")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Preparation and in vitro/in vivo characterization of enteric-coated nanoparticles loaded with the antihypertensive peptide VLPVPR
Authors Sun H, Liu D, Li Y, Tang X, Cong Y
Received 16 October 2013
Accepted for publication 6 December 2013
Published 3 April 2014 Volume 2014:9(1) Pages 1709—1716
DOI https://doi.org/10.2147/IJN.S56092
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Haiyan Sun, Dong Liu, Yan Li, Xuwei Tang, Yanli Cong
Shenzhen Key Laboratory of Fermentation, Purification and Analysis, Shenzhen Polytechnic, Guangdong, People's Republic of China
Abstract: Our previous study revealed that the peptide Val-Leu-Pro-Val-Pro-Arg (VLPVPR), which was prepared using deoxyribonucleic acid recombinant technology, effectively decreased the blood pressure of spontaneous hypertensive rats; however, the effect only lasts 6 hours, likely due to its low absorption in the gastrointestinal tract. To overcome this problem, the purpose of this study was to characterize (methoxy-polyethylene glycol)-b-poly(D,L-lactide-co-glycolide)-b-poly(L-lysine) nanoparticles as in vitro and in vivo carriers for the effective delivery of VLPVPR. In our study, the VLPVPR nanoparticles were prepared using a double emulsion method, coated with Eudragit S100, and freeze-dried to produce enteric-coated nanoparticles. The optimized parameters from the double emulsion method was obtained from orthogonal experiments, including drug loading (DL) and encapsulated ratio (ER) at 6.12% and 86.94%, respectively, and the average particle size was below 100 nm. The release experiment demonstrated that the nanoparticles were sensitive to pH: almost completely released at pH 7.4 after 8 hours, but demonstrated much less release at pH 4.5 or pH 1.0 in the same amount of time. Therefore, the nanoparticles are suitable for enteric release. In vivo compared with the untreated group, the medium and high doses of orally administered VLPVPR nanoparticles reduced blood pressure for more than 30 hours, demonstrating that these nanoparticles have long-lasting and significant antihypertensive effects in spontaneously hypertensive rats.
Keywords: mPEG-PLGA-PLL, in vivo studies, Val-Leu-Pro-Val-Pro-Arg peptide, enteric-coated, nanoparticle, antihypertensive peptide
Introduction
Hypertension is defined as a sustained elevation of systolic blood pressure above 140 mmHg and/or diastolic blood pressure above 90 mmHg. Overall, the prevalence of hypertension appears to be around 30%–45% of the general population, with a steep increase with aging.1 The cause of hypertension is variable, such as increased peripheral vascular smooth muscle tone, which leads to increased arteriolar resistance and reduced capacitance of the venous system.2 Angiotensin-converting enzyme (Enzyme Commision (EC) 3.4.15.1) plays an important role in blood pressure maintenance by regulating the renin–angiotensin system. It does that by converting angiotensin I to angiotensin II, which constricts the vessels. During the past two decades, numerous physiologically active peptides have been discovered in the hydrolysates of various food proteins. Among them, antihypertensive peptides (AHPs) have received considerable attention because they are potent angiotensin-converting enzyme inhibitors with acceptable antihypertensive effects and could serve as alternative therapeutics for patients with certain hypertension.3–5 To exert their antihypertensive effects in vivo, these peptides must remain intact when absorbed across the intestinal epithelium. Our previous study revealed that the AHP Val-Leu-Pro-Val-Pro-Arg (VLPVPR), which was prepared on a large scale using deoxyribonucleic acid recombinant technology, effectively decreased the blood pressure of spontaneously hypertensive rats, but the effect only lasts 6 hours, likely because this AHP was poorly absorbed in the gastrointestinal tract.6,7
To overcome this problem, we prepared enteric-coated nanoparticles loaded with the antihypertensive peptide VLPVPR. Nanoparticles have better properties for transporting protein drugs and improved pharmacokinetic profiles in vivo because their nanoscale size helps them penetrate tissues efficiently through capillaries and epithelial linings.8,9 In addition, because of VLPVPR’s high hydrophilicity, we utilized (methoxy-polyethylene glycol)-b-poly(D,L-lactide-co-glycolide)-b-poly(L-lysine) (mPEG-PLGA-PLL) as the entrapping material. The polymer mPEG-PLGA-PLL is widely used in the preparation of microparticles because it is nontoxic, well tolerated by the human body, biodegradable, and biocompatible.10,11 The double emulsification method was utilized to encapsulate proteins in this study. AHP is better absorbed in the ileum and the large intestine than in the jejunum. Thus, a polymer that would release the drug at pH >7 would be suitable for oral AHP delivery; the polymer Eudragit S100 has this characteristic.12 When utilized to entrap VLPVPR in nanoparticles, it would be expected to protect the peptide from degradation by gastric juices and allow it to be released in regions of the gastrointestinal tract with pH >7, such as the large intestine or the colon where proteolytic enzymes are scant. The enteric-coated nanoparticles were characterized by shape (scanning electron microscopy), size (laser diffraction method), and drug loading. Their in vitro release behavior was investigated in phosphate buffer at various pH values, and the in vivo bioactivity of the nanoparticles was studied in rats.
