")
Back to Journals » International Journal of General Medicine » Volume 15
Preliminary Screening of a Familial Tuberous Sclerosis Complex Pathogenic Gene
Authors Wang Y, Hu S, Tan X, Sang Q, Shi P, Wang C, Sang D
Received 5 February 2022
Accepted for publication 16 May 2022
Published 26 May 2022 Volume 2022:15 Pages 5247—5252
DOI https://doi.org/10.2147/IJGM.S359702
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Yuting Wang,1 SongNian Hu,2 XinYu Tan,2 Qingqing Sang,3 Peng Shi,3 Chun Wang,3 Daoqian Sang3
1Department of Neurology, The Third Affiliated Hospital of Anhui Medical University, Heifei, People’s Republic of China; 2State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing, People’s Republic of China; 3Department of Neurology, The First Affiliated Hospital of Bengbu Medical College, Bengbu, People’s Republic of China
Correspondence: Daoqian Sang, Department of Neurology, The First Affiliated Hospital of Bengbu Medical College, 287, Changhuai Road, Bengbu, 233004, People’s Republic of China, Email [email protected]
Purpose: The aim of this study was to screen the possible pathogenic genes of one family with tuberous sclerosis complexes (TSCs).
Patients and Methods: All family members were examined through detailed clinical evaluations, auxiliary examinations and CT. Then, we selected five members from this TSC family as the test samples. They were analysed by a new exon group sequencing method. Single nucleotide polymorphisms (SNPs) were screened by using databases, such as dbSNP and HAPMAP, and then the candidate genes were selected. Genes were analysed, and finally, the most likely mutation sites were screened. The results were examined by Sanger sequencing.
Results: In this TSC family, we identified c.913+2T>G, a splicing site mutation in the 9th intron region of TSC1. Family members without TSC did not have this mutation.
Conclusion: The mutations in the intron regions cannot be ruled out as a pathogenic factor for TSC.
Keywords: pathogenic genes, tuberous sclerosis complex, whole exon sequencing, intron mutation
Introduction
Tuberous sclerosis complex (TSC) is a neurocutaneous syndrome that is inherited in an autosomal dominant manner. TSC is associated with hamartoma formation in multiple organ systems. The lesions are often formed in embryonic period. The clinical features of TSC are facial sebaceous adenoma, seizures and cognitive disabilities. The pathogenesis of this disease is mainly related to two genes: TSC1 and TSC2. TSC1 and TSC2 encode the proteins hamartin and tuberin, respectively. No gene mutation hotspot has been found for these genes. Finding pathogenic mutations of TSC1 or TSC2 in the DNA of normal tissue is sufficient to make a definitive diagnosis of TSC.1 The pathogenesis of TSC is related to the activity of the mTOR pathway, and continuous excitation of the mTOR pathway is the molecular basis of TSC. Antagonism of the mTOR pathway with rapamycin may provide therapeutic options for TSC patients.2,3 Therefore, gene mutation detection can help to improve the early diagnosis rate, provide targeted treatment as soon as possible, and improve the prognosis of TSC patients to the greatest extent.
Most of the pathogenic genes of TSC are exonic gene mutations, whole-exome sequencing combined with Sanger sequencing may be a effective method for TSC diagnosis.4 In the current study, we used the Ion Torrent Proton sequencing platform for whole-exome scanning to locate and determine the possible pathogenic genes of one family with TSC.
Materials and Methods
Patients and Families
In our early clinical work, we identified a family with genetic inheritance of TSC. This family consisted of 13 people (6 males and 7 females), and five of these family members were TSC patients (1 male and 4 females) (Figure 1). After the patients and their families received and signed the informed consent form, five blood samples (2 males and 3 females) were collected from subjects II3, II4, III3, III4, and III5. From these subjects, 3–5 mL of venous blood were collected in EDTA anticoagulant tubes and preserved by the national genetic laboratory. All members of the family were followed up continuously. Written informed consent was obtained from participants under the research protocols approved by the Bengbu Medical College ethics review board (BYYFY-2012KY04).
