")
Back to Journals » International Journal of General Medicine » Volume 16
Prenatal Diagnosis of Retinoblastomas: A Scoping Review
Authors Rodriguez A, Kelley C, Patel A, Ramasubramanian A
Received 24 December 2022
Accepted for publication 23 March 2023
Published 27 March 2023 Volume 2023:16 Pages 1101—1110
DOI https://doi.org/10.2147/IJGM.S380634
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 8
Editor who approved publication: Dr Scott Fraser
Aurora Rodriguez,1 Caitlin Kelley,1 Anjali Patel,1 Aparna Ramasubramanian2
1School of Medicine, Creighton University, Phoenix, AZ, USA; 2Ophthalmology Department, Phoenix Children’s Hospital, Phoenix, AZ, USA
Correspondence: Aparna Ramasubramanian, Ophthalmology Department, Phoenix Children’s Hospital, 1919 E Thomas Road, Phoenix, AZ, 85016, USA, Tel +1 602-933-3937, Fax +1 602-933-2409, Email [email protected]
Purpose: The objective of this review is to explore the prenatal diagnosis of retinoblastoma and the recommended screening practices.
Patients and Methods: An electronic literature search on prenatal diagnosis of retinoblastoma was conducted on the PubMed database. Publications within the last 20 years that matched the inclusion criteria were selected. The literature search included the following keywords: retinoblastoma, prenatal, diagnosis, screening, and associated synonyms to increase search sensitivity. Nine studies were included for investigation and extracted to identify prenatal diagnostic and screening techniques for retinoblastoma, their associated impact, and the target population that should receive prenatal screening for retinoblastoma.
Results: Familial retinoblastoma has an autosomal inheritance pattern and 90% penetrance. Therefore, future parents with a family history of retinoblastoma are strongly advised to get tested for retinoblastoma (Rb) gene mutations; if one of the parents is positive for a mutated allele of the RB1 gene, there is a 45% chance that their child will inherit a mutated allele of the retinoblastoma gene, rendering the allele non-functional in all of the cells of the individual and predisposing the child to a higher risk of developing retinoblastoma as well as other secondary cancers. Thus, prenatal screening and diagnosis of retinoblastoma is crucial for early diagnosis and optimal treatment.
Conclusion: Prenatal testing for retinoblastoma in high-risk families is important for everyone in the family. For the parents, prenatal screening has been shown to improve their family planning decisions and psychological well-being as they can mentally prepare beforehand and make informed decisions. More importantly, these practices have shown to yield better treatment and vision outcomes in the newborn.
Keywords: retinoblastoma, prenatal, diagnosis, screening, imaging
Introduction
Retinoblastoma is the most common, pediatric primary intraocular malignancy. It arises from a mutated Retinoblastoma gene (Rb), a tumor suppressor gene found in chromosome 13 that regulates the cell cycle. A non-mutated retinoblastoma gene contains two non-mutated RB1 alleles. Since tumor suppressor genes only need one copy of the allele to function, tumorigenesis happens when both copies of the RB1 allele are mutated in the same retinal cell (two-hit hypothesis). The tumor cells of a person with retinoblastoma have a nonfunctional RB1 gene, this allows for dysregulation of the cell cycle and unregulated growth in those cells.1–3 The mutations rendering an RB1 gene nonfunctional can be either sporadic or inherited.4 Sporadic mutations are de novo modifications in somatic cells, while inherited mutations are modifications in gamete or sex cells that can be passed onto offspring. In heritable or familial retinoblastoma, a dysfunctional RB1 allele is directly passed down from the biological mother or father to the embryo. In this case, a child has already inherited a mutated RB1 allele, and only an additional, single sporadic mutation is needed to induce tumorigenesis.
Risk Factors and Incidence
Retinoblastoma is the most common pediatric intraocular malignancy with an incidence of 1 case per every 15,000 to 20,000 live births and 9000 newly diagnosed patients every year worldwide.2 The presence of a mutated RB1 allele predisposes the child to developing retinoblastoma as well as other secondary cancers (Osteosarcoma, fibrosarcoma, and melanoma are the most common secondary cancers).5 In heritable retinoblastoma, an RB1 allele mutation has an autosomal inheritance pattern, and 90% penetrance. Thus, a child born to a parent with familial retinoblastoma has a 45% chance of inheriting a copy of a dysfunctional RB1 allele and is considered “high risk”.4
It is important to identify high-risk populations because retinoblastoma is a highly aggressive tumor. In fact, retinoblastoma has up to a 70% mortality in some countries (located in Asia and Africa). However, in Europe, Canada, and the United States, the mortality rates are much lower (3–5%). The substantial difference between the two mortality rates has been associated with a delay of diagnosis of 6 months from the first clinical signs of retinoblastoma.2 Thus, early diagnosis is crucial worldwide. In highly developed countries, such as Canada and the United States where survival rates are high, a prompt diagnosis is still important because it can render better vision outcomes for the child.