Materials and methods
Materials
The recombinant antihypertensive peptide VLPVPR was prepared using genetic engineering technology in our laboratory (Shenzhen Key Laboratory, Shenzhen, People’s Republic of China). Eudragit S100 was purchased from Shanghai Chineway Pharmaceutical Tech Co, Ltd (Shanghai, People’s Republic of China). Poly(vinyl alcohol) (PVA) was obtained from Sigma-Aldrich (St Louis, MO, USA) with a molecular weight (MW) of 130–230 kD, and 87%–89% hydroxylated. mPEG-PLGA-PLL, with a polyethylene glycol MW of 2,000 and a glycolic acid:lactic acid (GA:LA) of 8:2, was prepared by Yourong Duan’s Lab at the Shanghai Cancer Institute, Shanghai, People’s Republic of China. Pure chromatographic acetonitrile was utilized. Other reagents were of analytical grade.
Experimental animals
Twelve-week-old male spontaneously hypertensive rats (SHR) with a body weight of 250–300 g were purchased from Shanghai SLAC Laboratory Animal Co, Ltd, Shanghai, People’s Republic of China. Each group included six rats. The rats were fed standard low-fat chow and had free access to water in a 25°C±1°C environment with 55%±5% relative humidity and regular light. Rats entered the study when their systolic blood pressure (SBP) rose above 180 mmHg. The experiments were performed after 1 week of training for the rats in the animal facility. All procedures were in accordance with the Regulations of Experimental Animal Administration issued by the Ministry of Science and Technology of the People’s Republic of China (http://www.most.gov.cn). The animal study protocols were approved by the Institutional Animal Care and Use Committee at Shenzhen Polytechnic, Shenzhen, People’s Republic of China.
Methods
Characterization of VLPVPR
The concentration of VLPVPR was determined by high-performance liquid chromatography (HPLC) (Agilent 1200 Variable Wavelength Detector; Santa Clara, CA, USA) with a C18 chromatographic column (Phenomenex Synergi; Torrance, CA, USA, 4.6 × 250 mm, 5 μm). The mobile phase was acetonitrile:water:trifluoroacetic acid 18:82:0.1.
Preparation of nanoparticles
Double-emulsify method
mPEG-PLGA-PLL was utilized to form nanoparticles using the double-emulsify method.13,14 VLPVPR (5 mg) was dissolved in 0.2 mL water to form the inner water phase. mPEG-PLGA-PLL (50 mg) was dissolved in 1 mL dichloromethane to form the oil phase. A 1% PVA solution was utilized as the outer water phase. The inner phase was added to the oil phase and emulsified with ultrasound (200 W, 5 seconds ×5) to obtain a single emulsion. This emulsion was poured into the outer water phase and emulsified with ultrasound (200 W, 5 seconds ×2) to obtain the double emulsion. With constant stirring, the double emulsion was immediately dispersed into 0.3% PVA. The mixture was stirred until the dichloromethane was removed and then was freeze dried.15 The drug loading (DL) and encapsulated ratio (ER) percentages were determined by HPLC and calculated per the following equation. The particle size was measured using a Nicomp particle sizer (380ZLS; PSS-Nicomp, Port Richey, FL, USA).

Optimization of the double emulsion method
The method was optimized in the L(9)34 orthogonal experiment (Table 1). VLPVPR was dissolved in the inner water phase. mPEG-PLGA-PLL (100 mg) was dissolved in 1 mL dichloromethane. The single emulsion was prepared by ultrasound (200 W, 5 seconds ×5). The outer water phase contained 1% PVA. The DL, ER, and particle size were determined as detailed above.
