business Media; 2001. | ||
Nagalaxmi V, Mahipal Reddy L, Manikanta Kumar A, Manjunath Shetty KS. Formulation and evaluation of controlled release matrix tablets of finasteride. Int J Pharm Dev Technol. 2014;4(1):63–70. | ||
Kurakula M, Ahmed TA. Co-delivery of atorvastatin nanocrystals in PLGA based in situ gel for anti-hyperlipidemic efficacy. Curr Drug Deliv. 2016;13:1–11. | ||
Gao Y, Qian S, Zhang J. Physicochemical and pharmacokinetic characterization of a spray-dried cefpodoxime proxetil nanosuspension. Chem Pharm Bull (Tokyo). 2010;58(7):912–917. | ||
Lee J. Drug nano- and microparticles processed into solid dosage forms: physical properties. J Pharm Sci. 2003;92(10):2057–2068. doi:10.1002/jps.10471. | ||
Sahu BP, Das MK. Nanosuspension for enhancement of oral bioavailability of felodipine. Appl Nanosci. 2013;4(2):189–197. doi:10.1007/s13204-012-0188-3. | ||
Maria O, Santos M, Laura A, Santos A, Pereira GR, Bonfilio R. A dissolution test for finasteride in immediate-release capsules. Dissolution Technol. 2013;20(3):25–33. | ||
El-Say KM, El-Helw A-RM, Ahmed OA, et al. Statistical optimization of controlled release microspheres containing cetirizine hydrochloride as a model for water soluble drugs. Pharm Dev Technol. 2015;20(6):738–746. doi:10.3109/10837450.2014.920353. | ||
Ahmed TA, El-Say KM, Mahmoud MF, Samy AM, Badawi AA. Miconazole nitrate oral disintegrating tablets: in vivo performance and stability study. AAPS PharmSciTech. 2012;13(3):760–771. doi:10.1208/s12249-012-9798-z. | ||
Hui Y, Huang NH, Ebbert L, et al. Pharmacokinetic comparisons of tail-bleeding with cannula- or retro-orbital bleeding techniques in rats using six marketed drugs. J Pharmacol Toxicol Methods. 56(2):256–264. doi:10.1016/j.vascn.2007.05.006. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.