")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Preparation and characterization of Tamoxifen citrate loaded nanoparticles for breast cancer therapy
Authors Maji R, Dey NS, Satapathy BS, Mukherjee B , Mondal S
Received 5 March 2014
Accepted for publication 14 April 2014
Published 25 June 2014 Volume 2014:9(1) Pages 3107—3118
DOI https://doi.org/10.2147/IJN.S63535
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Ruma Maji, Niladri Shekhar Dey, Bhabani Sankar Satapathy, Biswajit Mukherjee, Subhasish Mondal
Department of Pharmaceutical Technology, Jadavpur University, Kolkata (Calcutta), India
Background: Four formulations of Tamoxifen citrate loaded polylactide-co-glycolide (PLGA) based nanoparticles (TNPs) were developed and characterized. Their internalization
by Michigan Cancer Foundation-7 (MCF-7) breast cancer cells was also investigated.
Methods: Nanoparticles were prepared by a multiple emulsion solvent evaporation method. Then the following studies were carried out: drug-excipients interaction using Fourier transform infrared spectroscopy (FTIR), surface morphology by field emission scanning electron microscopy (FESEM), zeta potential and size distribution using a Zetasizer Nano ZS90 and particle size analyzer, and in vitro drug release. In vitro cellular uptake of nanoparticles was assessed by confocal microscopy and their cell viability (%) was studied.
Results: No chemical interaction was observed between the drug and the selected excipients. TNPs had a smooth surface, and a nanosize range (250–380 nm) with a negative surface charge. Drug loadings of the prepared particles were 1.5%±0.02% weight/weight (w/w), 2.68%±0.5% w/w, 4.09%±0.2% w/w, 27.16%±2.08% w/w for NP1–NP4, respectively. A sustained drug release pattern from the nanoparticles was observed for the entire period of study, ie, up to 60 days. Further, nanoparticles were internalized well by the MCF-7 breast cancer cells on a concentration dependent manner and were present in the cytoplasm. The nucleus was free from nanoparticle entry. Drug loaded nanoparticles were found to be more cytotoxic than the free drug.
Conclusion: TNPs (NP4) showed the highest drug loading, released the drug in a sustained manner for a prolonged period of time and were taken up well by the MCF-7 breast cancer cell line in vitro. Thus the formulation may be suitable for breast cancer treatment due to the good permeation of the formulation into the breast cancer cells.
Keywords: polylactide-co-glycolide nanoparticle, PLGA, breast cancer, multiple emulsion
Introduction
About one-fifth of cancer patients suffer from breast cancer worldwide.1 Various chemotherapeutic agents are used to treat the breast cancer. The existing anticancer agents do not greatly differentiate between the cancerous and normal cells, leading to systemic toxicity and adverse effects. This greatly limits the maximum permissible dose of the drug. Drug permeation into the cancer cells from the conventional formulation is very poor due to less distribution and quick elimination. The extensive distribution and rapid elimination from targeted organs result in a greater requirement of the drug by the tissue, which causes undesirable toxicity as well as being economically unsound.2 Polymeric nanoparticles play an important role in delivering such kinds of chemotherapeutic agents in a controlled manner. Delivering the drugs through the nanoparticles makes it possible to achieve the desired concentration of drug in the specific site, thus minimizing the side effects and reducing the toxicity, dose dumping, etc. Polylactide-co-glycolide (PLGA) is one of the polymers approved by the US Food and Drug Administration and European Medicine Agency for various kinds of drug delivery system in humans due to its biodegradability and biocompatibility.3,4 This is a copolymer of lactic acid and glycolic acid and these two monomers are endogenous compounds and easily metabolized by the body via the Kreb’s cycle.5 Depending on the molecular weight and copolymer ratio, the degradation time of PLGA can vary from several months to years.3,6 Different anticancer drugs, including Tamoxifen citrate, were loaded in the PLGA nanoparticles by many researchers.7–10 Tamoxifen citrate, an antiestrogenic compound, is the first choice for hormonal treatment of breast cancer in both post and premenopausal women for the last few decades. It is often used as an adjuvant therapy following primary treatment of early stage breast cancer. Depending upon the dose and tissue, Tamoxifen citrate can act as an antiestrogenic or as an estrogenic agent. For breast cancer it shows an antiestrogenic effect, and on the uterus it shows an estrogenic effect. Depending upon the dose and the concentration it has several side effects, such as endometrial carcinoma for postmenopausal women. Other side effects include liver cancer, venous thrombosis, pulmonary emboli, and an ocular effect includes retinopathy and corneal opacities.11–14
To overcome such severe side effects, in our research work we have mainly concentrated on the parenteral sustained release delivery of Tamoxifen citrate in nanoparticles so that they can penetrate in the tumor tissue and can be taken up well by endocytosis into the affected cells.
Materials and methods
Tamoxifen citrate (Sigma-Aldrich Co., St Louis, MO, USA), polyvinyl alcohol (PVA, MW 125,000; S.D. Fine Chem. Pvt. Ltd., Mumbai, India), PLGA (MW 50,000–75,000; lactide-co-glycolide ratio 85:15; Sigma-Aldrich Co.), hydroxypropyl-β-cyclodextrin (HPβCD; HiMedia Laboratories, Mumbai, India) and fluorescein isothiocyanate 98% (FITC) (HiMedia Laboratories) were used in the study. Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum and tetrazolium dye 3-(4,5-dimers dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were obtained from Sigma-Aldrich Co. All other chemicals used were of analytical reagent grade.
Preparation of nanoparticles
Tamoxifen citrate nanoparticles (TNPs) were prepared by a multiple-emulsion solvent evaporation method, with some modifications of the reported methods.15–17 The compositions of the different formulations are given in Table 1. Tamoxifen citrate and subsequently PLGA (85:15) (~200 mg) were dissolved in 0.5 mL of methanol and 1.5 mL of dichloromethane mixture (phase 1). PVA was dissolved in water (2.5% weight per volume [w/v]) (phase 2), and 0.5 mL of phase 2 was added drop-wise into phase 1 with homogenization at an optimized speed (22,500 rpm) using a high-speed homogenizer (IKA Laboratory Equipment, Model T10B Ultras-Turrax, Staufen, Germany). The prepared primary emulsion was then added slowly into 75 mL of 1.5% (w/v) PVA solution (phase 3) with a continuous homogenization (22,500 rpm), which produced a secondary emulsion. The secondary emulsion was then placed on a magnetic stirrer and stirred overnight for evaporation to remove organic solvent and solidification of the particles. The nanoparticles were then first separated by centrifugation at 5,000 rpm for 5 minutes to separate larger particles and then the supernatant was collected and recentrifuged at 15,000 rpm for 45 minutes. The solid particles, thus separated, were resuspended in Milli-Q water (Millipore Corp., Billerica, MA, USA), and centrifuged to wash the particles to remove the excess PVA attached on the surface of the nanoparticles and to remove the free drug.15 The washing was repeated three times. The separated nanoparticles were frozen at -40°C and lyophilized (Laboratory Freeze Dryer, Instrumentation India Ltd., Kolkata, India) for 7–8 hours to obtain a solid product. The product obtained after lyophilization was kept overnight in a desiccator for the removal of the remaining moisture, then the lyophilized samples were stored in an airtight container at 4°C.