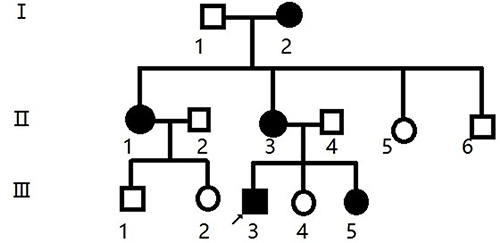 |
Figure 1 Family pedigree. Black symbols indicates affected individuals; open symbols indicates unaffected individuals; square indicates male; circle indicates female; arrow indicates proband. |
Methods
Genomic DNA was extracted from the blood samples of this family according to the instructions of the AxyPrep blood genomic DNA small-volume kit (Axygen, Union City, CA, USA). A Qubit® 2.0 fluorescence quantitative instrument (Invitrogen; Thermo Fisher Scientifific, Inc., Waltham, MA, USA) was used to test the quality of the genomic DNA samples. The Ion Proton system was used to complete whole exon group sequencing. Whole exon group sequencing was performed using multiple PCR expansions to amplify and capture the whole exon group sequence of patients’ genomic DNA from the blood samples. An Ion Proton sequencer (Thermo Fisher Scientifific, Inc.) was used to sequence the obtained DNA libraries to obtain a large amount of total original data. The variation type and location of the whole exon group in the DNA from the blood samples were identified by using mutation site recognition software. Using Ion Reporter (Thermo Fisher Scientifific, Inc.), mutation site interpretation software was applied to interpret the mutation site and provide important predictions and analysis results of the mutation consequence. Meaningless mutations were filtered, possible mutation sites were obtained, and the most likely pathogenic mutations were identified in combination with references from the literature. The mutation sites were verified by Sanger sequencing.
Results
Genetic Characteristics of the Family with Tuberous Sclerosis Complex
The family enrolled in this study consisted of three generations of tuberous sclerosis with consecutive onset, and there was no intergenerational inheritance phenomenon. The clinical manifestations were mainly skin damage, cognitive impairment, and seizures, all of which met the diagnostic criteria of TSC. The clinical characteristics of these patients are listed in Table 1.
 |
Table 1 Clinical Findings of Patients with TSC |
The proband was a 24-year-old male who began to have facial angiofibroma at approximately 4 years of age and had seizures at approximately 15 years of age. The seizure type was generalized tonic clonic seizures (GTCSs). His clinical manifestations are shown in Figure 2. The proband has provided informed consent for the image to be published.
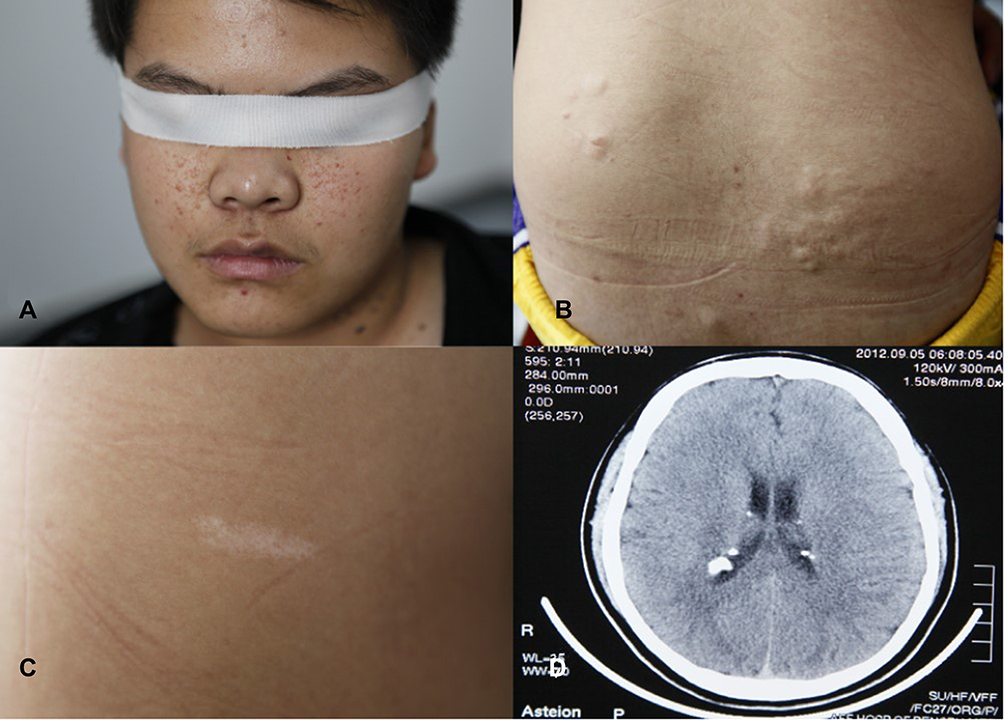 |
Figure 2 Clinical manifestations of proband. (A) Facial angiofibroma. (B) Shagreen patch. (C) Hypopigmented macules. (D) Subependymal nodules. |
Sequencing Results
We performed whole-exome sequencing of five family members. The sequencing results are shown in the Table 2.