Clinical Presentation
While retinoblastoma may occur at any age, the majority of cases present in children younger than two years of age.1 The most common presenting sign of retinoblastoma is leukocoria, which presents as an abnormal white fundal reflex, instead of the normal red-eye reflex, when illuminating the pupil. If retinoblastoma is identified three to six months after the first sign of leukocoria, then the malignancy is most likely curable and intraocular. Other clinical signs include strabismus, exophthalmia, orbital swelling, proptosis, hypopyon, and hyphema.2,4 Furthermore, due to the aggressive nature of retinoblastoma, symptoms can appear acutely, and worsen over a short period of time.
Some patients may have no presenting signs other than a positive family history.2 Therefore, it is important for families with a history of retinoblastoma to receive genetic testing and counseling to make informed family planning and reproductive decisions. If the couple decides to have children, it is strongly recommended for them to undergo prenatal retinoblastoma screening and diagnosis of the child to enhance treatment efforts for the preservation of vision and the globe of the newborn. Prenatal screening and testing techniques include non-invasive prenatal diagnosis (NIPD), preimplantation genetic diagnosis (PGD), chorionic villus sampling (CVS), amniocentesis, fetal ultrasound and prenatal MRI.6–8
If the embryo has a positive diagnosis of retinoblastoma, healthcare providers along with the family can work together on designing the best course of treatment for the child. Early intervention measures could lead to an early induction of the fetus to prevent continuous growth of the tumor, and provide earlier curative treatment.2,3 Earlier interventions have been associated with increased success in preserving vision and avoiding enucleation (removal of the eye).3,7 This review will further expand on the techniques and practices associated with each of the options available and the impact that it renders on the child and family members. Furthermore, this review will highlight the need for more clinical studies regarding prenatal diagnostic measures to increase implementation in clinical practice and public awareness.
Materials and Methods
An electronic literature search was conducted on the PubMed database on prenatal diagnosis of retinoblastoma within the last 20 years using the combination of following MeSH terms in title, abstract, or as other terms to maximize sensitivity:
