")
Back to Journals » International Journal of Nanomedicine » Volume 14
Preparation and optimization of lidocaine transferosomal gel containing permeation enhancers: a promising approach for enhancement of skin permeation
Authors Omar MM , Hasan OA, El Sisi AM
Received 12 January 2019
Accepted for publication 26 January 2019
Published 26 February 2019 Volume 2019:14 Pages 1551—1562
DOI https://doi.org/10.2147/IJN.S201356
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Mahmoud M Omar,1,2 Omiya Ali Hasan,1,2 Amani M El Sisi3
1Department of Pharmaceutics and Industrial Pharmacy, Deraya University, El-Minia, Egypt; 2Department of Pharmaceutics, Sohag University, Sohag, Egypt; 3Department of Pharmaceutics and Industrial Pharmacy, Beni-Suef University, Beni-Suef, Egypt
Aim: To develop the topical gel containing transferosomal lidocaine as alternative to painful local anesthetic injection.
Materials and methods: The transfersomes were prepared by film hydration technique using soybean phosphatidylcholine and cholesterol. The prepared transfersomes were evaluated for the morphology, drug loading, %EE, particle size and in vitro release. The transferosomal gel of lidocaine was prepared using HPMC k15 as gelling agent and propylene glycol, dimethyl sulfoxide (DMSO), and polyamidoamine dendrimer third generation (PAMAM G3) solutions were used as permeation enhancer. The formulated gels were evaluated for pH, viscosity, drug content and ex-vivo permeation of the gel. The analgesic effect of the formulation was tested using tail flick test.
Results: The transfersomes showed that transfersomes (F4) had the highest entrapment efficiency (%EE) approaching 79.87±2.35, low particle size 179.5 nm, and zeta potential of -43.5±4.74 mV. According to the rat tail flick test, the AUC0–90 minutes of the control formulation (lidocaine solution, A) was 352.32±5.87 seconds minutes. While the maximum AUC0–90 minutes value was found to be 570.5±6.81 seconds minutes for gel formulation (F) containing transfersomal lidocaine with PAMAM G3 dendrimer as permeation enhancer. In this case, the local anesthetic efficacy was increased by 1.62-folds as compared to control formulation.
Conclusion: From the present study, it can be concluded that the topical gel loaded with transfersomal lidocaine shows enhanced skin permeation effect along with increase in local anesthetic action of lidocaine.
Keywords: transfersome, local anesthesia, lidocaine, permeation enhancement, gelling agent
Introduction
Local anesthetics can reversibly block nerve endings without damage because of their binding with sodium channel of skin layers. Hence the nervous impulse is not propagated as a result of blocking of sodium ions influx.1 One of these is lidocaine, which is an amide-type local anesthetic fundamentally used in mucosal, dermal, and topical dosage forms. Local anesthetic action of lidocaine is rapid but short. Moreover, low anesthetic intensity of lidocaine topical formulation is the main disadvantage as a result of its poor percutaneous absorption.2
Lidocaine in pastes, creams, and ointment are the safest and most convenient mode of application for topical drug delivery system but these can be easily removed by wetting, movement, and contact, which is the main pitfall of these systems. So the new formulation strategy with bioadhesive preparation incorporated with enhanced local anesthetic effects is requested for the topical administration. Vesicular systems are fundamentally utilized as a vehicle for topical, mucosal, dermal, and transdermal drug delivery. Many literatures show benefits of vesicular system in improvement of drug permeation.3,4
Transfersomes have been found as one of the superior drug-delivery systems for topical application as compared to the conventional topical systems. They are characterized with ultraflexible bilayer membranes, which enable vesicles to be highly elastic and deformable. So, transfersomes can escape from narrow pores in the stratum corneum (one-tenth of their own diameter) under nonocclusive conditions. Moreover, transfersomes represent multilateral delivery for enhancing stability and using as a carrier of various drugs.
The major limitation of the transfersomes using topically is its liquid character. To overcome this, transfersomes are incorporated into suitable vehicle in which the original structure of the vesicles is preserved. It has been well-known fact that the transfersomes are compatible with the gel systems made from the polymers such as HPMC.5 So in this present investigation, HPMC k15 was used as gelling agent and that was used as a vehicle for the incorporation of the transfersomes for topical delivery system. HPMC was intentionally used due to its nontoxic nature, good swelling properties, and accommodation of high levels of drug.
The aim of the present work is to formulate HPMC k15 gel incorporating transferosomal lidocaine and to study the entrapment efficiency, in vitro drug release, skin retention, and its analgesic effect. Nanotransformes have been used as platforms to percutaneous administration for local anesthetic. Moreover, dimethyl sulfoxide (DMSO), propylene glycol (PG), and polyamidoamine dendrimer third generation (PAMAM G3) dendrimer have been used as skin permeation enhancers in gels. The local anesthetic effects of the formulated gels containing DMSO, PG, and PAMAM G3 dendrimer were evaluated using the tail flick anesthetic test.
Materials and methods
Lidocaine was obtained as a kind gift from Shouguang Fukang Pharmacy Factory (Shandong, China). Pluronic® F-127 (poloxmer 407) was purchased from BASF (Hanover, Germany). Soybean phosphatidylcholine and dipalmitoyl phosphatidylcholine were kindly gifted by Lipoid GmbH (Ludwigshafen, Germany). Disodium hydrogen phosphate, potassium dihydrogen phosphate, sucrose, and chloroform were purchased from Shanghai Chemical Co. (Shanghai, China). Other chemicals were of reagents or analytical grade.
Preparation of lidocaine-loaded transfersomes by thin-film hydration method
Lidocaine-loaded transfersomes formulations were prepared by thin-film hydration method. Soybean phosphatidylcholine, cholesterol, sodium cholate (SC), span 80 (sorbitan monostearate), and brij 35 with different molar ratios were dissolved in 10 mL of a mixture of three organic solvents (methanol:chloroform:ethanol) at (2:2:1) v/v/v ratio, as represented in Table 1.
