")
Back to Journals » The Application of Clinical Genetics » Volume 13
Prenatal Diagnosis of Pfeiffer Syndrome Patient with FGFR2 C.940-1G>C Variant: A Case Report
Authors Torres-Canchala L , Castaño D, Silva N, Gómez AM , Victoria A , Pachajoa H
Received 27 February 2020
Accepted for publication 28 May 2020
Published 11 August 2020 Volume 2020:13 Pages 147—150
DOI https://doi.org/10.2147/TACG.S251581
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Laura Torres-Canchala,1 Daniela Castaño,2 Nathalia Silva,2 Ana María Gómez,2 Alejandro Victoria,3 Harry Pachajoa4,5
1Centro de Investigaciones Clínicas, Fundación Valle del Lili, Cali, Colombia; 2Newborn Intensive Care Unit, Fundación Valle del Lili, Cali, Colombia; 3Obstetrical Intensive Care Unit, Maternal-Infant Department, Fundación Valle del Lili, Cali, Colombia; 4Centro de Investigaciones en Anomalías Congénitas y Enfermedades Raras, Universidad Icesi, Cali, Colombia; 5Genetics Service, Fundación Valle del Lili, Cali, Colombia
Correspondence: Laura Torres-Canchala
Centro de Investigaciones Clínicas, Fundación Valle del Lili, Cali, Colombia
Tel +57-331-9090 ext 4022
Email [email protected]
Background: Pfeiffer syndrome (PS) is an autosomal dominant disorder caused by mutations in fibroblast growth factor receptor FGFR1 and FGFR2 genes, occurring in approximately 1:100,000 live births. PS has a wide range of clinical expression and severity, so early prenatal diagnosis is difficult and genetic counseling is desirable. We describe a PS newborn with her ultrasound and molecular studies.
Case Report: We describe a female term newborn with cloverleaf-shaped skull, facial hypoplasia, low ears, exophthalmos and wide, broad and deviated thumbs and hallux. The patient was diagnosed by ultrasound at 29 WGA and referred to a tertiary care hospital for her follow-up. Molecular test revealed a heterozygous pathogenic variant in intron 8 of the FGFR2 gene (FGFR2: c.940– 1G>C). It was a de-novo mutation. At 17 days of life, craniosynostosis correction and a Lefort-III frontomaxillary advancement were performed.
Conclusion: Pfeiffer syndrome is a devastating genetic disorder. Prenatal diagnosis according PS morphological features in prenatal ultrasound allows timely genetic counseling, early referral to third-level centers, and close follow-up in the prenatal and postnatal stages.
Keywords: Pfeiffer syndrome, acrocephalosyndactylia, craniosynostosis, fibroblast growth factor receptor 2
Introduction
Pfeiffer syndrome (PS) was firstly described by Rudolph Pfeiffer as an acrocephalosyndactyly: bicoronal craniosynostosis, middle facial hypoplasia, digital anomalies of hands and feet, presenting in some cases, partial cutaneous syndactyly.1 PS prevalence is one of 100.000 live births, without sex preference.2 PS has an autosomal dominant inheritance pattern, generated by two types of mutations affecting the 1 or 2 gene of the fibroblast growth factor receptor (FGFR1 or FGFR2).2 There are three PS phenotypes to determine neurodevelopment and survival prognosis.3 In type 1, the patient presents mild physical manifestations without neurodevelopmental impairment. Types 2 and 3 are more frequent and severe. Type 1 is generally associated with FGFR1 and Type 2 and 3 with FGFR2. They could develop craniosynostosis and hypertelorism with external proptosis and cloverleaf skull in case of type 2.4,5
The diagnosis in PS patients is usually made after the birth. It is difficult a comprehensive management that could worsen the quality of life of the patients and their family. Prenatal diagnosis in utero is essential for an early approach in a tertiary care center. We describe a Type 2 PS patient diagnosed in prenatal stage and molecularly confirmed after birth through a new generation sequencing panel in a high-complexity hospital in Colombia.