“Retinoblastoma”, “retinal cancer”, “RB”, “retinal tumor”, “retinal malignancy”, “Prenatal”, “Pre-birth”, “Diagnosis”, “Screening”.
Inclusion/ Exclusion Criteria
Of the 74 studies found on PubMed, 9 studies were included in this literature review based on inclusion criteria and relevance. Studies were included if they examined the prenatal diagnosis of retinoblastoma, were a peer-reviewed journal publication, and were a study design of the following types: observational, cross-sectional study, case-control study, cohort study, randomized controlled trials or quasi experiments. Only studies in English were included. Articles were excluded if they were case reports, literature reviews, poster abstracts, unpublished doctoral theses, or published in non-peer reviewed journals (See Table 1).
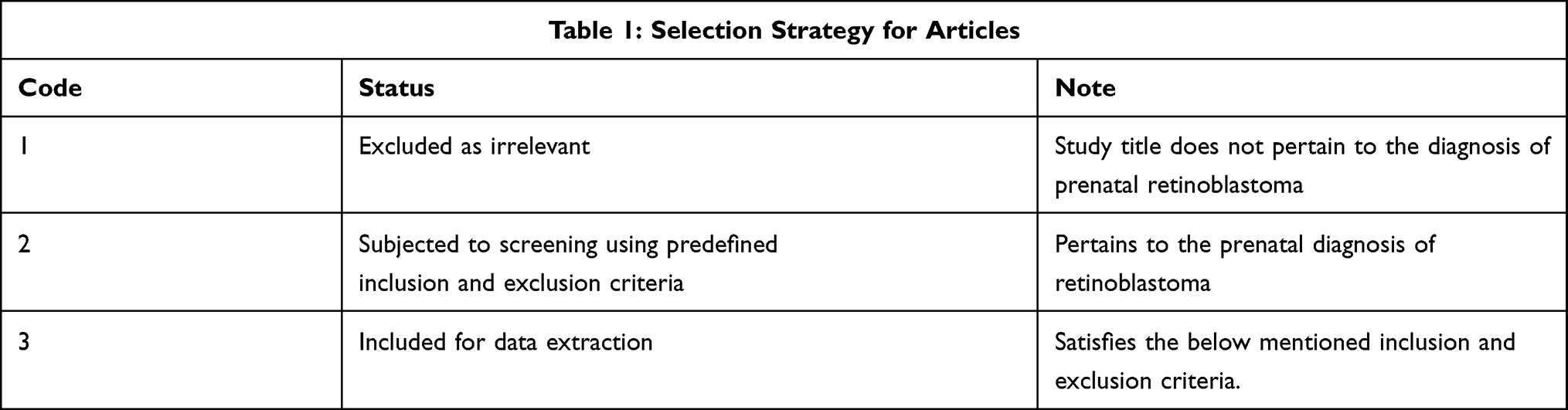 |
Table 1 A Summary of the Selection Strategy Applied to the Resultant PubMed Database Search |
Search Language
Process: Diagnosis
Outcome: Retinoblastoma
Population: Pregnant Women
Concept 1: “Retinoblastoma”
Key words: “retinoblastoma*”[tw] OR “retinal cancer*”[tw] OR “RB”[tw] OR “retinal tumor*”[tw] OR “retinal malignancy*”[tw]
Mesh: “Retinoblastoma”[Mesh]
Concept 2: Prenatal Diagnosis
Key Words: “prenatal diagnosis”[tw] OR “prenatal screening*”[tw] OR “prenatal screen*”[tw] OR “prenatal test*”[tw] OR “prenatal testing*”[tw]
MeSH: “Prenatal Diagnosis”[Mesh]
PubMed search results with final search language as follows:
“Retinoblastoma” [Mesh] OR “retinoblastoma*”[tw] OR “retinal cancer*”[tw] OR “RB” [tw] OR “retinal tumor*” [tw] OR “retinal malignanc*” [tw] AND “Prenatal Diagnosis” [Mesh] OR “prenatal diagnosis” [tw] OR “prenatal screening*” [tw] OR “prenatal screen*” [tw] OR “prenatal test*” [tw] OR “prenatal testing*” [tw].
Results
Data Extraction
A total of nine studies of good quality and content of high relevance on prenatal testing of retinoblastoma were included in the final sample for this review (See Table 2).
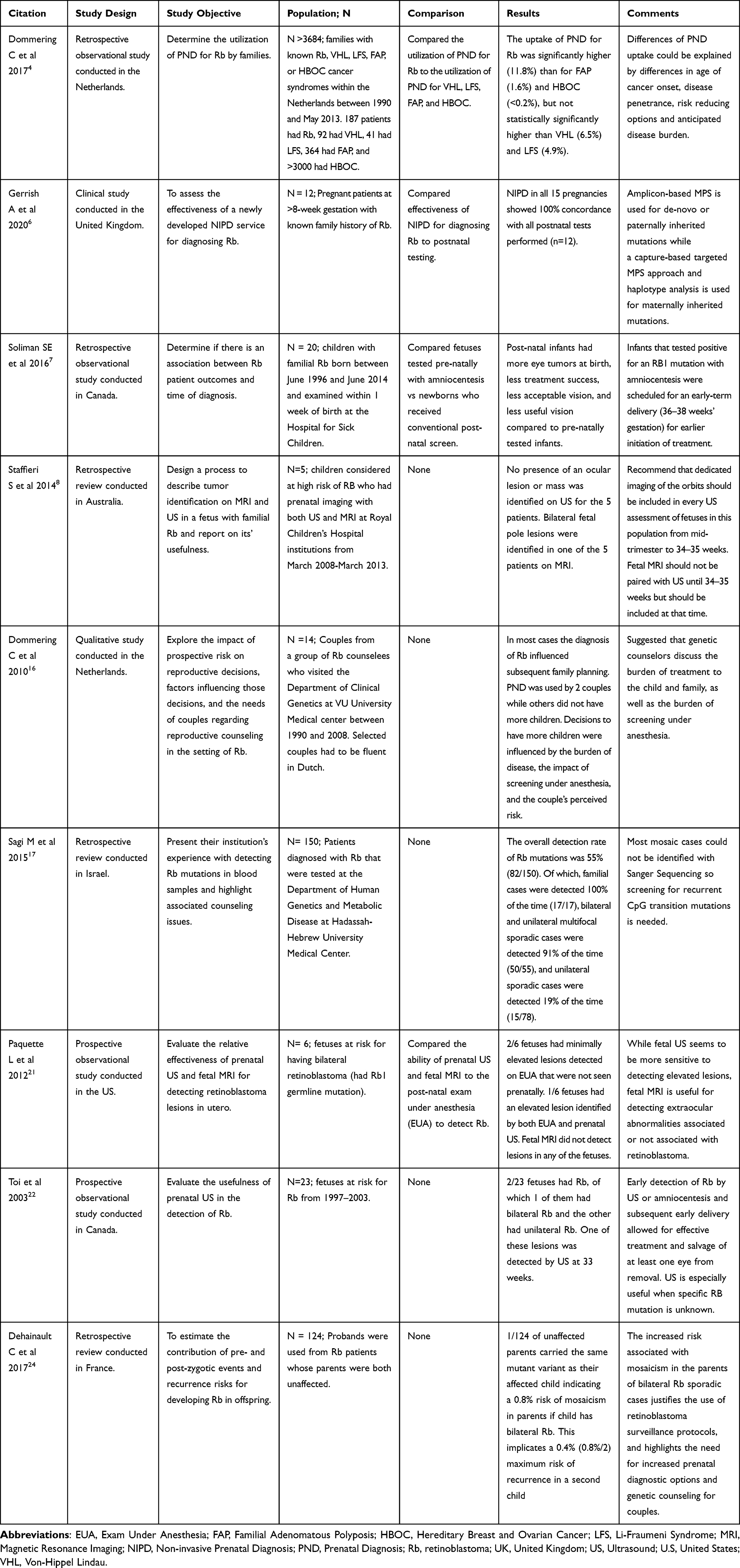 |
Table 2 Summary of Included Studies Investigating Prenatal Diagnosis of Retinoblastoma |
Techniques Available for Prenatal Diagnosis (See Table 3)