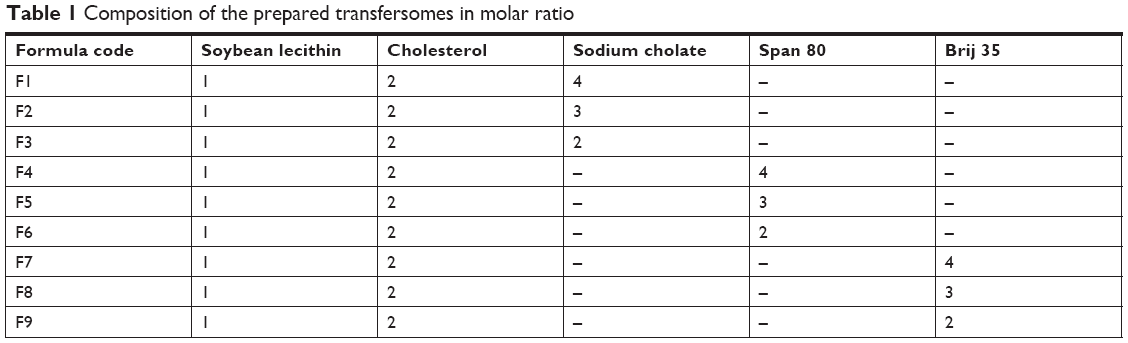 | Table 1 Composition of the prepared transfersomes in molar ratio |
Using rotary evaporator (type Hei-VAP manufactured by Heidolph Instruments GmbH & Co. KG, Schwabach, Germany), thin lipid film on the internal surface of the round-bottomed flask was formed. Lidocaine (100 mg) was dissolved in 20 mL of an isotonic phosphate buffer (pH 5.8). Lidocaine solution was used to hydrate the prepared thin film by rotation at 100 rpm for 2 hours. To form large multilamellar vesicles, the resulting suspensions were kept for 24 hours at 25°C. To form smaller vesicles, the transferosomal dispersions were sonicated for 30 minutes.
The lidocaine transfersomes were separated from the entrapped lidocaine by high-speed centrifugation at 20,000 rpm for 3 hours at −5°C using cooling ultracentrifuge (High-Speed Refrigerated Centrifuge, CR22N; Hitachi Ltd., Tokyo, Japan). To separate the untrapped lidocaine, clear supernatant was carefully taken out after the centrifugation. The transfersomes remained as precipitate containing the entrapped lidocaine. The precipitate was resuspended in 10 mL of isotonic phosphate buffer (pH 5.8) in order to be evaluated. The transferosomal dispersions (free from the untrapped lidocaine) were kept at a constant temperature of 4°C within glass vials. Laminar air flow hood was used for conducting experimental procedures under aseptic conditions (horizontal laminar flow hood, BZ Series, model BZ-3SS RX; Germfree, Ormond Beach, FL, USA).
Characterization of lidocaine-loaded transfersomes: determination of the particle size and zeta potential of the transferosomal dispersions
Aliquots of transferosomal dispersion (150 μL) were diluted with double-distilled water (850 μL) to examine the zeta potential and the particle size of the transfersomes. The zeta potential and particle size were measured by the method of dynamic light scattering using Malvern Zetasizer (Malvern Instruments Corp; Nano ZS ZEN 3600, Worcestershire, UK).
Drug loading
The percentage of lidocaine loading in transfersome was determined by using 4.0 mL of dispersion. Free lidocaine was separated from the transferosomal dispersions by subjecting the transfersomes to a high-speed centrifugation at 21,000 rpm at 10°C model T-70BL (Laby Instrument Industry, Haryana, India) for 3 hours. The supernatant was siphoned-off and free lidocaine concentration was measured at λmax=263 nm using UV spectrophotometer (Shimadzu, Kyoto, Japan). The transferosomal preparations were performed under aseptic conditions using a laminar air flow hood (horizontal laminar flow hood, BZ Series, model BZ-3SS RX; Germfree).
Determination of transfersomes lidocaine entrapment efficiency
The precipitate separated from supernatant was redispersed in 4 mL of isotonic phosphate buffer (pH 7). To perform the lysis of transfersomes for liberating the encapsulated lidocaine molecules, a 500 μL was diluted ten times with methanol (HPLC grade, ≥99.9%). The concentration of drug was determined spectrophotometrically at λmax=263 nm.
Entrapment efficiency could be defined as follows,
|
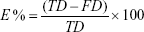
where E % is the percent encapsulation, TD is the total drug amount, and FD is the amount of free drug.
Investigation of the morphology of the transfersomal dispersions
The morphology of transferosomal dispersions was investigated with the field transmission electron microscopy (TEM; model JEM-1400 microscope; JEOL, Tokyo, Japan), where a 200 μL of freshly prepared transferosomal dispersions was diluted with distilled water (1.8 mL). A drop of the diluted dispersion was loaded on carbon-coated copper grid. For contrast enhancement, the deposit was stained by applying aqueous solution of uranyl acetate (10% w/w) for 1 minute. After drying of deposite using air, the grid was evaluated using TEM.6
In vitro release study
In vitro release study from the transfersomes
The in vitro release study was performed via a dialysis membrane according to Hao’s method. Briefly, an equivalent amount of 10 mg lidocaine-loaded transferosomal dispersion was introduced into dialysis bags (SERVA Electrophoresis GmbH, Heidelberg, Germany) with a molecular weight cutoff 12,000 kDa. The dialysis bags were suspended in an isotonic buffer solution (250 mL, pH 6.8, 37°C±2°C) at speed of rotation 1,500 rpm and placed within the dissolution flask of the USP dissolution apparatus. The samples (5 mL) were withdrawn and analyzed spectrophotometrically at λmax=263 nm every 45 minutes for 12 hours. The withdrawn samples were replaced with the same volume of fresh an isotonic buffer solution (pH 6.8). The concentration percentage of lidocaine at time (t) was estimated as follow:
|
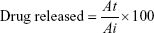
To explain the equation, Ai and At are the initial amounts of lidocaine entrapped in the transfersomes and the amounts of lidocaine released at time t, respectively.
Preparation of transfersomal gel of lidocaine with permeation enhancers
According to the results of characterization of the prepared lidocaine transferosomal preparations, formula F4 was chosen to be integrated into gel dosage form. The gel was prepared by the same procedures described by Schmolka (1972).7 In brief, in 10 mL distilled water, a required quantities of poloxamer 407 and HPMC k15 were added slowly and stirred with the help of magnetic stirrer at 50 rpm for 1 hour. To ensure the maximum dissolution of polymers, the prepared solution was left in the quiescent state for 12 hours in a refrigerator. Then, the solution (poloxamer with HPMC k15) was stirred slowly at 5°C for 5 hours until a gel was formed. Various formulations were prepared as shown in Table 2.
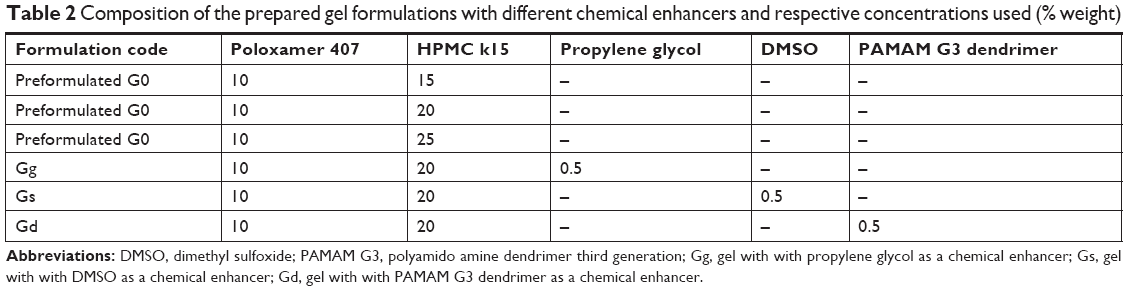 | Table 2 Composition of the prepared gel formulations with different chemical enhancers and respective concentrations used (% weight) |
A 50 mg equivalent lidocaine transfersomal suspension (from the prepared optimized transfersomal batch, F4) was added to the poloxamer dispersions. The proper amount of the used permeation enhancers (DMSO, PG, PAMAM G3 dendrimer) was added to the lidocaine–transfersomes–poloxamer dispersions. The prepared system was stirred for 45 minutes with the help of magnetic stirrer to form homogeneous and transparent gel. A lidocaine-containing gel was prepared using the same procedure.8 Drug content, drug release, and some organoleptic properties (such as texture, transparency, and color) of the prepared gel were determined. Finally, the prepared gel dosage forms containing permeation enhancers (PG, DMSO, PAMAM G3 dendrimer) were coded with Gg, Gs, and Gd, respectively.