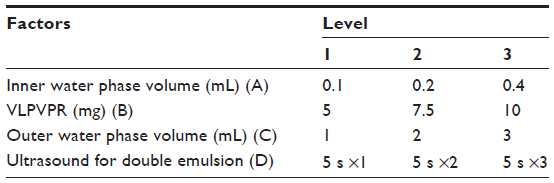 | Table 1 Orthogonal design factors and levels for the double emulsion method |
Preparation of enteric-coated nanoparticles
The nanoparticles were prepared via the optimized double emulsion method. Eudragit S100 was dissolved in dehydrated alcohol at 1 mg/mL and then was added dropwise to the mPEG-PLGA-PLL nanoparticle solution with constant stirring. The ratio of mPEG-PLGA-PLL to Eudragit S100 was 100:3.5, 100:7.0, or 100:10.5, respectively. The alcohol was removed by the decompression–volatilization method. The solution was finally freeze dried, and the DL was determined as previously described.16
In vitro release of nanoparticles
The enteric-coated nanoparticles were dispersed in 20 mL water. Four milliliters of the nanoparticle solution was sealed in dialysis tubing (MW cut off =7,000). The tube was dipped in 46 mL of 0.1 mol/L HCl or 0.05 M phosphate buffered saline (pH =4.5 or 7.4), and the release was determined at 37°C. Samples (200 μL) were removed to detect the amount of VLPVPR released from the enteric-coated nanoparticles, and the same volume of fresh medium was added. The concentration of VLPVPR was determined by HPLC.
In vivo bioactivity of nanoparticles
The SBP of the SHR was measured to confirm the in vivo bioactivity of the nanoparticles. Rats that had been given an oral dose of the peptide (400 μg VLPVPR/kg) or the nanoparticles (3.25, 6.5, and 13 mg/kg, respectively) were kept at 45°C for 3 minutes, and the SBP was measured using a tail cuff with a programmed noninvasive blood pressure controller (model ML125; AD Instruments Pty Ltd, Dunedin, New Zealand). A 0.9% (W/W) NaCl solution served as the negative control, and the antihypertensive effect of the nanoparticles dissolved in this saline solution was measured. At 0, 1, 2, 4, 6, 8, 10, 12, 16, 20, 24, 30, 36, and 48 hours after the oral administration of the nanoparticles, the SBP of the SHR was measured. At each time point, SBP was measured five times to get the average value. Data are presented as the mean ± standard deviation and were analyzed statistically using one-way analysis of variance test (StatView Software; Abacus Concepts, Inc, Piscataway, NJ, USA) with a significance level of P<0.05, followed by Scheffé’s post hoc test.
Results
Characterization of VLPVPR
The peak corresponding to VLPVPR was well separated from the other components. The signal to noise ratio (S/N) equaled 9.7 for 2 μg/mL VLPVPR. The correlation between peak area and VLPVPR concentration was greater than 0.9995 in the concentration range of 2–200 μg/mL. The precision, reproducibility, recovery, and stability were all acceptable for a quantitative method.
Optimization of the double emulsion method
The experimental data obtained from the orthogonal design are shown in Table 2. K1-K3 were the average DL and ER of nanoparticles under the various investigated conditions, and the maximum value was the optimum value. In addition, according to the largest donating rule, the factor with the largest range value (Kmax–Kmin) had the greatest effect on the preparation of nanoparticles.