Techniques revolving around Rb diagnosis include preimplantation genetic diagnosis, early prenatal testing, and late prenatal screening. Preimplantation genetic diagnosis can accurately diagnose the presence of a mutated RB1 gene before embryo transfer, and the couple may select an embryo without the inherited mutation for implantation. Early prenatal screening can be divided up into non-invasive and invasive methods. Non-invasive methods include using cell-free fetal DNA within the mother’s blood. It can be done as early as 8 weeks of gestation and is a definitive diagnosis.6 Invasive tests include chorionic villi sampling (CVS) or amniocentesis which can be done at 11 or 16 weeks of gestation, respectively.6,7,9 Amniocentesis is a procedure that removes amniotic fluid from the uterine cavity using a needle. It is performed transabdominally with ultrasound guidance.10 CVS can be performed with a transabdominal approach, which includes a spinal needle being inserted into the uterus under continuous ultrasound, or a transcervical approach, which involves access to fluid through the cervix.11 Risks identified with CVS, as with amniocentesis, include pregnancy loss, bleeding, infection, rupture of membranes, and uncertain results, although pregnancy loss rates have declined with use of ultrasound.11,12 Late prenatal screening includes fetal ultrasound or fetal MRI. It is important to note that late prenatal testing can only confirm Rb if it appears in utero but gives no indication as to whether the child has risk of developing retinoblastoma in the future.
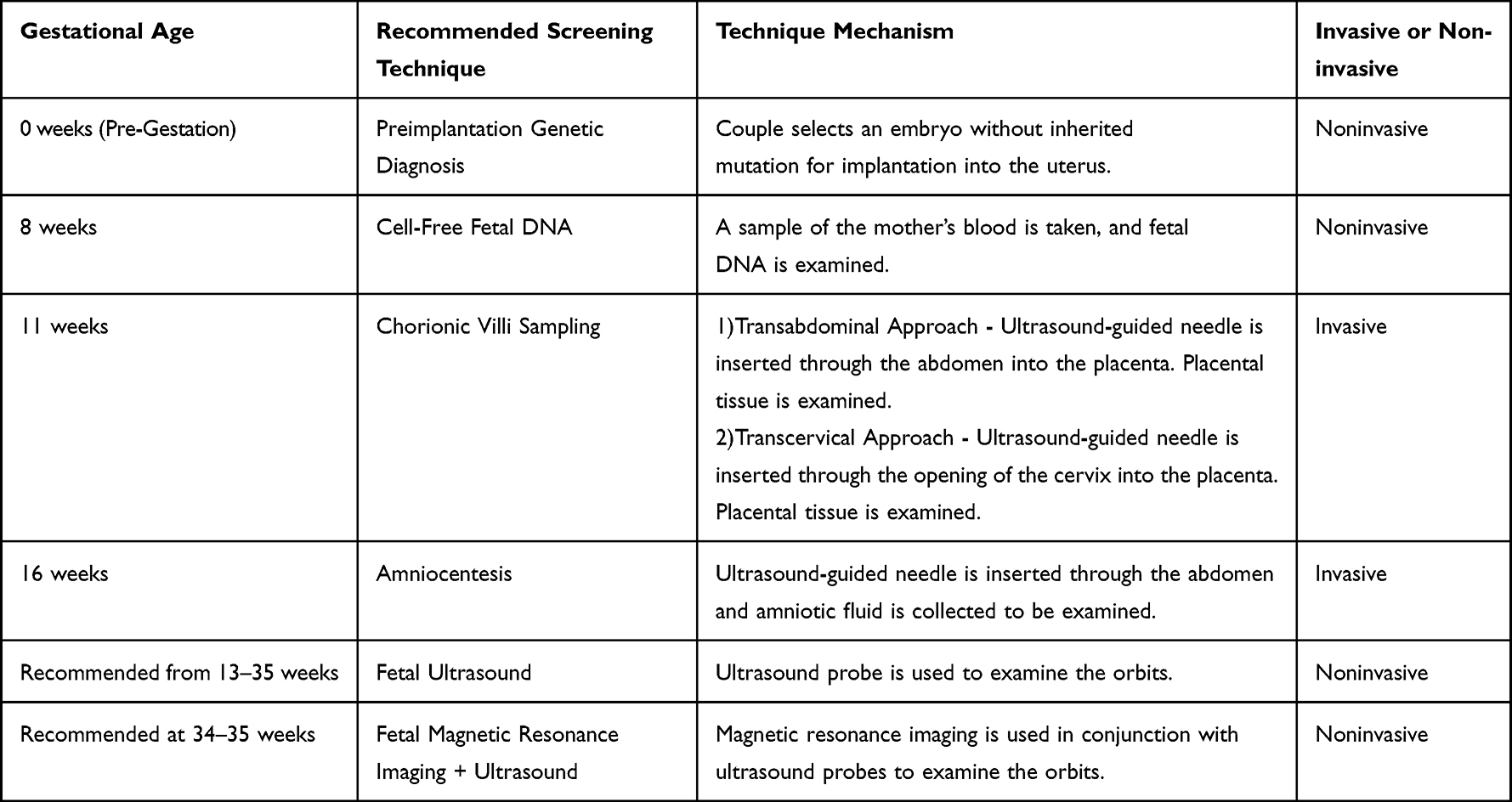 |
Table 3 Summary of Retinoblastoma Screening Techniques |
Impact on Treatment and Prognosis
Throughout the articles included in the literature review, multiple reasons regarding the need for detecting Rb prenatally were identified. First, there was a better chance to treat the disease early on, and thus, yield better results. The treatment of choice depends on the staging and size of the tumor, status of the contralateral eye and orbital structures, and resources available to the family. Small, less threatening tumors are often treated with focal therapy (including cryotherapy, laser photocoagulation, transpupillary thermotherapy, transcleral thermotherapy and plaque brachytherapy) while larger tumors are targeted with local (intra-arterial chemotherapy, enucleation, external beam radiation) and systemic therapies (intravenous chemotherapy). A combination of the previously mentioned treatments (such as using focal therapy as an adjunct to systemic therapy) can also be used.13,14