Characterization of the prepared gel
Rheological study of the gel
The viscosity of the prepared gels was measured using Brookfield Viscometer (Brookfield Engineering Laboratories, Inc. Middleboro, MA, USA) with S18 spindle, at speed 10 rpm. Rheological study of the gel was conducted in the shear rate range of 1–400 second−1. Constant readings of the viscosity were taken after a specified time of 5 minutes and noted in centipoises.
pH of the gel
The pH of all prepared gels containing lidocaine transfersomes was measured by using pH meter (SP-701, Suntex Company, New Taipei City, Taiwan).
Drug content uniformity of the gels
Lidocaine uniformity from the prepared gels was conducted to confirm uniform dispersion or distribution of lidocaine throughout the prepared gel. To conduct the content uniformity of prepared gels, samples taken from three to four different places from the container were analyzed spectrophotometrically at λmax=263 nm. In case of transfersomal lidocaine gel, it was mixed with sufficient quantity of Triton X-100 to release the drug and then analyzed spectrophotometrically at λ=263 nm.
Stability study
Stability study of the lidocaine-loaded transfersomes
To examine the aggregation of transfersomal dispersion and leakage of lidocaine from the prepared vesicles throughout storage, physical stability studies were conducted. A protocol of stability study developed by Du Plessis et al was carried out with some modifications.9 The prepared transfersomal dispersions were stored in tightly sealed amber vials (15 mL) at different temperatures (4°C±1°C, 25°C±1°C, and 37°C±1°C) for 3 months. Aliquots (1 mL) from each transfersomal preparation were withdrawn at specified time to measure each of the particle size and encapsulation efficiency of the transfersomal vesicles. The physical appearance of transfersomal dispersion was evaluated for visual observing any signs of sedimentation or creaming.
Stability study of the final gel formulations
The prepared gel formulations (Gg, Gs, and Gd) were stored in opaque bottles with cap covered with plastic sheet. The stored bottles were kept at three different temperatures (4°C±1°C, 25°C±1°C, and 40°C±1°C), 75% relative humidity (RH) ±5% for a period of up to 3 months. Lidocaine content in the stored gel formulations was determined periodically. Any change in the appearance of the stored gel formulations was recorded.
Ex vivo permeation study
Preparation of rat skin
Female Sprague Dawley rats (180–200 g) were purchased from the animal house of the Faculty of Medicine, Beni-Suef University. The rats were anesthetized with fluthane and executed humanely by decapitation. Removal of their hair was carried out carefully with an electric clipper and the skin of rat abdomen was enucleated. Any fat and debris was removed from the rat skin. The rate skin was stored at −20°C until further need. All steps were approved by the Committee of Animal Ethical in the Faculty of Medicine, Beni-Suef University. All animal experiments were carried out in accordance with the UK Animals (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments.
Permeation studies
Ex vivo permeation studies of lidocaine from gel was conducted on specially developed glass diffusion cell. The skin samples were immersed in cold bubbled Meduna’s mixture (95:5 oxygen/carbon dioxide). The treated skin samples were spread around one end of the designed diffusion cell. The surface area of the skin membrane was 2.5 cm2 with 100 mL, of receptor cell volume. The receptor cell was submerged 1 cm into a receptor compartment containing isotonic phosphate buffer (pH 6.4). The diffusion cells were loaded with 20 mg equivalent gel containing lidocaine. The receptor phase was stirred at 100 rpm and kept at 37°C±0.5°C. Sink condition was maintained throughout the permeation experiments. Aliquots (4 mL) were taken at specified time. Percent permeation of lidocaine was determined by spectrophotometric analysis of the taken samples at 263 nm. Taken samples were replaced with a fresh isotonic phosphate buffer. For comparison, lidocaine solution, lidocaine-containing gel, transfersomal lidocaine-containing gel, transfersomal lidocaine-containing gel with PG, transfersomal lidocaine-containing gel with DMSO, and transfersomal lidocaine-containing gel with dendrimer were evaluated.10–13 All determinations were accomplished in triplicate, and the findings were represented as the mean ± SD.
Tail flick test
Protocol of the conducted experiments was accepted by the Animal Ethical Committee of the Faculty of Medicine, Beni-Suef University. In the present study, 35 male Sprague Dawley rats (Charles River, Microkatu, Kuopio, Finland) weighing 290±25 g were distributed into seven groups (n=5). All rats were kept at specified environmental conditions (temperature: 20°C, humidity 40% RH) while conducting the experiment. The rats were accommodated to the specified environmental conditions for 10 days before carrying out tail flick test. To carry out the experiment, specified area of rat tail was exposed to radiant heat source, while the rats were loosely fixed.
Baseline latency for each rat was detected, which was defined as the mean time of four measurements before application of any formulation.14 The rats whose baseline latency mean time was more or less than the time range 2–4 seconds were excluded from the experiment.
As flicking of rat tail was occurred, the required time was recorded. A 10-second cutoff time was applied to keep away from tissue thermal injury.15 Tail flick latency (TFL) time was determined as the time from the application of the heat exposure to the flicking of the tail. Seven groups of the rats were handled gently. These groups of the rats were treated with 20 mg equivalent of lidocaine solution (first group), 20 mg equivalent of lidocaine-containing gel (second group), 20 mg equivalent of transfersomal lidocaine-containing gel (third group), 20 mg equivalent of transfersomal lidocaine-containing gel with PG as permeation enhancer (fourth group), 20 mg equivalent of transfersomal lidocaine-containing gel with DAMSO as permeation enhancer (fifth group), 20 mg equivalent of transfersomal lidocaine-containing gel with PAMAM G3 as permeation enhancer (sixth group), and saline solution (control group). Time course of lidocaine response of individual formulation was represented by plotting the mean of latency times as a function of time.