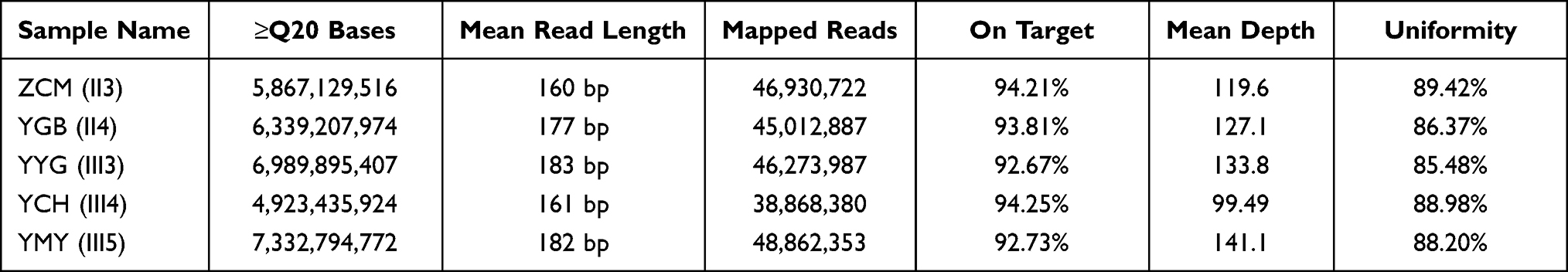 |
Table 2 Statistical Results of Sample Sequencing Data |
Variation of the Mutation Site
After quality filtration and interpretation, sample II3 obtained 38,449 variation sites, II4 obtained 37,824 variation sites, III3 obtained 37,754 variation sites, III4 obtained 38,108 variation sites, and III5 obtained 38,001 variation sites (including SNVs and indels).
Results of the Sequencing of the Sanger Validation Results
In this study, no suspected pathogenic mutations were found in the exon regions of the members of this family. However, IVS9ds+2T>G (c.913+2T>G), a splicing site mutation, was found in the 9th intron region of the TSC1 gene of II3. Family members without TSC (II4) did not have this mutation site (Figure 3). The splicing site mutation in the 9th intron region of TSC1 (IVS9ds+2T>G) has not been catalogued by the ClinVar database.
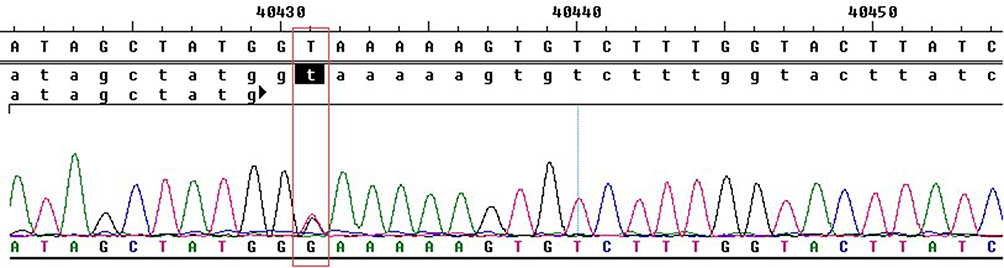 |
Figure 3 The splicing site mutation in the 9th intron region of TSC1 (c.913+2T>G). |
Discussion
TSC is an autosomal dominant genetic disease that is one of the neurocutaneous syndromes. Currently, studies have found that pathogenic mutations are located in the TSC1 and TSC2 genes. Harmatin is encoded by the TSC1 gene, and tuberin is encoded by the TSC2 gene. Harmatin and tuberin combine to form complexes through relevant functional domains. They play a role in the process of regulating cell morphology, growth and differentiation, participate in the regulation of the cell cycle, and function as tumour suppressor genes.5–7 Some studies have examined the consequences of intronic mutations that lead to the abnormal production of tuberin and ultimately promote tumorigenesis in TSC.8,9
Most of the pathogenic genes of TSC are exonic gene mutations, including missense mutations, nonsense mutations, splice site mutations, and small deletions. However, because of the different detection methods, the detection rate of genetic mutations is often different. Genetic mutations are not found in approximately 10%~25% of TSC patients by traditional genetic testing.10–12 Genetic diagnostic criteria were reaffirmed at the 2018 World TSC Conference, highlighting recent findings that some individuals with TSC are genetically mosaic for variants in TSC1 or TSC2.13 In addition, many intronic variants have been identified during mutational screening of both TSC genes, and they account for approximately 8% in TSC1 and 12% in TSC2.14
In this study, exome sequencing was performed on 3 patients with clear clinical diagnosis and 2 people without TSC in this family. The pathogenic mutations in this family were screened, and the results were examined by Sanger sequencing. Unfortunately, we failed to find possible mutation sites in the exon regions, but IVS9ds+2T>G (c.913+2T>G), a splicing site mutation, was found in the 9th intron region of the TSC1 gene of II3. Members of this family that do not have TSC do not have this mutation site. We hypothesized that the pathogenic mutation site of this family may be located in the intron region.