Additionally, knowing when the onset of the disease takes place plays a role in determination of treatment. Thus, being aware of the risk beforehand contributes to higher rates of eye salvage as it promotes better surveillance and, therefore, earlier detection of onset.7,11 When retinoblastoma is detected in-utero, families have the option to induce early late-term delivery to ensure earlier initiation of treatment. In turn, that earlier treatment has shown to yield better vision preservation and the utilization of less invasive therapies.7 Furthermore, the doubling time of retinoblastoma is thought to be around 15 days and hence there is a significant impact to early diagnosis and treatment.15 Most neonatal retinoblastoma tumors occur in the posterior pole region close to the macula (central vision) and hence the consequences on long term vision are profound.
Another theme repeatedly mentioned was that prenatal diagnosis of retinoblastoma leads to a greater ability to have a better guided family planning process if families at risk are given information about the status of their potential child’s risk.7,16 Negative impacts such as over testing or misdiagnoses were not discussed in the articles reviewed.
Abbreviations
A list of abbreviations used in this review regarding prenatal diagnosis of retinoblastoma was included (See Table 4).
 |
Table 4 A List of Abbreviations Used in the Article with Their Respective Explanation |
Discussion
Retinoblastoma, the leading primary ocular malignancy in children, is a fast-growing tumor that can start in utero. The majority of fetal retinoblastoma cases have a germline RB1 mutation; out of those cases, some of them have inherited a dysfunctional RB1 allele directly from one of their biological parents. This is because familial retinoblastoma has an earlier onset than sporadic retinoblastoma.9 Thus, children with prenatal retinoblastoma often have other relatives with previous or current retinoblastoma. Therefore, high-risk groups include those with a positive family history of retinoblastoma.6
High-risk families should undergo genetic testing to confirm the presence of a dysfunctional RB1 allele. If the molecular testing result is positive, these families should undergo genetic counseling. Obtaining genetic counseling is beneficial for family planning, which involves making informed reproductive decisions on whether to have children, minimize the number of live births, refrain from having children, or adopt.16,17 The screening for the RB1 gene also allows the parents to prepare emotionally for the possibility of encountering the disease in their offspring.16 If the parents decide to have children, they should undergo prenatal screening and testing to maximize their probability of early diagnosis and treatment.