To calculate the area under the effective curve (AUC) using linear trapezoidal rule, the mean of latency times for each group was plotted as a function of time.16
Following application of the prepared gel, reaction times were determined at 0, 10, 20, 30, 40, 50, 60, 75, 90, 105, and 120 minutes. The AUC was determined considering time of TFL from 10 to 120 postapplication using trapezoid rules.17
|
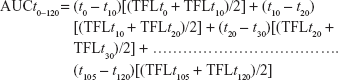
Statistics
The determined results were written as mean ± mean standard error. A one-way ANOVA was applied for statistical analysis of the data. The level of confidence was considered at P<0.05.
Result and discussion
Characterization of the nanotransfersomes
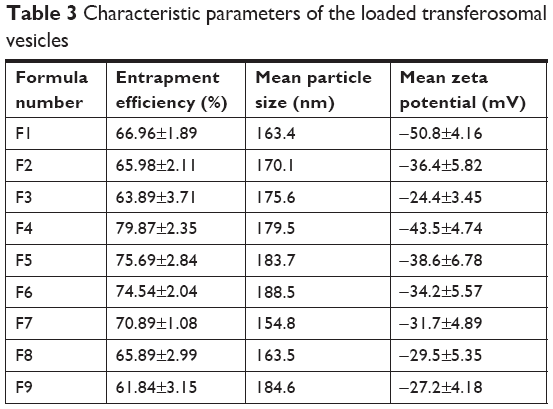
Table 3 showed the main properties of the prepared transferosomal formulations, which encapsulate lidocaine. The entrapment efficiency percent of deformable vesicles was detected to be in the range of 61.84%±3.15% to 79.87%±2.35%. The formula F4 showed significantly the highest percent of entrapment efficiency approaching 79.87% (P<0.05). All transferosomal formulations were found to be in the submicron to nanosize range. The formula F7 had the smallest size of 154.8 nm while the formula F6 had the largest size of 188.5 nm. The zeta potential values of the formulations were detected to be in the range of −50.8±4.16 mV to −27.2±4.18 mV. The formula F4 showed the highest percent encapsulation entrapment (79.87%±2.35%), small particle size (179.5 nm), and good release pattern. Accordingly, the formula F4 was used to be incorporated to formulate gel. Drug content of the prepared gel was within 97.4%–101.4% with insignificant difference of micrometers.
 | Table 3 Characteristic parameters of the loaded transferosomal vesicles |
Transfersomes have better permeation ability than liposomes because transferosomal vesicles are ultradeformable elastic and flexible vesicles. Hence they are formulated for noninvasive transdermal drug delivery.18 Single-chain surfactants (edge activators) were incorporated into the prepared transferosomal vesicles to destabilize the transferosomal vesicles and improve the deformability of the vesicular bilayer as a result of reducing the interfacial tension.8,19
Percent encapsulation entrapment is a great factor in case of transferosomal preparations because it may have a great effect on release of the drug. The formula F4 showed significant high percent encapsulation entrapment (P<0.05). This finding could be attributed to the formulated span 80 that improves elasticity and flexibility of the vesicle. Hence the ability of the transferosomal membrane to encapsulate large amounts of the drug was improved.20 Cholesterol may play a role in preventing the leakage of the drug from the prepared vesicles.21 The particle size of the prepared transferosomal vesicles varied significantly (P<0.05) due to the reduced interfacial tension of the prepared vesicles.19 The measured zeta potential values were found in a range of −27.2±4.18 to −50.8±4.16 mV. Those results approved that all transferosomal vesicles were colloidally stable. The stability could be attributed to avoid aggregation of vesicles as a result of strong electrostatic repulsion between them. Those findings are concurrent with the papers reported in the literature.22
The transmission electron micrograph
The transmission electron micrograph of examined transfersome (F4) represented the outline and the core of the spherical vesicles proving the vesicular characteristics of the prepared transfersome. The examined transfersome (F4) has vesicular shape with large internal aqueous core, with 200–150 nm in diameter. The transmission electron micrographs of the examined prepared (F4) vesicles are shown in Figure 1.
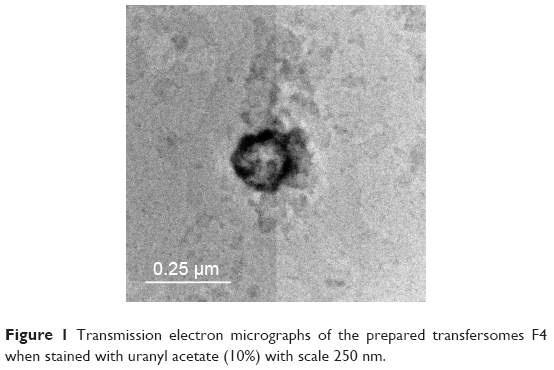 | Figure 1 Transmission electron micrographs of the prepared transfersomes F4 when stained with uranyl acetate (10%) with scale 250 nm. |
In vitro drug release from the prepared nanotransfersomes
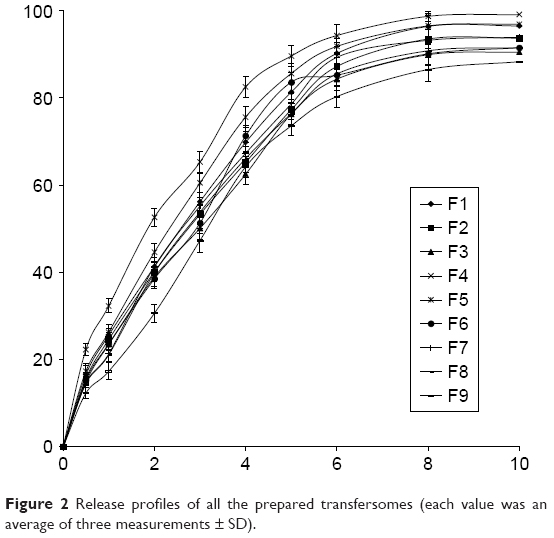
The release profile of lidocaine from the prepared transferosomal vesicles in phosphate buffer solution (0.1 M, pH 6.4) at 37°C was represented in Figure 2.
 | Figure 2 Release profiles of all the prepared transfersomes (each value was an average of three measurements ± SD). |
More than 80% of the drug content of all the prepared vesicles was released after 6 hours.
Statistical examination proved that there is no significant difference in the release values of lidocaine from all the formulae (P>0.05). All the transferosomal formulations released their drug contents according to a first-order kinetic model (R2>0.97).
As predicted, formulation of lidocaine as transfersome led to a controlled release rate because of reservoir effect of transfersome. The release figures of lidocaine from the prepared transferosomal vesicles were seemingly biphasic processes. The burst effect over the first 4 hours was obviously detected, followed by a slower release step. This finding could be explained as a result of a property of the bilayer or transfersome structure or reflect the loss of surface-associated material. Moreover, disability of lipid particles of the transferosomal vesicles to accommodate larger amounts of lidocaine may be another explanation for the burst effect.23 Also, the drug separation from the surface of the prepared vesicles followed by sustained release drug from the inner core was reported in the literatures.24,25
Finally, kinetic treatment of the release data proved that the drug release from the prepared vesicles was driven the according to first-order model was the best fit (R2>0.97).