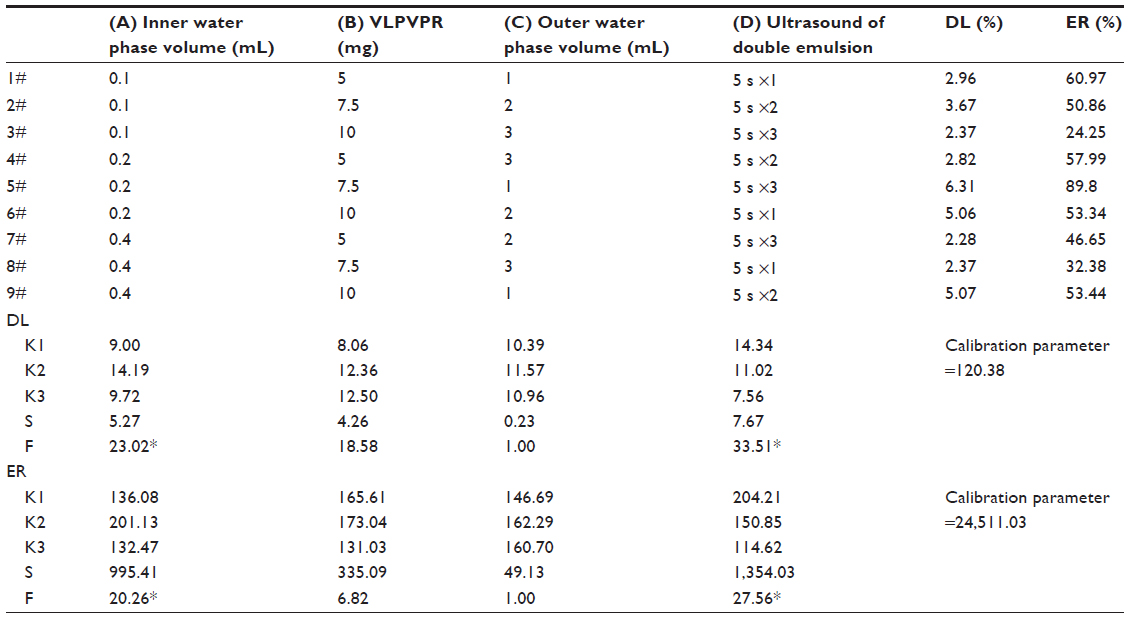 | Table 2 Results of the orthogonal experiment |
In this experiment, the particles were all smaller than 100 nm. Because the particle size was not critical for the optimization, it was not considered. The DL and ER results were analyzed according to the orthogonal experiment (Table 2). The hierarchy of the factors was D>A>B>C. Both D (ultrasound for double emulsion) and A (inner water phase volume) significantly affected DL and ER. The best combination for achieving the highest DL was A2B3C2D1, whereas A2B2C2D1 realized the highest ER.
Nanoparticle preparation methods
The nanoparticles prepared by the optimized double emulsion method had an average diameter of 75.2 nm and a narrow dispersion with PI =0.171 (Figures 1 and 2), ER =86.94%, and DL =6.12% (Table 3).
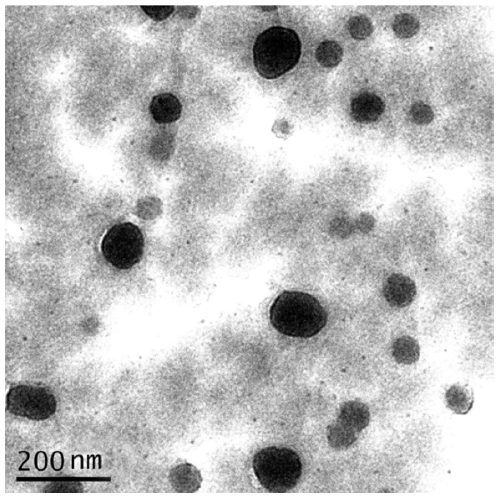 | Figure 1 Typical SEM images of mPEG-PLGA-PLL nanoparticles coated with Eudragit S100; bar, 200 nm. |
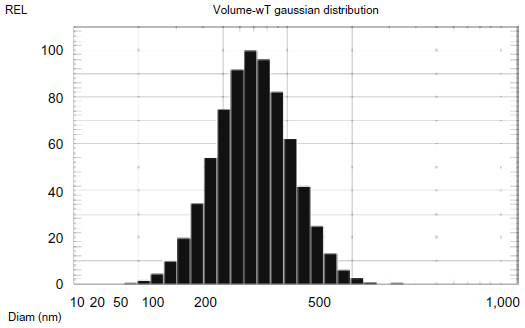 | Figure 2 Particle size of nanoparticles prepared using the double emulsion method. |
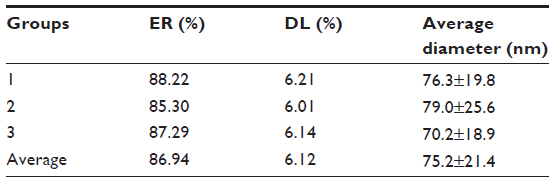 | Table 3 Characterization of nanoparticles prepared by the optimized double emulsion method |
In vitro release of nanoparticles
The release of free VLPVPR at 2 hours was approximately 80% (Figure 3), indicating that the dialysis tube did not hinder drug release. VLPVPR release from the enteric-coated nanoparticles was pH-sensitive (Figures 4–6). At pH 1.0, the nanoparticles sustained the release compared with free VLPVPR. Among the three enteric-coated nanoparticles, the third one (mPEG-PLGA-PLL:Eudragit S100 100:10.5) possessed the best pH-sensitive profile; that is, the enteric-coated nanoparticles released drug mostly in the small intestine, where pH value is about 7.4, to protect VLPVPR from degradation by proteolytic enzymes. At pH 7.4, drug release was slower than that of the free drug, whereas the drug release from the other two nanoparticles was similar at pH 4.5 and 7.4.