Preimplantation genetic testing is an option for couples with a confirmed RB1 mutation. Preimplantation diagnosis happens in conjunction with in-vitro fertilization (IVF) and allows parents to know the RB1 inherited mutation status before the implantation of the embryo.6,18 This process is the earliest interventional procedure that reduces the incidence of heritable retinoblastoma in the family.4
Other forms of early prenatal testing include chorionic villus sampling (CVS), and amniocentesis in order of availability based on fetal age.6 Each of the early prenatal testing procedures includes a small incision and subsequent sampling of chorionic tissue or amniotic fluid respectively. The tissue or fluid collection is then genetically analyzed for the presence of an RB1 mutation.10,11 However, some families choose to not undergo testing. A Dutch review stated that a minority of patients with family history of retinoblastoma undergo CVS or amniocentesis for retinoblastoma.2 This could be partially explained by the invasive nature of most prenatal diagnostic techniques, and the associated risk for complications such as infection, limb abnormalities or miscarriage.12 However, the incidence of complications is low. A systematic review that analyzed 12 controlled studies for amniocentesis and 7 for CVS concluded that the procedure-related risks for fetal loss with a 95% confidence interval was 0.30% and 0.20%, respectively.19 Additionally, a 7.5 year study period focused on amniocentesis-related complications yielding urgent fetus delivery, had an incidence rate of 0.7% percent (95% CI, 0.16, 1.24).20 Thus, the benefits from early diagnosis and enhanced treatment of retinoblastoma seem to outweigh the perceived risks. However, if a couple is concerned with invasive prenatal screening techniques, non-invasive measures are also available.
Non-invasive prenatal diagnosis (NIPD) is the earliest form of non-invasive testing for retinoblastoma. NIPD takes a blood sample of the mother and analyzes the cell-free fetal DNA within the blood using amplicon-based massively parallel sequencing (MPS) for de-novo or paternally inherited mutations or capture-based targeted MPS approach and haplotype analysis in maternally inherited mutations. Genetic analysis from NIPD is diagnostic and does not require further testing. In fact, a recent study demonstrated that the result from NIPD of retinoblastoma matched 100% with postnatal retinoblastoma screening and diagnostic results. Additionally, NIPD is available by 8 weeks of gestation. The early diagnostic and non-invasive nature of NIPD can be an attractive option for parents concerned with the complications associated with more invasive procedures. Furthermore, NIPD seems to be a cost-effective option for patients and healthcare settings. This is particularly important in developing countries that have a high burden of disease and mortality due to scarce resources that make screening and early detection rates difficult. Nevertheless, NIPD possesses its own risks and limitations. For instance, both amplicon- and capture-based MPS require a fetal fraction greater than 4% in the mother’s blood in order to get the most reliable test result. The authors of the study estimate that a minimum gestational age of 8 weeks is needed to ensure the fetal-derived cfDNA requirement. Since fetal fraction increases with maternal gestation, couples are able to receive NIPD testing closer to their delivery date if they choose to do so. Additionally, most NIPD analytical techniques have been designed to detect paternal or de novo mutations because of the challenge in detecting maternally inherited alleles in the presence of cell-free DNA of maternal origin in maternal plasma. When attempting to detect a maternally inherited allele, capture-based targeted sequencing with relative haplotype dosage is used. While this allows for the analysis of maternal mutations in the cfDNA, a previous child (regardless of retinoblastoma status) is required in order to phase the mutant haplotype. Furthermore, since cfDNA is naturally found in fragments of 140–160 bp, amplicon-based NIPD is not capable of detecting RB1 variations greater than 10 bp in length. While data on the incidence of paternal variations greater than 10 bp is limited, microarray analysis can be used to assess the size of deletions in the RB1 gene. If microarray analysis detects a variation greater than 10 bp, relative haplotype dosage (RHDO) analysis can be done as long as the couple has a previous child for haplotype phasing.6 Other non-invasive testing techniques include fetal ultrasound and MRI.