Case Report
A 32-yeat old pregnant woman with no relevant past medical history underwent a routine ultrasound scan showing a foetus of 29 weeks of gestational age (WGA). Findings included frontal prominence and a cloverleaf skull suggestive of craniosynostosis and hypoplastic nasal bone (Figure 1). In this ultrasound scan, feet and hands had no abnormalities. The woman was considered high-risk pregnancy and was referred to a tertiary care center for her weekly follow-up and the caesarean was scheduled at 38 WGA. After delivery, the female newborn had APGAR scores of 8, 9 and 10 at minutes 1, 5 and 10, respectively. Birth weight was 2862 grams, height was 48 cm and hear circumference was 33 centimeters. The physical exam showed a cloverleaf skull, hypertelorism, facial hypoplasia, low ears, exophthalmos, sacral dimple, and wide, short and deviated thumbs and hallux (Figure 2).
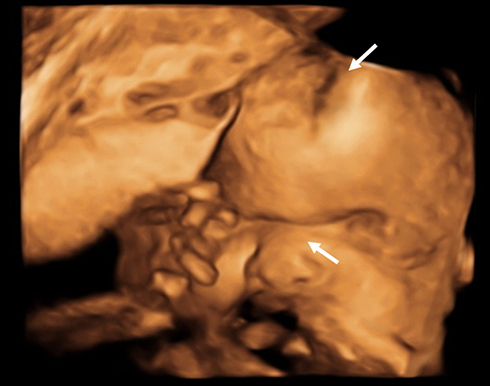 |
Figure 1 3D-ultrasound at 29 WGA. The foetus has frontal prominence and a cloverleaf skull suggestive of craniosynostosis and hypoplastic nasal bone (white arrows). |
The 3D CT head scan confirmed the cloverleaf skull, complete bilateral frontoparietal craniosynostosis, incomplete craniosynostosis of the sagittal and bilateral lambdoid suture, orbital alteration with bilateral proptosis secondary to hypertelorism and trans ependymal edema (Figure 2). The neonatal cranial sonography showed dysgenesis of corpus callosum with tentorium cerebelli high implantation. She also had persistent arterial ductus with bidirectional shunt without hemodynamic instability. There were no renal and ophthalmic malformations.
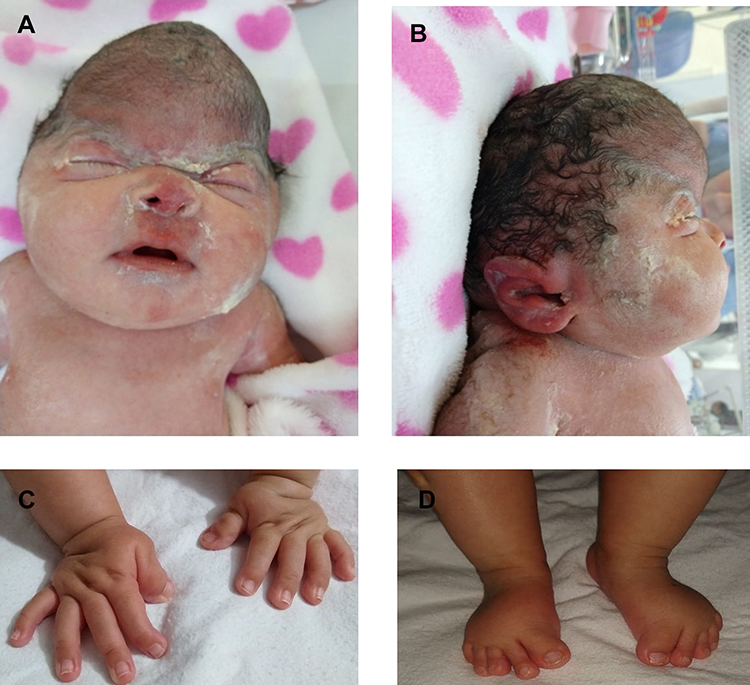 |
Figure 2 Physical findings at birth. (A and B). Female newborn with cloverleaf skull, hypertelorism, facial hypoplasia, low ears, exophthalmos, (C) short and deviated hallux and (D) thumbs. |
A new generation sequencing panel study was performed. It studies 241 genes potentially responsible for one or more Mendelian diseases related to hereditary osteodysplasia, collagen diseases and short stature. We found a heterozygous pathogenic variant in intron 8 of the FGFR2 gene (FGFR2: c.940–1G>C). The finding was confirmed using an ABI 3500 sequencer (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) by Sanger sequencing. Bioinformatic analysis of the variant was performed; FATHMM classified the variant as “damaging,” while MutationTaster classified it as “causing disease”. Currently, it is classified as “pathogenic” according to the American College of Medical Genetics and Genomics (ACMG) guidelines (PVS1, PM2, PP3).6 It was a de-novo mutation as the parents were unaffected. At 17 days old, a craniosynostosis correction and a Lefort-III frontomaxillary advancement were performed. In the postoperative period, she had hydrocephaly and ventricular dilatation requiring a ventriculoperitoneal catheter (VPC). The VPC was discontinued after 6 days and the patient was discharged at 24 days-old with a multidisciplinary outpatient follow-up.