Characterization of lidocaine transferosomal gel
The optimized formulation of transferosomal suspension (F4) was to be incorporated into the prepared gel. PG, DMSO, and PAMAM G3 were used as skin permeation enhancers. The permeation enhancers were used to maximize the permeation of lidocaine through the skin so as to enhance the local anesthetic effect of lidocaine. The transferosomal lidocaine along with permeation enhancers would show better local anesthetic activity as compared to the general formulations.
pH of the gel
The pH of the formulated gels of HPMC k15 and different permeation enhancers was found between 5.3 and 7.6. The results of various pH values with different permeation enhancers revealed that all examined gels were compatible with skin. The gel containing PAMAM G3 dendrimer as permeation enhancer showed the pH very closer to the pH of the human skin so it could be assumed that no skin irritation will occur after application of dendrimer transferosomal gel while gel prepared with DMSO as a permeation enhancer had pH of 5.3. This pH represents the acidic characteristics so there would have been chances of skin irritation. Moreover, the negative charge of the transferosomal vesicles enhanced the transdermal drug permeation. The charge of skin surface is a slight negative. Hence, a slight enhancing transdermal drug permeation was reported as result to electrostatic repulsion between the charge of the skin surface and the optimized gel.26
Rheological properties
Rheograms of the prepared gel are represented in Figure 3. All the examined gel samples exhibited a thixotropic pseudoplastic behavior, which was a preferable property in some pharmaceutical preparations such as gel.
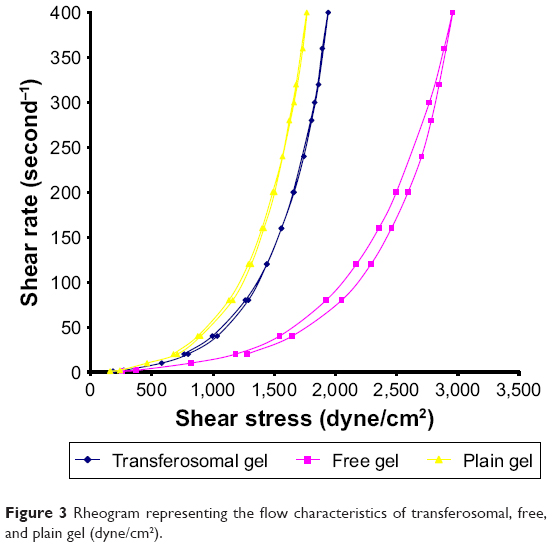 | Figure 3 Rheogram representing the flow characteristics of transferosomal, free, and plain gel (dyne/cm2). |
Self-alignment of colloidal structure in the parallel directions of applied shear may be the cause of the pseudoplasticity results.27 The prepared gel samples were exposed to different rates of shear. From the obtained results, the rate of shear was plotted against shearing stress as shown in Figure 3. Thixotropic behavior was determined in all samples of the examined gels. By applying ascending shearing stress on the examined gel samples, long chains of HPMC polymer may be positioned as weak construction of parallel lines, leading to more and more ease of flow (upcurve). On contrary, by applying descending shearing stress on the gel samples, the construction begin to reform and gradual restoration of viscosity occurs (down-curve). All viscosity values on down-curve were less than on upcurve at the same shearing stress. Moreover, the loop area surrounded by upcurve and down-curve is a measure of the thixotropic behavior. As shown in Figure 3, all the examined gel samples were characterized with a distinct area of loop.28
Effect of HPMC k15 concentration on bioadhesive forces and viscosity
To optimize the bioadhesive forces, various concentrations of HPMC k15 gels (1.5%, 2.0%, and 2.5%) were prepared. The viscosity of the HPMC k15 gels at 1.5%, 2.0%, and 2.5% were 10.7, 17.1, and 23.9 Pa s, respectively; Viscosity was determined in the shear rate range of 1–400 second−1 and shearing rate 200 dyne/cm2. The bioadhesive force of HPMC k15 at 15%, 20%, and 25% were 79.5±3.1, 108.7±2.8, and 143.2±2.4 gf, respectively. Accordingly, the increase in the viscosity of HPMC k15 gel led to the increase in bioadhesive forces.
Analysis of the texture profile of the gel system was also evaluated. The evaluation of texture profile included mechanical characteristics of the gel such as hardness, adhesiveness and cohesiveness. The hardness of gel containing 15% HPMC k15 was 14.0±0.71 g and cohesiveness of HPMC k15 gel containing transferosomal dispersion was 3.05±0.16 g. It was found that as the concentration of transferosomal dispersion in gel increased, the cohesiveness reduced. Accordingly the transferosomal gel was less coherent. During stress sweep of 15% gel, it was observed that there was linearity between stress and strain produced all over the applied stress range, which was the indication for the system working in correct range. Also the lidocaine uniformity in case of 20% gel was found to be 98.97%–101.25%. This range of the uniformity was higher than others. Based on the viscosity, bioadhesive force, lidocaine uniformity, hardness, and cohesiveness, the concentration 20% gel was chosen as the optimized concentration for the HPMC k15.
Stability study
The determinations of entrapment efficiency and particle size of different transfersomes, which were stored at 4°C, 25°C, and 37°C over a period of 3 months, were represented as shown in Tables 4 and 5. In refrigeration condition (4°C), insignificant changes (P>0.05) in entrapment efficiency and particle size of the stored transfersomes were detected. Transferosomal vesicles stored at refrigeration condition were physically stable. No color change or sedimentation was observed. However, there were significant changes (P<0.05) in entrapment efficiency and particle size of the transfersomes stored at 25°C and 37°C. Dramatic increase in particle size of transferosomal vesicles stored at 25°C and 37°C proved physical instability of the stored transfersomes at these conditions. These findings may be ascribed to the swelling or aggregation of vesicles. Significant decrease in entrapment efficiency was detected representing leakage of lidocaine from different transfersomes. Decrease in entrapment efficiency may be ascribed to raised temperature that led to enhanced fluidity of lipid bilayer of vesicles. Moreover chemical degradation of phospholipids may be deteriorated leading to membrane packaging defect. Pale yellow and slight sedimentation were observed. Stability study of the transferosomal lidocaine-containing gel with different permeation enhancers (Gp, Gd, and Gp) showed no significant decrease in drug content over 3 months for the stored formulations at 4°C. However, there are significant decreases in drug content over 3 months for the stored formulations at 25°C and 37°C, as shown in Table 6. The findings of stability study suggested that storage of transfersome at 4°C may enhance stability of transfersomes. Embedding transfersome into gel may support stability of transferosomal vesicles as a result of increasing viscosity of the carrier, reducing possibility of infusion.