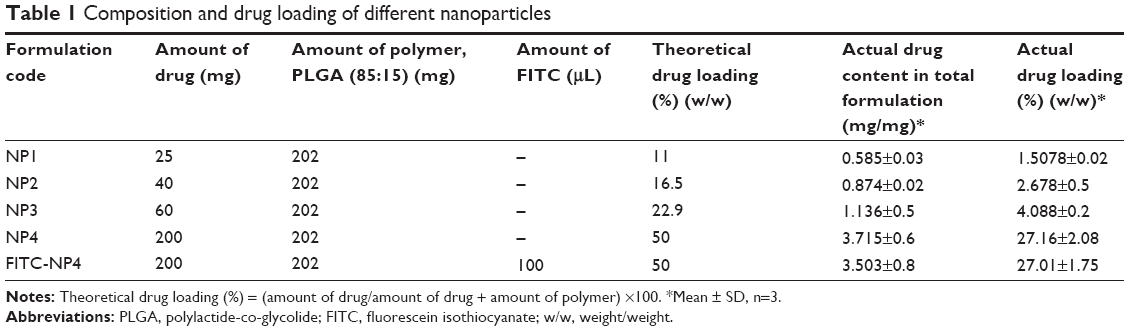 | Table 1 Composition and drug loading of different nanoparticles |
FITC was used as a fluorescent marker to visualize TNPs. A stock solution of FITC (0.4% w/v) in ethanol: chloroform (1:3) was prepared, and 100 μL of this stock solution was added into phase 1 during the emulsification process, when necessary.
Drug-excipients interactions by Fourier transform infrared spectroscopy
A Fourier transform infrared (FTIR) spectroscope (Magna-IR 750, Series II, Nicolet Instruments Inc., Madison, Wisconsin, USA) was used to obtain FTIR spectra of Tamoxifen citrate, PLGA, PVA, their physical mixture (1:1), and the prepared lyophilized formulation with and without the drug. The sample for analysis was mixed with potassium bromide (1:100 ratio) and compressed into a pellet by using a hydraulic press. The pellet was scanned under the FTIR spectroscope.
Physicochemical characterization of nanoparticles
Determination of drug loading
For the determination of drug loading, an accurately weighed amount of TNPs (2 mg) was placed into a centrifuge tube, and 2 mL of 5% SLS-NaOH solution was added. It was continuously shaken for 3–4 hours at 37°C in an incubator shaker. The dispersed phase was separated from the continuous phase by centrifugation. Then the supernatant was collected and the released drug was assayed spectrophotometrically at 278 nm. The percentage of drug loading was calculated using Equation 1:
|
|
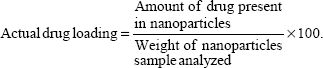
Surface morphology and particle size measurement using field emission scanning electron microscopy
Particle size and external morphology of the nanoparticles were studied by field emission scanning electron microscopy (FESEM) (Model-JSM-6700F; JEOL, Tokyo, Japan). The samples were coated with platinum before observation at an acceleration voltage of 5 kV.
Average particle sizes, polydispersity indices, and zeta potentials of different formulations were determined by a dynamic light scattering method using Data Transfer Assistance (DTA) software by Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK) at 25°C. Samples were diluted with Milli-Q (Millipore Corp.) water before measurement.
Transmission electron microscopy
The morphology and drug distribution in the experimental nanoparticles were examined by the transmission electron microscope (TEM) (FEI type FP5018/40 Tecnai G2 Spirit Bio TWIN). The nanoparticle suspension in Milli-Q water was dropped on standard carbon coated copper grid (mesh) and air dried for 5 hours. TEM images were taken and analyzed.
Drug release study
Drug release from the nanoparticles was studied in 1% HPβCD in phosphate buffered saline pH 7.4. Five milligrams of each formulation were taken in separate tubes, and 1.5 mL of the required medium was added separately in each tube. The samples were kept at 37°C in an incubator and were shaken slowly at 120 rotation/minute. After the scheduled time intervals, the tubes were centrifuged and 0.5 mL of supernatant was collected. The same volume of the respective fresh medium was replaced in the sample tube and incubated under the same condition as mentioned above. The collected samples were suitably diluted, if required, and analyzed spectrophotometrically at 234 nm.17
Energy dispersive X-ray analysis
Encapsulation of the drug within the nanoparticles was confirmed by energy dispersive X-ray (EDX) analysis. It is a technique by which the elemental composition of the sample can be identified. The EDX analysis system works as an integrated feature of a scanning electron microscope (JSM 60, JEOL, Tokyo, Japan). Briefly, lyophilized formulations were spread on the metal stub and platinum coating was carried out. The samples were then examined under a scanning electron microscope. From the samples or from different areas of the sample, the elemental compositions were determined.
Cellular uptake study
Confocal laser scanning microscopy was used to visualize the uptake of the polymeric nanoparticles within the cancer cells. For fluorescence imaging of cellular uptake, MCF-7 cells (at 104 cells/mL) were cultivated for 24 hours on cover slips in six well culture plates (3 mL/well). TNP (NP4) suspensions, 50 μl/mL and 100 μl/mL, were then added to the cell culture medium at a concentration of 300 μg/mL. The cells were washed three times after incubation for 3 hours and then fixed using 4% paraformaldehyde aqueous solution. After fixing for 15 minutes, they were rinsed with phosphate buffered saline (pH 7.4) solution. After that the cover slips were taken out carefully and placed on the slide and air-dried. Finally, they were observed using a confocal laser scanning microscopy system (Andor Spinning Disc Confocal Microscope; Andor Technology Ltd., Belfast, Northern Ireland, UK).18
MTT assay for in vitro cell viability studies
MCF-7 cells were cultured in DMEM without phenol red and supplemented with 10% fetal bovine serum. The cell culture medium was maintained at 37°C in a humidified incubator containing 5% CO2 atmosphere. Trypsinized confluent cell monolayers were grown (75%–80%) and the cells in the exponentially growing phase were used for cytotoxicity experiments.