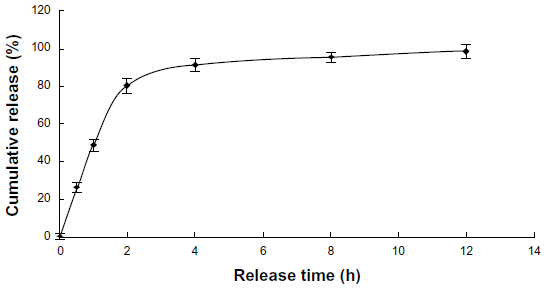 | Figure 3 Cumulative release of free VLPVPR in PBS (pH =7.4). |
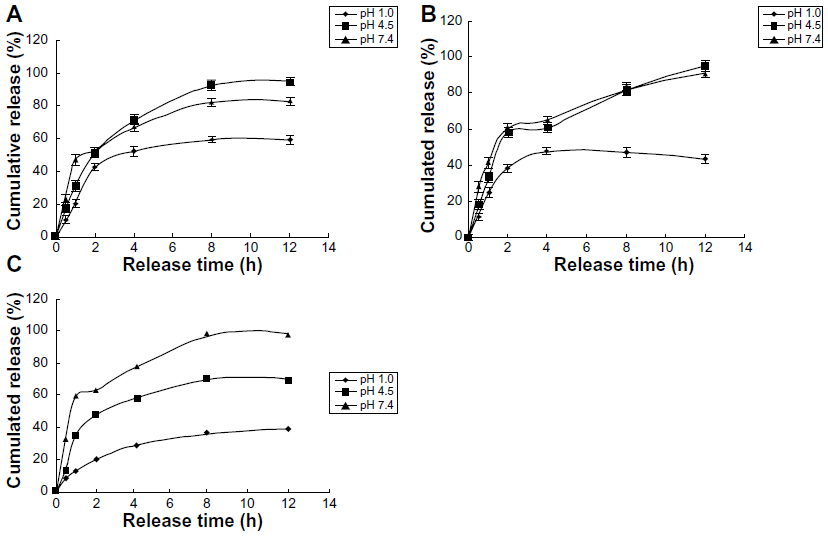 | Figure 4 Cumulative release of three enteric-coated nanoparticles in different pH media. |
In vivo bioactivity of the nanoparticles
At 0, 1, 2, 4, 6, 8, 12, 16, 20, 24, 30, 36, and 48 hours after oral administration of the recombinant AHP, the SBP of SHR was measured (Figure 5). The SBP of spontaneously hypertensive rats was dramatically decreased by recombinant AHP at an oral dose of 0.4 mg AHP/kg body weight. The antihypertensive effect was obtained 2–6 hours after oral administration, with a peak during the second hour (−43±7.9 mmHg).
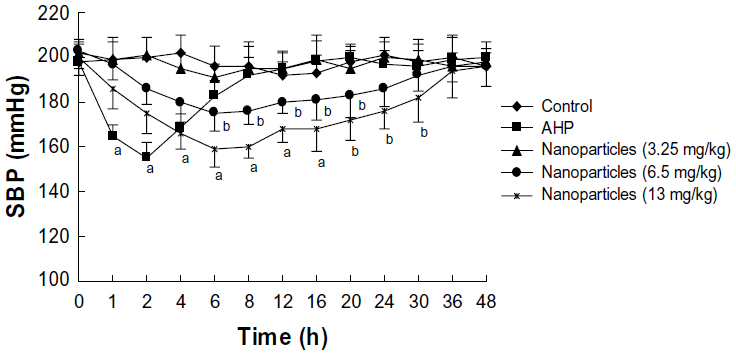 | Figure 5 Effects of a single oral dose of mPEG-PLGA-PLL nanoparticles on SBP in SHR. |
Different doses of AHP-loaded nanoparticles (3.25, 6.5, and 13 mg/kg) were given orally to SHR, and the effects were compared with the control group. As shown in Figure 5, the SBP did not significantly decrease at the 3.25 mg/kg dose. However, 6.5 mg/kg and 13 mg/kg significantly decreased SBP in a dose-dependent manner compared with the negative control group. The effect peaked at 6 hours and lasted for approximately 30 hours. The SBP did not change in the negative control rats in the 48 hours after drug administration.