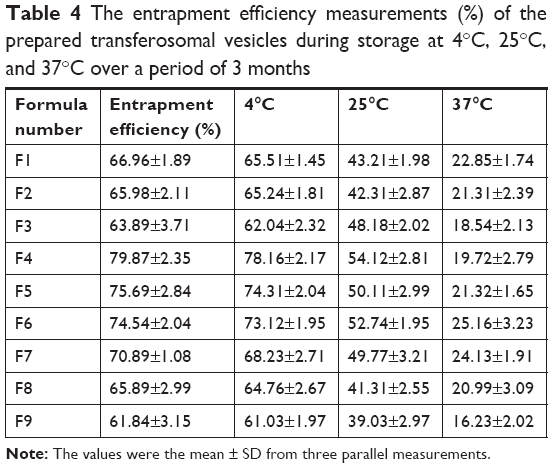 | Table 4 The entrapment efficiency measurements (%) of the prepared transferosomal vesicles during storage at 4°C, 25°C, and 37°C over a period of 3 months |
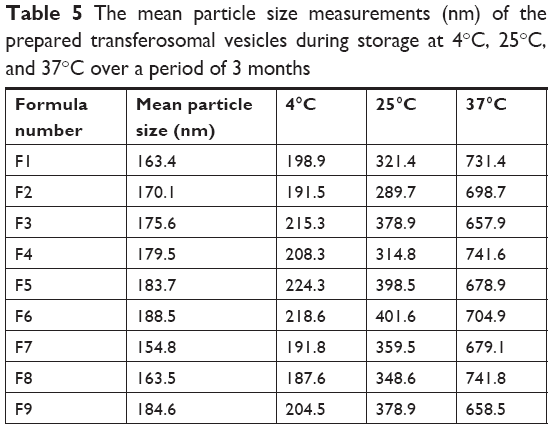 | Table 5 The mean particle size measurements (nm) of the prepared transferosomal vesicles during storage at 4°C, 25°C, and 37°C over a period of 3 months |
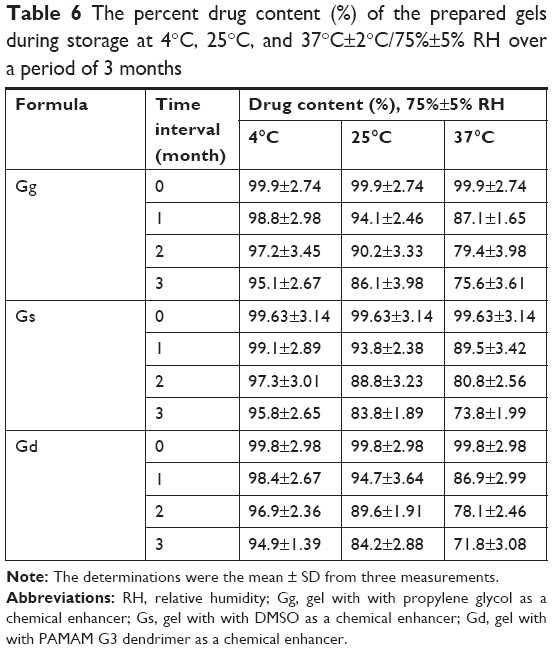 | Table 6 The percent drug content (%) of the prepared gels during storage at 4°C, 25°C, and 37°C±2°C/75%±5% RH over a period of 3 months |
Effect of permeation enhancers on lidocaine across the rat skin
Skin is the outermost layer of the body providing many functions including homeostasis, protection, and barrier to the external environment. Stratum corneum is a rate limiting layer in transdermal permeation of the drugs. Many mechanisms showing different transdermal penetration pathways have been suggested.29,30 So in order to enhance the transdermal penetration of the lidocaine various permeation enhancers such as PG (nonionic surfactant), DMSO, and PAMAM G3 were incorporated in HPMC k15 transferosomal gel.
Results of in vitro drug permeation studies conducted with various formulations of lidocaine are shown in Table 7. The comparative permeation of lidocaine from lidocaine solution (A), lidocaine-containing gel (B), transferosomal lidocaine-containing gel (C), transferosomal lidocaine-containing gel with PG (D), transferosomal lidocaine-containing gel with DMSO (E), and transferosomal lidocaine-containing gel with PAMAM G3 (F) has also been shown in Table 7. Lidocaine-transferosomal gels containing permeation enhancers have significant increase in skin retention and permeation (P<0.05).
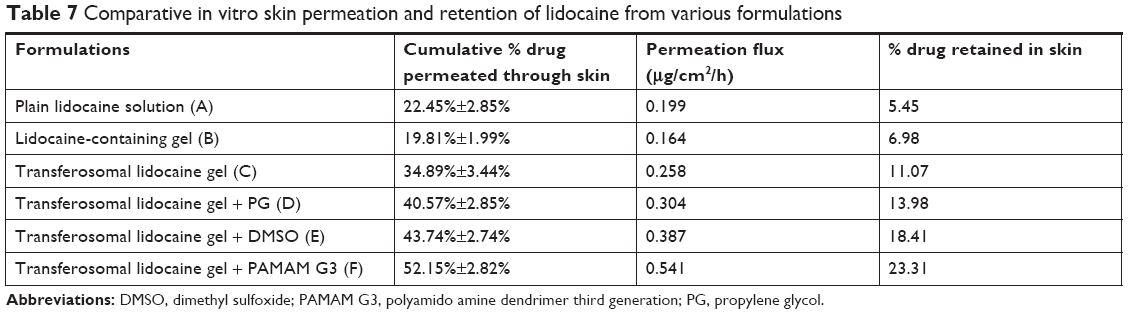 | Table 7 Comparative in vitro skin permeation and retention of lidocaine from various formulations |
The percent cumulative drug permeated through skin from lidocaine solution (A) and lidocaine in HPMC k15 gel (B) was found to be 22.45%±2.85% and 19.81%±1.99%, respectively. Moreover, when the lidocaine transferosomal gel (C) was applied, the percent cumulative lidocaine permeated through skin became 34.89%±3.44%. From these findings, transferosomal preparation showed significant improvement in the skin permeation of lidocaine. Further improvement in the skin permeation was detected when permeation enhancers were formulated in transferosomal gel. Furthermore, the skin permeation of lidocaine detected with transferosomal gel containing PG (D) and DMSO (E) permeation enhancers was 40.57%±2.85% and 43.74%±2.74%, respectively. The greatest skin permeation detected with transferosomal gel containing PAMAM G3 dendrimer (F) was 52.15%±2.82%.
The order of skin permeation of lidocaine through skin was found as F>E>D>C>A>B. So that concludes that transferosomal gel containing permeation enhancers played an important role in skin permeation of the lidocaine.
Higher values of permeation flux observed with formulation containing transferosomal lidocaine (C) as compared to lidocaine solution (A) and lidocaine in HPMC k15 gel (B). Further on, the permeation flux was enhanced in preparations containing permeation enhancers.