The cytotoxicity study of TNPs (NP4) was investigated in MCF-7 cells using the MTT assay method.19 The cytotoxicity of the nanoparticles was determined after 48 hours incubation with MCF-7 cells. To determine the cell cytotoxicity/viability, the cells were plated at a density of 5×103 cells/well (optimal seeding density) in 96 well plates and the plate was kept at 37°C in 5% CO2 atmosphere in a CO2 incubator (Model MCO-15AC; Sanyo Electric Biomedical Co. Ltd., Osaka, Japan). After 12 hours of incubation, the medium in the wells was replaced by a fresh medium containing nanoparticles (added to the medium just before its incorporation in the well) with varying concentrations. After 48 hours, MTT dye solution was added to each well. The incubation was continued for a further 4 hours at 37°C and 5% CO2 for exponentially growing cells. Then the medium in each well containing unbound MTT and death cells was removed by suction. The formazan crystals were solubilized with 100 μL dimethylsulfoxide, and the solution was vigorously mixed to dissolve the reacted dye. All the experiments were performed in triplicate. The absorbance of each well was read on a microplate reader (multimode plate reader, SpectraMax M5; Molecular Devices, CA, USA) at 540 nm. Two different experimental control media, one containing nanoparticles without a drug and the other containing a free drug, were used. The cell viability (%) of the drug containing nanoparticles related to the control wells containing nanoparticles without a drug and a free drug respectively, was calculated by absorbance of test sample/absorbance of the control sample ×100.
Results
FTIR study
The interactions, if any, between the drug and the polymers (PLGA and PVA), were investigated by FTIR spectroscopy. All the important peaks (Figure 1, A–G) of Tamoxifen citrate were found to exist namely, ketonic group at 1,741 cm−1, amine (N-H bend) at 1,588 cm−1, phenyl ring substitution at 768 cm−1 to 707 cm−1, methyl group at 1,306 cm−1, amine (C-N stretch) at 1,044 cm−1, at 844 cm−1 for aromatic ring out of plane bend in the physical mixture of the drug and the excipients. However, few minor shiftings of peaks of the excipients were detected in the wave numbers from 940 cm−1 to 920 cm−1 responsible for alkane, from 3,406 cm−1 to 3,402 cm−1 responsible for benzene ring or substituted benzene, and at 3,435 cm−1 responsible for the OH group. The shifting of such peaks might have taken place due to the weak physical interactions such as formation of weak H-bonding, van der Waals’ force of attraction, dipole–dipole interaction, etc, which could lead to the formation of a spherical structure of the nanoparticles. The blank formulation and the formulation with the drug had similar peaks, indicating that there was no free drug at the nanoparticle surface.
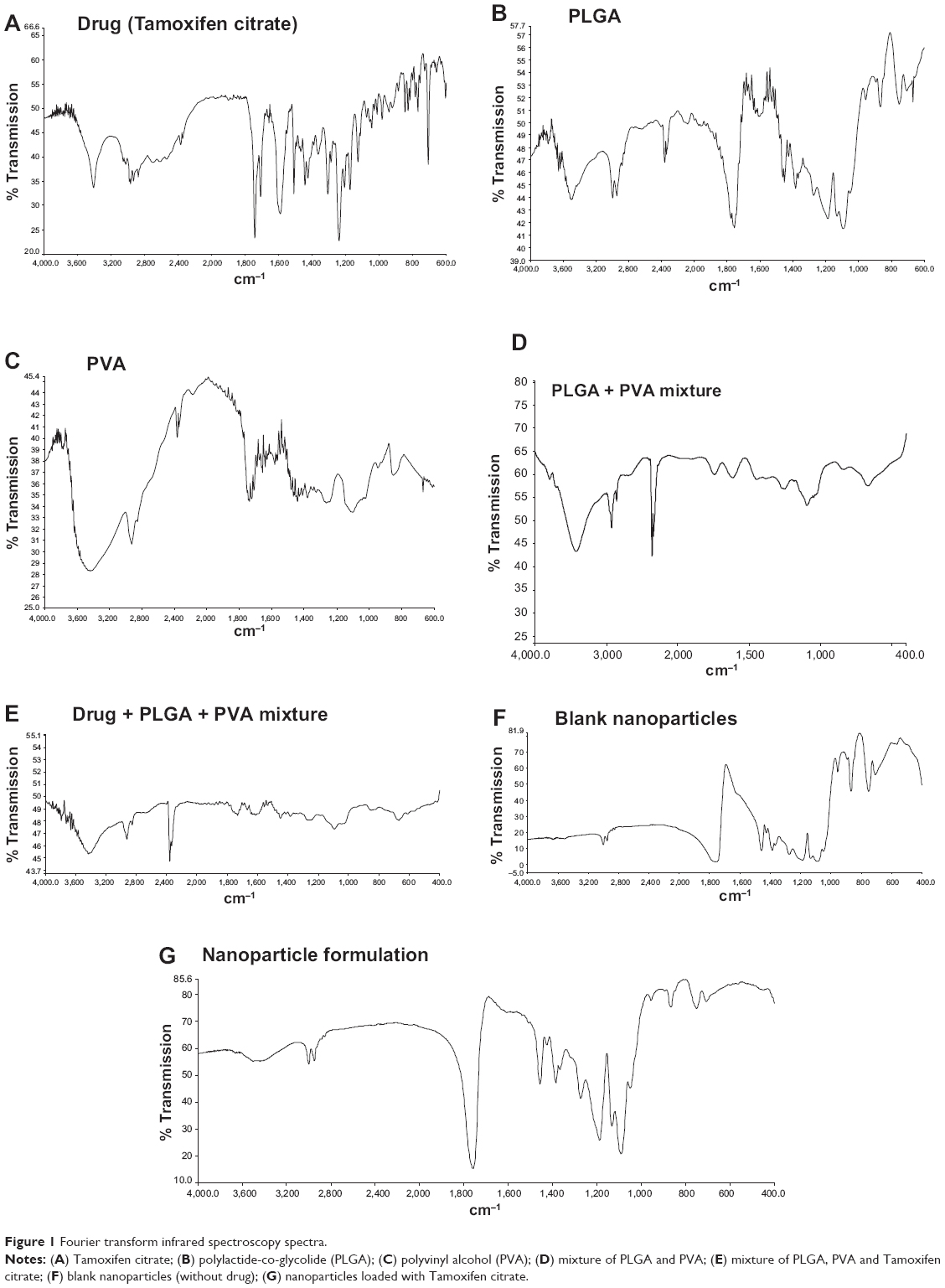 | Figure 1 Fourier transform infrared spectroscopy spectra. |
Drug loading
Drug loading of TNPs varied from 1.5%±0.02%−27.16%±2.08% (Table 1). It was found to be maximum for NP4 and minimum for NP1. Theoretical drug loading of different formulations, NP1 to NP4, varied from 11%, to 50%, respectively. Drug loading was found to be directly proportional to the amount of the drug added during the preparation of the different formulations up to an approximate drug: PLGA ratio of 1:3. However, at a 1:1 (drug:PLGA) ratio, drug loading was found to be enhanced seven times more, than drug loading at a 1:3 ratio.