The number of retinoblastoma cases detected in-utero by fetal ultrasound and/or MRI are very limited. Furthermore, studies comparing both imaging modalities have yielded different results for which method of imaging tends to detect retinoblastoma earlier and with more accuracy. Nevertheless, recent studies agree that both fetal ultrasound and fetal MRI are useful in detecting retinoblastoma in-utero. When fetal ultrasound and MRI are used as a measure for retinoblastoma screening, these are usually done in series and during the third trimester of pregnancy (starting at 28 weeks). However, Ultrasound can be started as early as 13 weeks, it does not possess any risks or threats to the mother or the child, and it has been shown to be effective in detecting prenatal retinoblastoma.8,18,21,22 Additionally, Doppler imaging with Ultrasound has been used to examine tumor vascularization and identify tumors from hemorrhage. On the other hand, fetal MRI has the ability to detect extraocular abnormalities that are either related or unrelated to retinoblastoma. The main limitation to using either imaging modality for the screening and diagnosis of retinoblastoma is that minimally elevated, small tumors (less than 2 mm) may go undetected until it grows bigger in size.21,22 Ultrasound also tends to be user dependent so the time of diagnosis can vary depending on the expertise of the person using the ultrasound probe.23 Furthermore, since fetal ultrasound and fetal MRI are mainly done during the last trimester of pregnancy, diagnosis of retinoblastoma is delayed and it could prevent the couple from carefully setting a proper treatment plan. Additionally, this could present a challenge for parents to emotionally and financially prepare for taking care of a child with retinoblastoma.6,7,16
A positive prenatal diagnosis of retinoblastoma is not only informative to the parents but is beneficial for both the parents and the child.6,7 In fact, a study done in 2016 compared the vision and globe outcome for children tested prenatally and postnatally. Soliman SE et al conducted an observational study on 20 children; eight of them were spontaneously delivered and had postnatal testing for retinoblastoma (cohort 1) while the remaining 12 underwent amniocentesis and were scheduled for an early term delivery (36–38 weeks of gestational age) due to a positive result of the RB1 mutant allele. Out of the two cohorts, the first group had a greater amount of vision threatening tumors (50% compared to 25%), worse treatment success defined as avoidance of enucleation and external beam radiation (37.5% compared to 92%), worse acceptable vision outcomes defined as better than 0.2 (50% compared to 87.5%) and 12.5% of them ended with legal blindness. On the other hand, cohort 2 had smaller and less vision-threatening tumors (75% compared to 12.5%) yielding greater treatment success and none of the children ended with legal blindness. Thus, the results from the study showed that children that had a positive prenatal diagnosis of retinoblastoma and were able to undergo an early-term delivery had better vision prognosis and health outcomes.7 This is in part because retinoblastoma is a rapidly growing malignancy that doubles in size approximately every 15 days.15 Therefore, early-term delivery on children with a positive RB1 mutant allele seems to be a safe, and favored option for families that desire to address retinoblastoma sooner. Overall, the children that underwent prenatal diagnosis were able to preserve better vision and avoid invasive therapies such as external beam radiation and enucleation when compared to children who received postnatal testing and diagnosis. Delay of treatment in fast growing tumors like retinoblastoma even by a few days to weeks is highly significant and can lead to worse globe and vision outcomes. Thus, implementation for surveillance protocols for retinoblastoma in clinical practice should be encouraged.24
The literature supports prenatal diagnosis and early treatment of familial retinoblastoma. Thus, healthcare professionals share the responsibility to communicate the benefits of genetic testing and prenatal diagnosis, as well as the risks of delayed treatment to couples with a positive history of retinoblastoma that want to conceive.
Conclusion
Familial retinoblastoma is a heritable form of retinoblastoma that has up to 90% penetrance and is passed down in an autosomal dominant pattern. Due to the disposition that retinoblastoma has to grow rapidly and impair vision, published literature on this ocular malignancy repeatedly stated that early diagnosis of retinoblastoma is crucial for globe salvage, preservation of visual ability, and optimization of treatment in the child. This literature review identified several prenatal screening and diagnostic techniques for retinoblastoma such as preimplantation genetic diagnosis, non-invasive prenatal diagnosis, chorionic villus sampling, amniocentesis, fetal ultrasound, and prenatal MRI. At this time, no diagnostic practice is devoid of increased risks and limitations. However, the risks associated with prenatal diagnostic practices are low, while delaying diagnosis of the child could lead to serious complications.
It is highly recommended that families at risk of retinoblastoma receive extensive counseling regarding the condition, the benefits of prenatal diagnosis, and the risks associated with delayed diagnosis and treatment. We also note the need for more clinical studies focused on prenatal screening and diagnosis so they can be more easily implemented into clinical practice. Additionally, literature written for the general population in the form of flyers, visuals, or papers readily accessible in clinics could serve as a means to educate families so they can make informed decisions regarding reproductive planning and the future wellbeing of their child.
Acknowledgments
The authors would like to thank their respective affiliations for supporting them to write this review. The authors used the “Preferred Reporting Items for Systematic reviews and Meta-Analyses extension for Scoping Reviews (PRISMA-ScR) Checklist” to produce and accurately write a comprehensive final report.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No financial support was provided for this publication.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Berrocal A, Mavrodrifes E, Murray T. Chapter 55- Retinoblastoma. Retinal Imaging. 2006;471–479. doi:10.1016/B978-0-323-02346-7.50060-0
2. Dimaras H, Kimani K, Dimba E, et al. Retinoblastoma. The Lancet. 2012;379(9824):1436–1446. doi:10.1016/S0140-6736(11)61137-9
3. Richter S, Vandezande K, Chen N, et al. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet. 2003;72(2):253–269. doi:10.1086/345651
4. Dommering CJ, Henneman L, van der Hout AH, et al. Uptake of prenatal diagnostic testing for retinoblastoma compared to other hereditary cancer syndromes in the Netherlands. Fam Cancer. 2017;16(2):271–277. doi:10.1007/s10689-016-9943-z