The permeation flux was found to be 0.304, 0.387, and 0.541 μg/cm2/hour in (D), (E), and (F), respectively. PAMAM G3 permeation enhancer demonstrated the greatest value of the flux compared to the others. Moreover, drug retention study clearly shows that the amount of lidocaine retained in the skin was considerably the highest in case of preparations containing transferosomal preparations with permeation enhancers than preparations with nontransferosomal and plain lidocaine preparation. From aforementioned study, it was obvious that the transferosomal gel formulated with PAMAM G3 dendrimer as permeation enhancer could not only improve the penetration of lidocaine but also boosted in the localization of skin tissue.
Tail flick test for local anesthetic action
Basal nociceptive level, the analgesic activity of therapeutic agent and tolerance formation can be evaluated by tail flick test. Table 8 showed the AUC0–90 minutes of the rat tail flick test for various preparations containing lidocaine. The AUC value of the control preparation (A) was 352.32±5.87 seconds minutes.
 | Table 8 Comparative maximum possible effect (%MPE; at 60 and 90 minutes) and AUC of various lidocaine-containing formulations |
The greatest AUC value was detected to be 570.5±6.81 seconds minutes for preparation (F) containing transferosomal lidocaine with PAMAM G3 dendrimer as permeation enhancer. Accordingly, the efficacy of local anesthetic was enhanced by 1.62-folds as compared to control preparation. Moreover, the efficacy of local anesthetic of preparation (C) containing transferosomal lidocaine in gel system was enhanced by 1.27-folds as compared to the control. And also, the efficacy of local anesthetic of preparations (D) and (E) was enhanced by 1.40- and 1.47-folds, respectively, as compared to the control. The comparative graphical representation is shown in Figure 4.
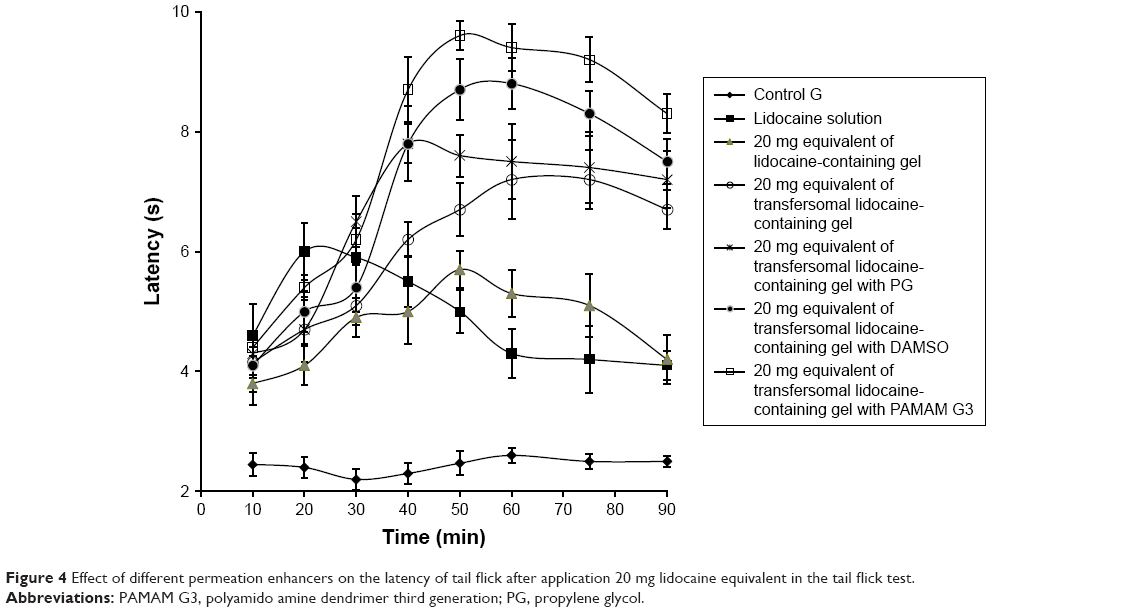 | Figure 4 Effect of different permeation enhancers on the latency of tail flick after application 20 mg lidocaine equivalent in the tail flick test. |
From above results it was clear that transferosomal lidocaine in gel system played vital role for the enhancing local anesthetic activity of the lidocaine. According to the aforementioned findings, transferosomal formulations of lidocaine in gel preparations played an important role for improving the local anesthetic activity of the lidocaine. Those findings could be elucidated according to negative charge of transfersomal gel. Hence, weak electrostatic repulsion between transfersomes and intercellular components of skin is generated. The generated repulsion leads to accelerate penetration of negatively charged transfersomes through follicles of different skin layers. Rapid penetration may suggest that transfersomes reach as intact part in basal area of follicles releasing their cargo into blood vessels.31 Moreover, lipoidal nature of transferosomal augmented the skin permeation of lidocaine, hence, the activity of local anesthetic was improved.32 Further on, the improvement of local anesthetic activity was greatly potentiated by formulating the skin permeation enhancers. PG enhanced transdermal absorption but the mechanism of its action is not a clear. However, role of PG as cosolvent is the cause of enhancing thermodynamic activity of the drug. Hence, driving force of lidocaine diffusion may be enhanced.33 DMSO may also enhance transdermal penetration by improving partitioning of drug into skin layers. DMSO may improve extraction of skin lipid and keratin interaction. Moreover, it enhances displacement of bound water and interactions with the lipid alkyl chains in the subcutaneous SC. The solubility properties of subcutaneous layer changed as result of a change intercellular aqueous domain. Facilitated drug partitioning from the vehicle into the SC may play a role in its action. PAMAM dendrimers may covalently bind with different drug molecule.34 PAMAM dendrimers may modify and promote the drug dissolution from the transfersomes. Penetration of skin through the follicular route could be another mechanism. Moreover, low-generation dendrimers may attenuate function of the subcutaneous barriers, especially in the presence of potent carrier such as transfersome.35 The formulated permeation enhancers within transferosomal lidocaine allowed the greatest skin permeation and boosted improvement of the local anesthetic activity.
Conclusion
Transfersomes are excellent drug carrier to permeate skin tissues. Embedding of transferosomal lidocaine into gel improves permeation of the drug. Moreover, stability of transferosomal vesicles is improved when they are embedded into gel dosage form. Use of certain skin permeation enhancers with transferosomal lidocaine gel is available and potentiates the permeation of the drug. This technique can serve as a potential tool for delivery of various topical drugs without altering the skin structure.
Disclosure
The authors report no conflicts of interest in this work.