Determination of particle morphology of different formulations using FESEM
FESEM photographs of different formulations are shown in Figure 2. Prepared nanoparticles were submicron in size, spherical in shape, and with a smooth surface.
 | Figure 2 Field emission scanning electron microscopic images. |
Determination of particle size and zeta potential
Particle sizes and zeta potentials of different formulations are shown in Figure 3 and Figure 4 respectively. The average particle sizes of different formulations were (Table 2) 242.5 nm, 331.8 nm, 375.6 nm, 382 nm, with the polydispersity indices of 1.00, 0.143, 0.016, 0.611 for the formulations from NP1 to NP4 respectively. The zeta potential of different formulations had values from −10.5 mV to −16.5 mV, indicating the value was decreasing with the increasing amount of drug incorporation in the formulation.
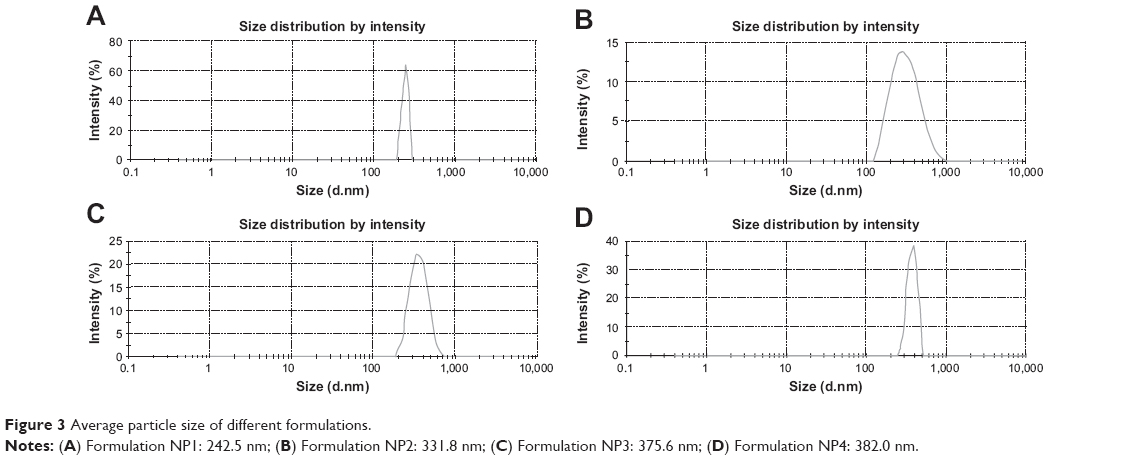 | Figure 3 Average particle size of different formulations. |
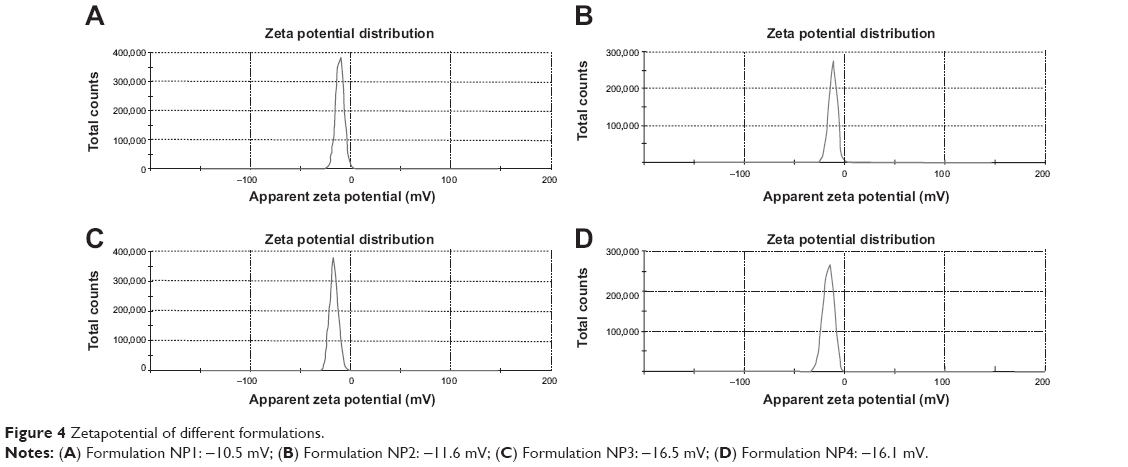 | Figure 4 Zetapotential of different formulations. |
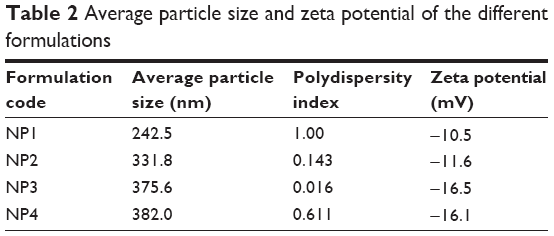 | Table 2 Average particle size and zeta potential of the different formulations |
Transmission electron microscopy
The morphology and size of nanoparticles obtained were examined by TEM. TEM photographs of the prepared nanoparticles (Figure 5) show that there was homogeneous molecular distribution of the drug in the polymer based nanoparticles and the drug was not distributed in the particulate form. There was no diffraction of transmission of electrons through the particles and that is why uniform dark particles were seen without any spots. Spotted particles support the presence of a drug in particulate form rather than its distribution in molecular form.
 | Figure 5 Transmission electron scanning microscopic images of experimental formulation NP4. |
EDX study
EDX analysis showed weight % and atomic % of various elements (C, O, and N) in various nanoparticle formulations (Figure 6 and Table 3). The weight % of C, O, and N in TNPs was 41.09%, 26% and 32.9% respectively; and the values for blank nanoparticles were 26.23%, 48.84%, and 24.92% respectively. The atomic % of C, O, and N in TNPs was 46.26%, 21.98%, and 31.76% respectively; in blank nanoparticles it was 31.13%, 43.51%, and 25.36% respectively. The differences in values of weight % and atomic % of elements were due to the presence of Tamoxifen citrate in the TNPs.