Discussion
VLPVPR is a water-soluble peptide. It is stable below 0°C for 30 days, at 4°C for 7 days, at 37°C for 48 hours, and at 60°C for 24 hours. It is stable between pH 5.0 and pH 9.2. It degraded after exposure to 200 W ultrasound for 15 minutes, 400 W for 7 minutes, or 600 W for 5 minutes (data not shown). Therefore, the nanoparticles should be prepared quickly, and high temperature, extreme pH, and intense ultrasound should be avoided. Methods to prepare peptide nanoparticles include double emulsion and ionic gelation.
The emulsion-solvent evaporation method is popular for preparing PLGA nanoparticles because it yields small, uniform, round particles with a smooth surface. Commonly, hydrophobic PLGA is dissolved in an organic solvent. If the drug is also hydrophobic, it is dissolved with the PLGA. This organic phase is emulsified with an aqueous phase to make an O/W (O, oil, W, water, O/W, oil-in-water emulsion) single emulsion. If the drug is hydrosoluble, it is dissolved in an aqueous phase W1, which is emulsified in the PLGA organic solution to make a W1/O emulsion. This emulsion is emulsified in a second aqueous phase W2 to form a W1/O/W2 double emulsion.17 VLPVPR is a water-soluble drug, therefore, the double emulsion method is preferable. PLGA is a biocompatible polymer, but because it is hydrophilic, it does not allow for substantial DL of water-soluble drugs. We chose mPEG-PLGA-PLL as the polymer to form the double emulsion because the hydrophilic block in this polymer was compatible with the peptide and the hydrophobic block could form the emulsion. The ion gelation method is also commonly used to prepare peptide nanoparticles.15
The nanoparticles were designed to be enteric targeting. Therefore, the encapsulating material should dissolve in neutral or basic media and release the drug, but remain intact in acidic media to protect the drug from gastric juices. mPEG-PLGA-PLL does not possess this enteric-dissolving characteristic. The nanoparticles should be made of an enteric-dissolving material or be coated with an enteric-coating material. Eudragit S100 is a popular coating material that dissolves at pH >7.0. It is an anionic polymer synthesized from methacrylic acid and methacrylic acid methyl ester and exhibits pH-dependent solubility. Furthermore, it is liposoluble with poor peptide compatibility and does not dissolve in low polarity and volatile organic solvents such as dichloromethane; therefore, it cannot form the double emulsion to entrap VLPVPR, but can be used to coat the outside of the nanoparticles.
The characteristics of the single emulsion (W1/O) are important for the stability of the double emulsion (W1/O/W2). The drug entrapped in the single emulsion can escape via several routes: molecular diffusion, micelle transport, and disruption of the oil drops.18–20 Therefore, a stable single emulsion with a small drop size is necessary to obtain a stable double emulsion with a high DL. We decided to prepare the single emulsion by ultrasound to obtain the smallest possible drop size. We also studied factors that affected the DL and ER. As shown in Tables 1 and 2, the inner water phase volume and the ultrasound parameters for the double emulsion had a significant influence on these particular variables.
A small inner water phase volume will yield a small drop size in a single emulsion. In the optimization experiment (Tables 1 and 2), the DL and ER increased when the inner water phase volume decreased from 0.4 mL to 0.2 mL, but dropped when it decreased further. Perhaps when the inner water phase volume fell too low, the drug pressure increased and disrupted the oil drop. The level in A2 was the best. The DL increased as the drug dose increased, with the result B1<B2≈B3, but for the ER, the result was B1≈B2>B3. This factor was not as important; therefore, we chose B2.
In this study, the DL did not change as the outer water phase volume increased. The ER exhibited a trend of increasing first and then stabilizing; this may be because the ratio of the volume of the outer water phase to that of the single emulsion affected the drop size of the double emulsion. Big double emulsion drops can entrap more inner water drops, but a greater outer continuous phase is more important for stability. Furthermore, if the outer water phase is too small, the resulting double emulsion will be too viscous, which will get more broadly dispersed. The level C2 was the best. The ultrasound parameters for the double emulsion significantly impacted the DL and ER. Ultrasound that was too strong resulted in drop disruption and drug leakage. Therefore, we chose D1.