At the time of the writing this report, the patient was 12 months and was diagnosed with psychomotor development retardation. A polysomnogram diagnosed sleep apnea-hypopnea syndrome. A nasolaryngoscopy showed multiple turbinoseptal synechiae and right choana atresia.
This case report was approved by the institutional IRB to publish the case details. Written informed consent was provided by the patient’s legal guardian to have the case details and any accompanying images published.
Discussion
Prenatal diagnosis of Pfeiffer syndrome is challenging in prenatal care. PS low incidence and the wide variability of morphological findings, which can also be related to other conditions, makes it difficult to suspect this syndrome early pregnancy stages.7 Pfeiffer syndrome is a clinically and autosomal dominant disorder caused by mutations in FGFR1 and FGFR2.2 Only 17 cases of diagnosis in the prenatal stage have been reported in the world.2 We present the clinical findings of a patient with SP diagnosed in-utero due to a variant in FRFR2 gene.8–13
Although most cases are diagnosed in their neonatal stage, prenatal diagnosis is possible. Three-dimensional obstetric ultrasound is the first-line diagnostic tool for suspected PS, being useful to verify suture closure in the third trimester of pregnancy for the most severe cases14 although the diagnosis with lower gestational ages has been reported.15 Adding to craniosynostosis, principal ultrasound prenatal findings suggestive to PS are thumbs deformations and big toes.7 Even when the cloverleaf skull is the most common finding in PS, and in our patient, it was the most important finding to perform PS diagnosis, the absence of this characteristic does not exclude the diagnosis, as well as its presence does not directly certify this syndrome, because other diseases can also present.7 The presence of wide fingers, sacral appendix, vertebral fusions and coronal clefts in the prenatal ultrasound should lead the clinician to perform prenatal molecular tests to rule out PS.2,7 Almost all cases of PS are a consequence of new mutations because the parents are unaffected. Nevertheless, there is a percentage where the abnormal gene can be inherited from either parent. In the cases were FGFR2 mutations could be inherited from the father, Cell-free DNA extracted from maternal plasma can provide valuable molecular information and identify the genetic diagnosis in utero.16
Our patient had a Pfeiffer syndrome type 2. Her cloverleaf skull it is a consequence of premature fusion of all sutures, except the metopic and squamous suture. This condition produces the frontal prominence of both middle fossae. The patient also presented hydrocephalus and dysgenesis of the corpus callosum. Our patient also had hypertelorism and severe ocular proptosis. This condition can lead to endophthalmitis and rupture of the eyeball, so periodic evaluation by the ophthalmologist is necessary. The upper airway is also compromised in PS type 2 patients given the facial hypoplasia that characterizes them. Although the patient had a wide nasal bridge, she had no obstruction of the upper airway.
The karyotype in all PS cases reported is normal so the cytogenetic study is not recommended.2 In PS, 94% of mutations occur in exon 8 or 10 of the FGFR2 gene, which is also associated with other syndromic craniosynostosis, often sporadic, such as Apert syndrome, Crouzon among others;17 In our patient, the variant detected in intron 8 of the FGFR2 gene, in a heterozygous state, is an intronic variant that alters the splicing of RNA whose alteration contributes to the presentation of the disease.
An early and accurate diagnosis is essential for multidisciplinary management. Prenatal diagnosis allows for rapid genetic counseling, early referral to third-level centers, and close follow-up in the prenatal and postnatal stages. Therefore, the implementation of new generation sequencing panels that include genes associated with genetic syndromes related to craniosynostosis is a useful strategy to improve the diagnosis. In addition to this, clinical suspicion is essential to detect more cases in the prenatal stage, as well as early referral to tertiary care centers for a multidisciplinary evaluation is necessary to improve the patient’s living conditions.