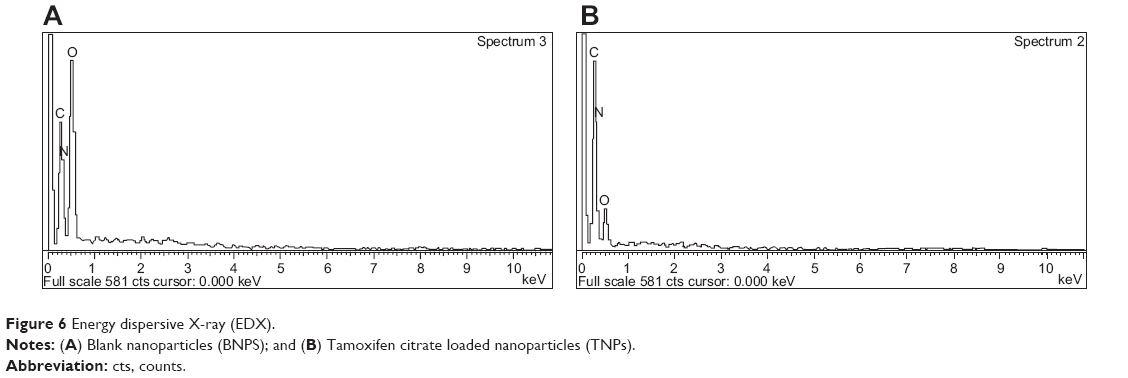 | Figure 6 Energy dispersive X-ray (EDX). |
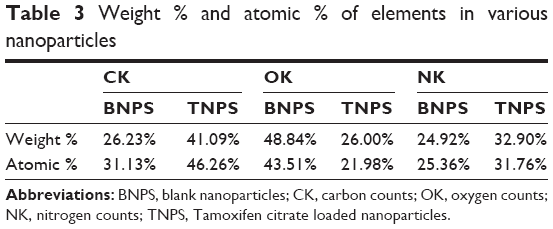 | Table 3 Weight % and atomic % of elements in various nanoparticles |
Drug release study
A drug release study was carried out to understand in vitro drug release pattern from the formulations. A sustained drug release pattern was observed. After 60 days of in vitro drug release, variable amounts (in terms of percentages) (mean ± SD; n=3)namely, 83.76%±0.43%, 55.21%±0.77%, 40.69%±0.11%, and 9.47%±0.1% of Tamoxifen citrate were released from NP1, NP2, NP3, and NP4, respectively (Figure 7). To evaluate the drug-release kinetic patterns, drug release data were assessed using zero order, first order, Korsmeyer–Peppas, and Higuchi kinetic models.20 Calculated R2 values for the kinetics were tabulated (Table 4). The corresponding plot (log cumulative percent drug release versus log time) (data not shown) for the Korsmeyer–Pappas equation indicates a good linearity (R2=0.9976, 0.9652, 0.9721, 0.9736) for NP1 to NP4, respectively. The release exponents (n-value) for the formulations, (NP1 to NP4) were 1.09, 0.914, 0.901, and 0.852, respectively.
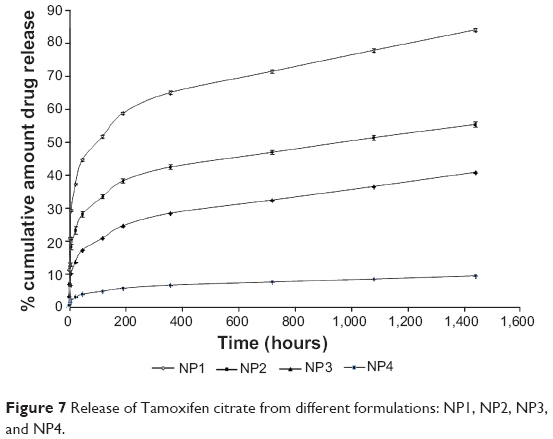 | Figure 7 Release of Tamoxifen citrate from different formulations: NP1, NP2, NP3, and NP4. |
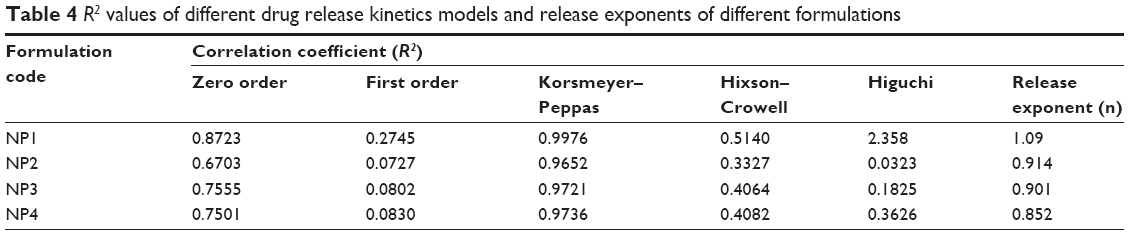 | Table 4 R2 values of different drug release kinetics models and release exponents of different formulations |
Cellular uptake study
To investigate the cellular uptake of the nanoparticles in MCF-7 cells, a short-term in vitro particle endocytosis test was carried out using FITC-TNPs. Figure 8 shows that TNPs penetrated the cell membrane and were distributed in the cytoplasm, but not in the nuclei. Moreover, the green fluorescent dots in the samples increased with the increasing concentrations of the nanoparticles. These images have demonstrated concentration dependent endocytosis of nanoparticles in MCF-7 cells.