According to the DL and ER results, we chose the optimized A2B2C2D1 method, which proved to be reproducible. Because the nanoparticles had been freeze-dried before the DL and ER were measured, the results may have been artificially high due to the instability of VLPVPR.
Compared with the drug release of free VLPVPR (Figure 3), the enteric-coated nanoparticles postpone drug release at different pH values (pH 1.0, 4.5, and 7.5). Figures 4A and B showed the enteric-coated nanoparticles (mPEG-PLGA-PLL:Eudragit S100 100:3.5, mPEG-PLGA-PLL:Eudragit S100 100:7) release drug more slowly at pH 1.0 in 12 hours than that of free VLPVPR, but in 4 hours the drug release still increase to 40%. For these two kinds of enteric-coated nanoparticles, the release curves are similar at pH 4.5 and 7.4, indicating that VLPVPR can also release in the duodenum (pH 4.5) where it may be degraded by gastric juices. Based on the release results, the most suitable pH sensitivity profile occurred at the ratio of mPEG-PLGA-PLL to enteric coat of 100:10.5 (Figure 4C). The human stomach (pH 1.0) emptying time is 1–3 hours. Orally administered drugs pass quickly through the duodenum, remain for 4–6 hours in the small intestine (pH 6.8), and spend approximately 12 hours in the colon (pH 7.4). According to the release results for the enteric-coated nanoparticles, the release ratio in the first several hours was low in the pH corresponding to the stomach or small intestine. Because VLPVPR is unstable, our release experiment was only performed for 12 hours, but in vivo it would be absorbed at the same time it is released.
Until now, we did not have sufficient pharmacokinetics data on VLPVPR because it is unstable and easily degraded. Therefore, the in vivo experiments were carried out to illustrate the slow-release effect of the enteric-coated nanoparticles compared with that of free VLPVPR. The results showed that nanoparticles significantly decreased SBP in a dose-dependent manner and lasted for approximately 30 hours, while the antihypertensive effect of free VLPVPR lasts for only 6 hours (Figure 5). A grant supported by the government on how to develop and validate pretreatment methods and ultra performance liquid chromatography-mass spectrometry. methods for the determination of the antihypertensive peptide is ongoing in our lab, which will shed light on drug metabolism and pharmacokinetics.
Conclusion
This study determined the best preparation of VLPVPR enteric-coated nanoparticles. mPEG-PLGA-PLL was utilized as the entrapping material. The nanoparticles were prepared using the double emulsion method, coated with Eudragit S100, and freeze dried.
The optimized double emulsion method was obtained from orthogonal experiments. The best method was the following: VLPVPR (10 mg) was dissolved in 0.2 mL water to form the inner water phase. mPEG-PLGA-PLL (100 mg) was dissolved in 1 mL dichloromethane for the oil phase. The single emulsion was prepared by ultrasound (5 seconds ×5 times). Two milliliters of a 1% PVA solution was utilized as the outer water phase. The double emulsion was prepared by a single 5-second ultrasound burst. The emulsion was quickly dispersed into 50 mL of a 0.3% PVA solution and stirred until the dichloromethane was removed. The Eudragit S100 was dissolved in ethanol and added dropwise into the nanoparticle solution at a ratio of mPEG-PLGA-PLL:Eudragit S100 of 100:10.5. Ethanol was removed by decompression, and the solution was freeze-dried to produce the enteric-coated nanoparticles.
The release experiment confirmed the pH sensitivity of the nanoparticles. VLPVPR was almost completely released at pH 7.4 after 8 hours, but much less was released at pH 4.5 or pH 1.0 in the same amount of time. The in vivo experiments carried out in SHR shows the slow-release antihypertensive effect of the nanoparticles either. A better method to detect VLPVPR concentration in plasma is needed to help us interpret and predict metabolism and pharmacokinetics of the VLPVPR-loaded nanoparticles in rats in further study. The data demonstrated that nanoparticles significantly decreased SBP, and the antihypertensive effect lasted for approximately 30 hours, which confirmed the slow-release of the enteric-coated nanoparticles in vivo.