References
Beydoun A, Backonja MM. Mechanistic stratification of antineuralgic agents. J Pain Symptom Manage. 2003;25(5 Suppl):S18–S30. | ||
Ben-David B, Demeo PJ, Lucyk C, Solosko D. A comparison of minidose lidocaine-fentanyl spinal anesthesia and local anesthesia/propofol infusion for outpatient knee arthroscopy. Anesth Analg. 2001;93(2):319–325. | ||
Rode A. Nanocarriers: a novel approach for enhanced drug delivery through skin. Asian J Pharm. 2018;12(1). | ||
El-Menshawe SF, Kharshom R, El Sisi A. Preparation and optimization of buccal propranolol hydrochloride nanoethosomal gel: a novel approach for enhancement of bioavailability. J Nanomed Nanotechnol. 2017;8(2):1000435. | ||
Shamma RN, Elsayed I. Transfersomal lyophilized gel of buspirone HCl: formulation, evaluation and statistical optimization. J Liposome Res. 2013;23(3):244–254. | ||
Lei W, Yu C, Lin H, Zhou X. Development of tacrolimus-loaded transfersomes for deeper skin penetration enhancement and therapeutic effect improvement in vivo. Asian J Pharm. 2013;8(6):336–345. | ||
Schmolka IR. Artificial skin. I. Preparation and properties of pluronic F-127 gels for treatment of burns. J Biomed Mater Res. 1972;6(6):571–582. | ||
Ali MF, Salem HF, Abdelmohsen HF, Attia SK. Preparation and clinical evaluation of nano-transferosomes for treatment of erectile dysfunction. Drug Des Dev Ther. 2015;9:2431–2447. | ||
Duplessis J, Ramachandran C, Weiner N, Muller D, Müller DG. The influence of lipid composition and lamellarity of liposomes on the physical stability of liposomes upon storage. Int J Pharm. 1996;127(2):273–278. | ||
Agarwal R, Katare OP, Vyas SP. Preparation and in vitro evaluation of liposomal/niosomal delivery systems for antipsoriatic drug dithranol. Int J Pharm. 2001;228(1–2):43–52. | ||
Singh P, Roberts MS. Skin permeability and local tissue concentrations of nonsteroidal anti-inflammatory drugs after topical application. J Pharmacol Exp Ther. 1994;268(1):144–151. | ||
Honeywell-Nguyen PL, Bouwstra JA. The in vitro transport of pergolide from surfactant-based elastic vesicles through human skin: a suggested mechanism of action. J Control Release. 2003;86(1):145–156. | ||
Singh S, Vardhan H, Kotla NG, Maddiboyina B, Sharma D, Webster TJ. The role of surfactants in the formulation of elastic liposomal gels containing a synthetic opioid analgesic. Int J Nanomed. 2016;11:1475–1482. | ||
Keyhanfar F, Shamsi Meymandi M, Sepehri G, Rastegaryanzadeh R, Heravi G. Evaluation of antinociceptive effect of pregabalin in mice and its combination with tramadol using tail flick test. Iran J Pharm Res. 2013;12(3):483. | ||
Gårdmark M, Höglund AU, Hammarlund-Udenaes M. Aspects on tail-flick, hot-plate and electrical stimulation tests for morphine antinociception. Pharmacol Toxicol. 1998;83(6):252–258. | ||
Field MJ, Oles RJ, Lewis AS, McCleary S, Hughes J, Singh L. Gabapentin (neurontin) and S-(+)-3-isobutylgaba represent a novel class of selective antihyperalgesic agents. Br J Pharmacol. 1997;121(8):1513–1522. | ||
Meymandi M, Sepehri G, Mobasher M. Gabapentin enhances the analgesic response to morphine in acute model of pain in male rats. Pharmacol Biochem Behav. 2006;85(1):185–189. | ||
Patel R, Singh SK, Singh S, Sheth NR, Gendle R. Development and characterization of curcumin loaded transfersome for transdermal delivery. J Pharm Sci Res. 2009;1(4):71–81. | ||
Shaji J, Lal M. Preparation, optimization and evaluation of transferosomal formulation for enhanced transdermal delivery of a COX-2 inhibitor. Int J Pharm Pharm Sci. 2014;6(1):467–477. | ||
Malakar J, Sen SO, Nayak AK, Sen KK. Formulation, optimization and evaluation of transferosomal gel for transdermal insulin delivery. Saudi Pharm J. 2012;20(4):355–363. | ||
Salem HF, Ahmed SM, Hassaballah AE, Omar MM. Targeting brain cells with glutathione-modulated nanoliposomes: in vitro and in vivo study. Drug Des Devel Ther. 2015;9:3705. | ||
Xu X, Yu Z, Zhu Y, Wang B. Effect of sodium oleate adsorption on the colloidal stability and zeta potential of detonation synthesized diamond particles in aqueous solutions. Diam Relat Mater. 2005;14(2):206–212. | ||
Mokhtar M, Sammour OA, Hammad MA, Megrab NA. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int J Pharm. 2008;361(1–2):104–111. | ||
Panwar P, Pandey B, Lakhera PC, Singh KP. Preparation, characterization, and in vitro release study of albendazole-encapsulated nanosize liposomes. Int J Nanomedicine. 2010;5:101–108. | ||
Nounou MM, El-Khordagui LK, Khalafallah NA, Khalil SA. In vitro release of hydrophilic and hydrophobic drugs from liposomal dispersions and gels. Acta Pharm. 2006;56(3):311. | ||
Mitkari BV, Korde SA, Mahadik KR, Kokare CR. Formulation and evaluation of topical liposomal gel for fluconazole. Indian J Pharm Educ Res. 2010;44(4):324–333. | ||
Wu L, Ohtani M, Takata M, et al. Magnetically induced anisotropic orientation of graphene oxide locked by in situ hydrogelation. ACS Nano. 2014;8(5):4640–4649. | ||
Dal Mas J, Zermiani T, Thiesen LC, et al. Nanoemulsion as a carrier to improve the topical anti-inflammatory activity of stem bark extract of Rapanea ferruginea. Int J Nanomed. 2016;11:4495. | ||
Walker RB, Smith EW. The role of percutaneous penetration enhancers. Adv Drug Deliv Rev. 1996;18(3):295–301. | ||
Zhao K, Singh J. Mechanisms of percutaneous absorption of tamoxifen by terpenes: eugenol, d-limonene and menthone. J Control Release. 1998;55(2–3):253–260. | ||
Ogiso T, Yamaguchi T, Iwaki M, Tanino T, Miyake Y. Effect of positively and negatively charged liposomes on skin permeation of drugs. J Drug Target. 2001;9(1):49–59. | ||
Salem HF, Ahmed SM, Omar MM. Liposomal flucytosine capped with gold nanoparticle formulations for improved ocular delivery. Drug Des Devel Ther. 2016;10:277. | ||
Williams AC, Barry BW. Penetration enhancers. Adv Drug Deliv Rev. 2012;64:128–137. | ||
Borowska K, Laskowska B, Magoń A, Mysliwiec B, Pyda M, Wołowiec S. PAMAM dendrimers as solubilizers and hosts for 8-methoxypsoralene enabling transdermal diffusion of the guest. Int J Pharm. 2010;398(1–2):185–189. | ||
Sun M, Fan A, Wang Z, Zhao Y. Dendrimer-mediated drug delivery to the skin. Soft Matter. 2012;8(16):4301–4305. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.