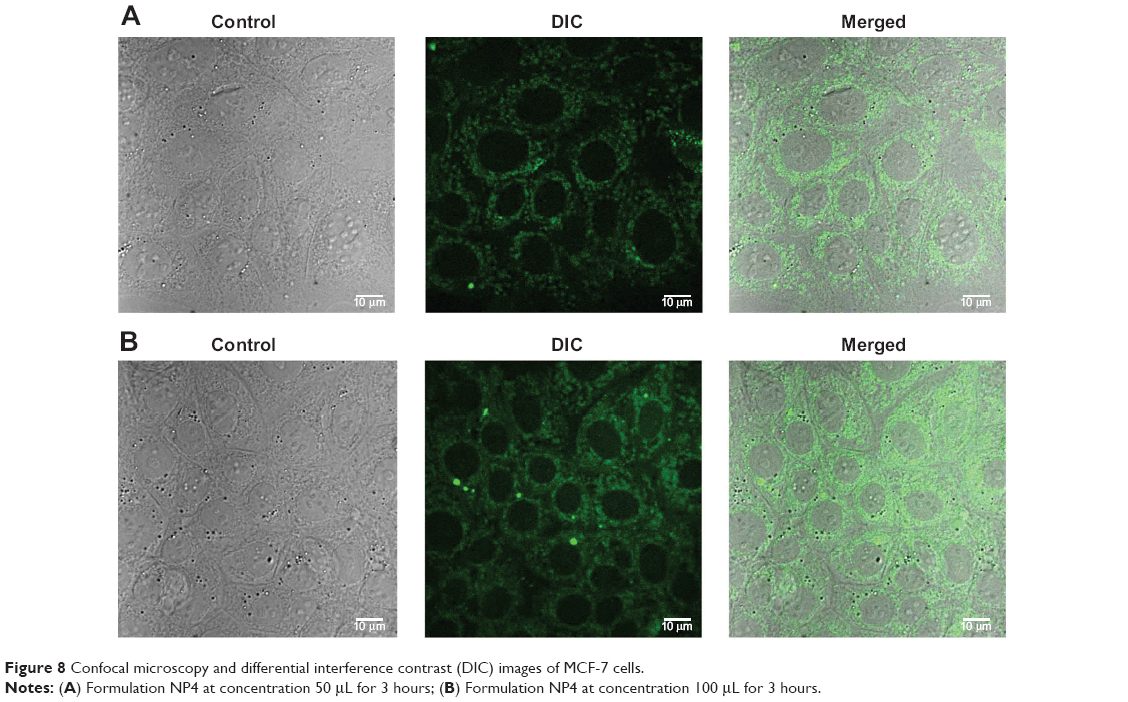 | Figure 8 Confocal microscopy and differential interference contrast (DIC) images of MCF-7 cells. |
In vitro cytotoxic assay
The proliferation/viability of MCF-7 cells was assessed by MTT assay after 48 hours of incubation with the free drug and nanoparticles with or without the drug, respectively (Figure 9). The cytotoxic effect of nanoparticles increased with an increase in Tamoxifen citrate concentration. The toxicity of Tamoxifen citrate increased as the drug concentration increased from 12.5 μM to 200 μM. The toxic effect of Tamoxifen citrate markedly decreased the cell, viability from 70.13% to 21.81%. In this cytotoxicity test, TNPs caused more death of viable cells than Tamoxifen citrate alone (free drug). Again, nanoparticles without the drug failed to produce any toxicity to viable cells and exhibited viability of 99.11%. This increased toxicity may be due to the preferential uptake of nanoparticles than that of the free drug (Tamoxifen citrate).
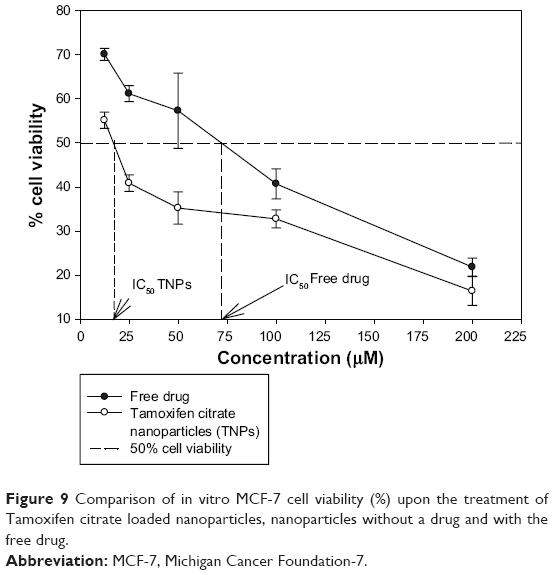 | Figure 9 Comparison of in vitro MCF-7 cell viability (%) upon the treatment of Tamoxifen citrate loaded nanoparticles, nanoparticles without a drug and with the free drug. |
Discussion
When FTIR spectra of the physical mixture of Tamoxifen citrate and the excipients were compared, no shifting of predominant peaks of the drug and the excipients was found, suggesting no chemical interaction had taken place. However, some physical interactions between the drug and the mixture of the excipients were observed. This might be due to the formation of weak bonds such as weak hydrogen bonding, van der Waals’ force of attraction, dipole–dipole interaction, etc. The nonreactive nature of PLGA with the Tamoxifen citrate has been reported earlier also.14,21,22 Absence of the peaks of the drug in the FTIR spectrum for the formulations indicates that no free drug was available on the nanoparticle surface. This could be due to molecular distribution of the drug in the polymeric molecular scaffold in the nanoparticles.
In the drug loading study, increasing the amount of the drug in the formulations was found to increase drug loading. This may be because of the ability of the polymer matrix to accommodate a large amount of Tamoxifen citrate molecules in the polymeric network until it reached its saturation point. However, the saturation point (that is the maximum amount of drug molecules which can be accommodated in the polymer matrix of a definite quantity) has not been determined in the present study.
FESEM images of the prepared nanoparticles show that all the particles prepared were spherical in shape, and had a smooth surface. The particles were found to be in submicron size with a relatively narrow range of distribution as supported by the PDI value. The particle size obtained from FESEM was further supported by the particle size obtained from the dynamic light scattering method. The average particle sizes of the formulations varied from 242 nm to 382 nm.
The zeta potential of the different batches varies from −10.5 mV to −16.5 mV. The zeta potential describes the nature of the electrostatic potential at the surface of a particle. An absolute zeta potential value of −30 mV to +30 mV suggests that the particles would remain in a suspended state for a longer period and are not susceptible to quick agglomeration in the liquid state.23,24 Thus, zeta potential values of the experimental TNPs suggest that the prepared nanoparticles should be stored in a lyophilized state and should be reconstituted only before injecting.