Acknowledgments
This study was supported by Science and Technology Planning Project of Guangdong Province (NO:2009B030803016). The project was supported by GDHVPS (2011), Science and Technology Planning Project of Guangdong Province (NO:2011B010500006) and Shenzhen Science and Technology Planning Project (NO:ZDSY20120619093923525).
Disclosure
The authors report no conflicts of interest in this work.
References
Pereira M, Lunet N, Azevedo A, Barros H. Differences in prevalence, awareness, treatment and control of hypertension between developing and developed countries. J Hypertens. 2009;27(5):963–975. | |
Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42:1206–1252. | |
Karaki H, Doi K, Sugano S, et al. Antihypertensive effect of tryptic hydrolysate of milk casein in spontaneously hypertensive rats. Comp Biochem Physiol C. 1990;96(2):367–371. | |
Yoshikawa M, Fujita H, Matoba N, et al. Bioactive peptides derived from food proteins preventing lifestyle-related diseases. Biofactors. 2000;12(1–4):143–146. | |
Iroyukifujita H, Eiichiyokoyama K, Yoshikawa M. Classification and antihypertensive activity of angiotensin i-converting enzyme inhibitory peptides derived from food proteins. J Food Science. 2000;65:564–569. | |
Liu D, Sun H, Zhang L, Li S, Qin Z. High-level expression of milk-derived antihypertensive peptide in Escherichia coli and its bioactivity. J Agric Food Chem. 2007;55(13):5109–5112. | |
Sun H, Liu D, Li S, Qin Z. Transepithelial transport characteristics of the antihypertensive peptide, Lys-Val-Leu-Pro-Val-Pro, in human intestinal Caco-2 cell monolayers. Biosci Biotechnol Biochem. 2009;73(2):293–298. | |
Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55(3):329–347. | |
Yuan W, Liu Z. Controlled-release and preserved bioactivity of proteins from (self-assembled) core-shell double-walled microspheres. Int J Nanomedicine. 2012;7:257–270. | |
Yin P, Wang Y, Qiu Y, et al. Bufalin-loaded mPEG-PLGA-PLL-cRGD nanoparticles: preparation, cellular uptake, tissue distribution, and anticancer activity. Int J Nanomedicine. 2012;7:3961–3969. | |
Liu P, Wang H, Wang Q, et al. cRGD conjugated mPEG-PLGA-PLL nanoparticles for SGC-7901 gastric cancer cells-targeted delivery of fluorouracil. J Nanosci Nanotechnol. 2012;12(6):4467–4471. | |
Jain D, Panda AK, Majumdar DK. Eudragit S100 entrapped insulin microspheres for oral delivery. AAPS PharmSciTech. 2005;6(1):E100–E107. | |
Yin Y, Chen D, Qiao M, Wei X, Hu H. Lectin-conjugated PLGA nanoparticles loaded with thymopentin: ex vivo bioadhesion and in vivo biodistribution. J Control Release. 2007;123(1):27–38. | |
He W, Jiang X, Zhang ZR. Preparation and evaluation of poly-butylcyanoacrylate nanoparticles for oral delivery of thymopentin. J Pharm Sci. 2008;97(6):2250–2259. | |
Xu D, Wu F, Chen Y, Wei L, Yuan W. pH-sensitive degradable nanoparticles for highly efficient intracellular delivery of exogenous protein. Int J Nanomedicine. 2013;8:3405–3414. | |
Zheng AP, Wang KS, Hao R. Study on preparation release in vitro and biological activity of thymopentin-loaded pH-sensitive chitosan nanoparticles for oral administration. Chin Pharm J. 2007;42:679–685. | |
Tewes F, Munnier E, Antoon B, et al. Comparative study of doxorubicin-loaded poly(lactide-co-glycolide) nanoparticles prepared by single and double emulsion methods. Eur J Pharm Biopharm. 2007;66(3):488–492. | |
Omotosho JA, Whateley TL, Florence AT. Methotrexate transport from the internal phase of multiple w/o/w emulsions. J Microencapsul. 1989;6(2):183–192. | |
Ficheux M-F, Bonakdar L, Leal-Calderon F, Bibette J. Some stability criteria for double emulsions. Langmuir. 1998;14(10):2702–2706. | |
Garti N, Magdassi S, Whitehill D. Transfer phenomena across the oil phase in water-in oil-in water multiple emulsion evaluated by Coulter Counter I. Effect of emulsifier 1 on water permeability. J Colloid Interface Sci. 1985;104(2):587–591. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.