Conclusion
Pfeiffer syndrome type 2 is a genetic disorder caused by mutations in the FGFR2 gene, which encodes the human fibroblast growth factor type 2 receptor that consequently generates a wide range of abnormalities, mainly craniosynostosis. Recognizing morphological features in prenatal ultrasound allow a timely and comprehensive management, improving the quality of life of the PS patients and their family.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Pfeiffer. Dominant hereditary acrocephalosyndactylia. Z Kinderheilkd. 1964;90(301):20.
2. Giancotti A, D’Ambrosio V, Marchionni E, et al. Pfeiffer syndrome: literature review of prenatal sonographic findings and genetic diagnosis. J Matern Neonatal Med. 2017;30(18):2225–2231. doi:10.1080/14767058.2016.1243099
3. Machado RA, Ferreira SB, Martins L, et al. A novel heterozygous mutation in FGFR2 gene causing Pfeiffer syndrome. Am J Med Genet Part A. 2017;173(10):2838–2843. doi:10.1002/ajmg.a.38389
4. Plomp AS, Hamel BC, Cobben JM, et al. Pfeiffer syndrome type 2: further delineation and review of the literature. Am J Med Genet. 1998;75(3):245–251. doi:10.1002/(SICI)1096-8628(19980123)75:3<245::AID-AJMG3>3.0.CO;2-P
5. Kalathia MB, Parikh YN, Dhami MD, Hapani PT. Pfeiffer syndrome. J Pediatr Neurosci. 2014;9(1):85–86. doi:10.4103/1817-1745.131499
6. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
7. Saliba S, Morel B, Gonzales M, et al. Variable prenatal presentation of Pfeiffer syndrome: suggested aids to prenatal sonographic diagnosis. Prenat Diagn. 2018;38(5):349–356. doi:10.1002/pd.5231
8. Teebi AS, Kennedy S, Chun K, Ray PN. Severe and mild phenotypes in Pfeiffer syndrome with splice acceptor mutations in exon IIIc of FGFR2. Am J Med Genet. 2002;107(1):43–47. doi:10.1002/ajmg.10125
9. Cornejo-Roldan LR, Roessler E, Muenke M. Analysis of the mutational spectrum of the FGFR2 gene in Pfeiffer syndrome. Hum Genet. 1999;104(5):425–431. doi:10.1007/s004390050979
10. Bessenyei B, Nagy A, Szakszon K, et al. Clinical and genetic characteristics of craniosynostosis in Hungary. Am J Med Genet Part A. 2015;167(12):2985–2991. doi:10.1002/ajmg.a.37298
11. Hibberd CE, Bowdin S, Arudchelvan Y, et al. FGFR-associated craniosynostosis syndromes and gastrointestinal defects. Am J Med Genet Part A. 2016;170(12):3215–3221. doi:10.1002/ajmg.a.37862
12. Chun K, Teebi AS, Azimi C, Steele L, Ray PN. Screening of patients with craniosynostosis: molecular strategy. Am J Med Genet. 2003;120A(4):470–473. doi:10.1002/ajmg.a.20258
13. Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19(10):1105–1117. doi:10.1038/gim.2017.37
14. Delahaye S, Bernard JP, Rénier D, Ville Y. Prenatal ultrasound diagnosis of fetal craniosynostosis. Ultrasound Obstet Gynecol. 2003;21(4):347–353. doi:10.1002/uog.91
15. Gómez-Gómez JL, Fernández-Alonso AM, Moreno-Ortega I, Mas-Greño L, Berenguel-Martínez J, Fiol-Ruiz G. Prenatal diagnosis of Pfeiffer syndrome prior to 20 weeks’ gestation. J Obstet Gynaecol (Lahore). 2013;33(3):309. doi:10.3109/01443615.2012.753424
16. Zhang J, Li J, Saucier JB, et al. Non-invasive prenatal sequencing for multiple Mendelian monogenic disorders using circulating cell-free fetal DNA. Nat Med. 2019;25(3):439–447. doi:10.1038/s41591-018-0334-x
17. Ko JM. Genetic syndromes associated with craniosynostosis. J Korean Neurosurg Soc. 2016;59(3):187–191. doi:10.3340/jkns.2016.59.3.187
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.