TEM photographs of the nanoparticles confirmed their morphology, which reveals that homogeneous and molecular drug distribution was observed in the nanoparticles and the drug was not distributed in particulate forms.
In EDX analysis, the difference in values of weight % and atomic % of elements was the proportional increase of the elements due to the presence of Tamoxifen citrate in the nanoparticles. The study suggests that Tamoxifen citrate was encapsulated in the nanoparticles.
Tamoxifen citrate release was in a sustained manner over a period of 60 days from all the experimental formulations. In the NP1 case, maximum drug release was seen 83.76%±0.43% over the 60 days and in the NP4 case, the drug release was minimal during the said period. Much slower drug diffusion from the formulations might be responsible for that. Drug release kinetics of the different formulations were best fitted for the Korsmeyer–Peppas equation. When the release exponent n value is ≤0.43, it is Fickian release. The n-value between 0.43 and 0.85 is defined as non-Fickian release. When n is ≥0.85, it is case-II transport.25 In this study n-values of all the four formulations (ie, NP1, NP2, NP3 and NP4), were greater than 0.85. Interestingly, particles with less average diameter in the nanosize range were found to release the drug in a more sustained manner. This suggests that all the experimental formulations had greater affinity to obey the Korsmeyer–Peppas kinetic model which involves a case II transport (relaxation-controlled release) mechanism. More linearity (as assessed by R2 values) of drug release data toward Korsmeyer–Peppas kinetics implies anomalous diffusion controlled drug release by more than one process.26 Due to long-term slow degradation of PLGA, release of drug molecules follows a coupling of diffusion and erosion mechanism for PLGA degradation by slow hydrolysis of ester linkage by three phases.27 The three predominant sequences by which PLGA degrades in vivo are: random polymeric chain scission causing a decrease in molecular weight without much loss of molecular weight along with the formation of soluble monomers; a decrease in polymeric molecular weight with a rapid loss of mass due to the formation of oligomers and soluble monomers; and the conversion of soluble oligomers into soluble monomers causing complete solubilization. This ultimately causes anomalous drug diffusion.
The cellular uptake of nanoparticles by MCF-7 breast cancer cells is influenced by nanoparticle shape, size, surface properties, and concentration of nanoparticles in the medium, incubation time, and temperature, etc.28 In the present study, nanoparticles uptake by MCF-7 cells was good. Localization of TNPs was in the cytoplasm but not in the nucleus. Further, the uptake was found to be dependent on the nanoparticle concentration in the study MCF-7 cellular media of the experiment of endocytosis in vitro.
The cytotoxicity of TNPs was more than that of Tamoxifen citrate alone. The possible mechanism underlying the enhanced efficacy of TNPs against MCF-7 may include the enhanced intracellular drug accumulation by nanoparticle uptake.29–31 However, the advantage of TNPs over Tamoxifen citrate free drug is that a single dose of TNPs will provide a much longer drug action (sustained) as compared to a single dose of free drug and may provide passive targeting due to the enhanced permeability and retention effect as reported earlier.32
Some literature has shown that TNPs have a greater potential than the free drug, however the present study is not just a similar mimicking document. The present study is predominantly different from those available reports.14,22,33 Mirzajani et al14 and Cirpanli et al33 have used a completely different variety of PLGA polymer in which the lactide-glycolide ratio was 50:50. In the present study, we used PLGA of lactide-glycolide ratio 85:15. Cirpanli et al33 used a completely different technique (nanoprecipitation method) than the multiemulsion solvent evaporation method used here to prepare the nanoparticles. Further, Cirpanli et al showed PLGA (50:50) Tamoxifen nanoparticles with positive zeta potentials, which have been reported to be cleared from the blood very quickly as compared to the nanoparticle with negative zeta value. In our study, the prepared nanoparticles had negative zeta potentials which generally allow them to be in the blood circulation for a longer time.34 Although Mirzajani et al used a similar technique to the present study, they used different homogenization speeds (13,500 rpm and 24,000 rpm) and stabilizer concentrations 2% and 3%. Earlier work from our laboratory22 has presented the fixed speeds of homogenization (16,000 rpm) and variable (different) speeds of centrifugation (5,000 rpm and 14,000 rpm). In the present study drug polymer ratios were different. Similarly the speed of homogenization (22,500 rpm) and separation by centrifugation were also varied. Last but the not least, none of the above mentioned studies have performed cellular uptake of the Tamoxifen citrate PLGA nanoparticle along with MTT assay, as investigated here, and the results of the study show concentration dependent cellular uptake of nanoparticle in vitro by MCF-7 breast cancer cells. Drug release also varied predominantly among the studies. Thus the present study has its own uniqueness.
Conclusion
The outcome of the present investigation proposes a novel formulation of TNPs, prepared by a multiple emulsion solvent evaporation technique. The polymeric particles in a nanosize range with a desired drug polymer ratio can be produced. The size, drug loading and the drug release kinetics can be optimally controlled. Further, TNPs were internalized well in breast cancer cells in vitro, suggesting their suitability in breast cancer treatment. Preferential uptake of nanoparticles rather than the free drug by MCF-7 cells causes the cells to be more viable to the free drug. However, in vivo studies are warranted with the nanoparticles.
Acknowledgment
Authors are indebted to the Indian Council of Medical Research for providing the financial grant (no. 45/20/2010-NAN/BMS) to conduct the study.
Disclosure
The authors report no conflicts of interest in this work.