5. Robert M, Kliegman MD. Retinoblastoma. Nelson Textbook of Pediatrics; 2020.
6. Gerrish A, Bowns B, Mashayamombe-Wolfgarten C, et al. Non-Invasive Prenatal Diagnosis of Retinoblastoma Inheritance by Combined Targeted Sequencing Strategies. J Clin Med. 2020;9(11):3517. doi:10.3390/jcm9113517
7. Soliman SE, Dimaras H, Khetan V, et al. Prenatal versus Postnatal Screening for Familial Retinoblastoma. Ophthalmology. 2016;123(12):2610–2617. doi:10.1016/j.ophtha.2016.08.027
8. Staffieri SE, McGillivray G, Elder JE, et al. Managing fetuses at high risk of retinoblastoma: lesion detection on screening MRI. Prenat Diagn. 2015;35(2):174–178. doi:10.1002/pd.4514
9. Castéra L, Gauthier-Villars M, Dehainault C, et al. Mosaicism in clinical practice exemplified by prenatal diagnosis in retinoblastoma. Prenat Diagn. 2011;31:1106–1108. doi:10.1002/pd.2837
10. Jindal A, Sharma M, Karena ZV, Chaudhary C. Amniocentesis. Treasure Island (FL): StatPearls Publishing; 2022.
11. Jones TM, Montero FJ. Chorionic Villus Sampling. Treasure Island (FL): StatPearls Publishing; 2022.
12. PHE Publications Gateway Number: GW-437. Chorionic Villus Sampling (CVS) and Amniocentesis: Information for Parents. London, UK: NHS Fetal Anomaly Screening Programme; 2017.
13. Fabian ID, Onadim Z, Karaa E, et al. The management of retinoblastoma. Oncogene. 2018;37:1551–1560. doi:10.1038/s41388-017-0050-x
14. Pandey AN. Retinoblastoma: an overview. Saudi J Ophthalmol. 2014;28(4):310–315. doi:10.1016/j.sjopt.2013.11.001
15. Abramson DH, Schefler AC, Beaverson KL, Rollins IS, Ruddat MS, Kelly CJ. Rapid growth of retinoblastoma in a premature twin. Arch Ophthalmol. 2002;120(9):1232–1233. doi:10.1001/archopht.120.9.1232
16. Dommering CJ, van den Heuvel MR, Moll AC, Imhof SM, Meijers-Heijboer H, Henneman L. Reproductive decision-making: a qualitative study among couples at increased risk of having a child with retinoblastoma. Clin Genet. 2010;78(4):334–341. doi:10.1111/j.1399-0004.2010.01484.x
17. Sagi M, Frenkel A, Eilat A, Weinberg N, Frenkel S. Genetic screening in patients with Retinoblastoma in Israel. Fam Cancer. 2015;14(3):471–480. doi:10.1007/s10689-015-9794-z
18. Neriyanuri S, Raman R, Rishi P, Govindasamy K, Ramprasad VL, Sharma T. Prenatal genetic diagnosis of retinoblastoma--clinical correlates on follow-up. Indian J Ophthalmol. 2015;63(9):741–742. doi:10.4103/0301-4738.170979
19. Salomon LJ, Sotiriadis A, Wulff CB, Odibo A, Akolekar R. Risk of miscarriage following amniocentesis or chorionic villus sampling: systematic review of literature and updated meta-analysis. Ultrasound Obstet Gynecol. 2019;54:442–451. doi:10.1002/uog.20353
20. Stark C, Smith R, Lagrandeur R, Batton D, Lorenz R. Need for Urgent Delivery After Third-Trimester Amniocentesis. Obstet Gynecol. 2000;95(1):p 48–50.
21. Paquette LB, Miller D, Jackson HA, et al. In utero detection of retinoblastoma with fetal magnetic resonance and ultrasound: initial experience. AJP Rep. 2012;2(1):55–62. doi:10.1055/s-0032-1316465
22. Toi A, Sutherland J, Gallie B, Gardiner J, Sermer M. Evaluation of the fetus at risk for retinoblastoma: what is the role of prenatal ultrasound? Ultrasound Med Biol. 2003;29:S137. doi:10.1016/S0301-5629(03)00562-3
23. Pinto A, Pinto F, Faggian A, et al. Sources of error in emergency ultrasonography. Crit Ultrasound J. 2013;5 Suppl 1:S1. doi:10.1186/2036-7902-5-S1-S1
24. Dehainault C, Golmard L, Millot GA, et al. Mosaicism and prenatal diagnosis options: insights from retinoblastoma. Eur J Hum Genet. 2017;25(3):381–383. doi:10.1038/ejhg.2016.174
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.