References
Malik AA, Wani KA, Ahmad SR. Breast conservative therapy. J Med Sci. 2012;15(1):7–14. | ||
Sinha R, Kim GJ, Nie S, Shin DM. Nanotechnology in cancer therapeutics: bioconjugated nanoparticles for drug delivery. Mol Cancer Ther. 2006;5(8):1909–1917. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Lü JM, Wang X, Marin-Muller C, et al. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev Mol Diagn. 2009;9(4):325–341. | ||
Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75(1):1–18. | ||
Prokop A, Davidson JM. Nanovehicular intracellular delivery systems. J Pharm Sci. 2008;97(9):3518–3590. | ||
Averineni RK, Shavi GV, Gurram AK, et al. PLGA 50:50 nanoparticles of paclitaxel: Development, in vitro anti-tumor activity in BT-549 cells and in vivo evaluation. Bull Mater Sci. 2012;35(3):319–326. | ||
Hassani SM, Bagheri S. Computational procedure for determining physicochemical properties of doxorubicin-PLGA nanoparticles and daunorubicin-PLGA nanoparticles. Int J Pharm Tech Res. 2012;4(3): 1242–1246. | ||
Pradhan R, Poudel BK, Ramasamy T, Choi HG, Yong CS, Kim JO. Docetaxel-loaded polylactic acid-co-glycolic acid nanoparticles: formulation, physicochemical characterization and cytotoxicity studies. J Nanosci Nanotechnol. 2013;13(8):5948–5956. | ||
Wohlfart S, Khalansky AS, Gelperina S, et al. Efficient chemotherapy of rat glioblastoma using doxorubicin-loaded PLGA nanoparticles with different stabilizers. PLoS One. 2011;6(5):e19121. | ||
The BIG 1-98 Collaborative Group. Letrozole therapy alone or in sequence with tamoxifen in women with breast cancer. N Engl J Med. 2009;361(8):766–776. | ||
Peters-Engl C, Frank W, Danmayr E, Friedl HP, Leodolter S, Medl M. Association between endometrial cancer and tamoxifen treatment of breast cancer. Breast Cancer Res Treat.1999;54(3):255–260. | ||
de Lima GR, Facina G, Shida JY, et al. Effects of low dose tamoxifen on normal breast tissue from premenopausal women. Eur J Cancer. 2003;39(7):891–898. | ||
Mirzajani F, Rafati H, Atyabi F. Fabrication of biodegradable poly (d, l-lactide-co-glycolide) nanoparticles containing tamoxifen citrate. Iranian Polymer Journal. 2010;19(6):437–446. | ||
Lamprecht A, Ubrich N, Hombreiro Pérez M, Lehr CM, Hoffman M, Maincent P. Biodegradable monodispersed nanoparticles prepared by pressure homogenization-emulsification. Int J Pharm. 1999;184(1): 97–105. | ||
Ueda M, Kreuter J. Optimization of the preparation of loperamide-loaded poly (L-lactide) nanoparticles by high pressure emulsification-solvent evaporation. J Microencapsul. 1997;14(5):593–605. | ||
Sinha B, Mukherjee B, Pattnaik G. Poly-lactide-co-glycolide nanoparticles containing voriconazole for pulmonary delivery: in vitro and in vivo study. Nanomedicine. 2013;9(1):94–104. | ||
Li X, Tian X, Zhang J, et al. In vitro and in vivo evaluation of folate receptor-targeting amphiphilic copolymer-modified liposomes loaded with docetaxel. Int J Nanomedicine. 2011;6:1167–1184. | ||
Fonseca C, Simõesa S, Gaspar R. Paclitaxel-loaded PLGA nanoparticles: preparation, physicochemical characterization and in vitro anti-tumoral activity. J Control Release. 2002;83(2):273–286. | ||
Rudra A, Manasadeepa R, Ghosh MK, Ghosh S, Mukherjee B. Doxorubicin-loaded phosphatidylethanolamine-conjugated nanoliposomes: in vitro characterization and their accumulation in liver, kidneys, and lungs in rats. Int J Nanomedicine. 2010;5:811–823. | ||
Beckett AH, Stenlake JB. Infrared spectrophotometry. In: Practical pharmaceutical chemistry. 4th ed. Part 2. CBS Publishers, India: New Delhi; 1997:384–390. | ||
Sahana B, Santra K, Basu S, Mukherjee B. Development of biodegradable polymer based tamoxifen citrate loaded nanoparticles and effect of some manufacturing process parameters on them: a physicochemical and in-vitro evaluation. Int J Nanomedicine. 2010;5:621–630. | ||
Meiβner T, Potthoff A, Richter V. Suspension characterization as important key for toxicological investigations. J Phys Conf Ser. 2009;170(1). | ||
Basu S, Mukherjee B, Chowdhury SR, et al. Colloidal gold-loaded, biodegradable, polymer-based stavudine nanoparticle uptake by macrophages: an in vitro study. Int J Nanomedicine. 2012;7:6049–6061. | ||
Nayak AK, Pal D. Development of pH–sensitive tamarind seed polysaccharide-alginate composite beads for controlled diclofenac sodium delivery using response surface methodology. Int J Biol Macromol. 2011;49(4):784–793. | ||
Pattnaik G, Sinha B, Mukherjee B, et al. Submicron-size biodegradable polymer-based didanosine particles for treating HIV at early stage: an in vitro study. J Microencapsul. 2012;29(7):666–676. | ||
Mukherjee B, Roy G, Santra K, Sahana B. Lactide-glycolide polymers as nanodimensional carriers for drugs. Int J Biomed Nanosci Nanotechnol. 2010;1:230–246. | ||
Kocbek P, Obermajer N, Cegnar M, Kos J, Kristl J. Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. J Contol Release. 2007;120(1–2):18–26. | ||
Li X, Li R, Qian X, et al. Superior antitumor efficiency of cisplatin-loaded nanoparticles by intratumoral delivery with decreased tumor metabolism rate. Euro J Pharm Biopharm. 2008;70(3):726–734. | ||
Zhang L, Yang M, Wang Q, et al. 10-Hydroxycamptothecin loaded nanoparticles: preparation and antitumor activity in mice. J Control Release. 2007;119(2):153–162. | ||
Zhang L, Hu Y, Jiang X, Yang C, Lu W, Yang YH. Camptothecin derivative-loaded poly(caprolactone-co-lactide)- b-PEG-b-poly(caprolactone-co-lactide) nanoparticles and their biodistribution in mice. J Control Release. 2004;96(1):135–148. | ||
Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153(3):198–205. | ||
Cirpanli Y, Yerlikaya F, Ozturk K, et al. Comparative evaluation of in vitro parameters of tamoxifen citrate loaded poly(lactide-co-glycolide), poly(epsilon-caprolactone) and chitosan nanoparticles. Pharmazie. 2010;65(12):867–870. | ||
Unger F, Wittmar M, Kissel T. Branched polyesters based on poly [vinyl-3-(dialkylamino) alkylcarbamate-co-vinyl acetate-co-vinyl alcohol]-graft-poly (d, l-lactide-co-glycolide): Effects of polymer structure on cytotoxicity. Biomaterials. 2007;28(9):1610–1619. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.