Abdelwahed et al15 performed a study on the pathogenicity of the splicing site mutation found in TSC2 in silico studies and mRNA analysis. Bioinformatics tools predicted that splicing site mutations have pathogenic effects on splicing machinery. RT-PCR followed by sequencing revealed that this mutation caused exon skipping and premature termination in a patient with TSC. It affects mRNA splicing, inducing the loss of TSC2 protein function and promoting tumorigenesis. Other researchers have made similar discoveries. Atypical mutation sites in the intron region may cause the skipping or retention of related exons in TSC1/2, leading to protein dysfunction.16 Niu et al17 found a c.2355+1G>C splicing variant of the TSC2 gene. Sequencing of cDNA confirmed that 62 bases have been inserted into the 3’ end of exon 21, which has caused a frameshift producing a truncated protein. They speculate this novel splicing variant of the TSC2 gene probably underlay the TSC in the proband.
Alternative splicing is universal in mammalian genes and involves nearly 95% of various regulatory processes. Thus, it plays a crucial role in hereditary diseases and tumorigenesis.18–20 Qiu et al18 reported that if a mutation changed the proportion of the two original splicing modes, it may also be a pathogenic mutation, even though it did not produce a new splicing pattern. This result indicates that the mutation changed the regulating factors.
Therefore, we speculate that the splicing site mutation in TSC1 (c.913+2T>G) may affect the expression of the TSC1-encoded protein hamartin through alternative splicing, which may potentially contribute to TSC susceptibility. All these findings support the notion that intron mutations may be disease-causing mutations in our patients. To some extent, this study increases the comprehension of the molecular mechanism of TSC pathogenesis. We look forwards to more discoveries of the pathogenesis at the transcriptional and translational levels in the future, which could provide another therapeutic option for this disease.
Conclusion
With the increasing recognition that splicing site mutations have pathogenic effects on splicing machinery and sequence variations frequently affect splicing by using mechanisms other than just disrupting splice sites, this form of gene mutation must now be considered in greater detail. We speculate that the mutations in the intron regions cannot be ruled out as a pathogenic factor for TSC.
Ethical Approval
This study was conducted in accordance with the Declaration of Helsinki and was approved by the Bengbu Medical College ethics review board (BYYFY-2012KY04).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was received.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Northrup H, Krueger DA, Northrup H. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 international Tuberous Sclerosis Complex consensus conference. Pediatr Neurol. 2013;49(4):243–254. doi:10.1016/j.pediatrneurol.2013.08.001
2. Peron A, Northrup H. Tuberous sclerosis complex. Am J Med Genet C Semin Med Genet. 2018;178(3):274–277. doi:10.1002/ajmg.c.31657
3. Okamoto SI, Prikhodko O, Pina-Crespo J, et al. NitroSynapsin for the treatment of neurological manifestations of tuberous sclerosis complex in a rodent model. Neurobiol Dis. 2019;127:390–397. doi:10.1016/j.nbd.2019.03.029
4. Wu S, Guo Y, Liu C, et al. Identification of a de novo TSC2 variant in a Han-Chinese family with tuberous sclerosis complex. J Chin Med Assoc. 2021;84(1):46–50. doi:10.1097/JCMA.0000000000000455
5. Dodd KM, Dunlop EA. Tuberous sclerosis–A model for tumour growth. Semin Cell Dev Biol. 2016;52:3–11. doi:10.1016/j.semcdb.2016.01.025
6. Feliciano DM. The neurodevelopmental pathogenesis of Tuberous Sclerosis Complex (TSC). Front Neuroanat. 2020;14:39. doi:10.3389/fnana.2020.00039
7. Wang MX, Segaran N, Bhalla S, et al. Tuberous sclerosis: current update. Radiographics. 2021;41(7):1992–2010. doi:10.1148/rg.2021210103
8. Li Y, Cao J, Chen M, et al. Abnormal neural progenitor cells differentiated from induced pluripotent stem cells partially mimicked development of TSC2 neurological abnormalities. Stem Cell Rep. 2017;8(4):883–893. doi:10.1016/j.stemcr.2017.02.020
9. Tyburczy ME, Wang JA, Li S, et al. Sun exposure causes somatic second-hit mutations and angiofibroma development in tuberous sclerosis complex. Hum Mol Genet. 2014;23:2023–2029. doi:10.1093/hmg/ddt597
10. Gao S, Wang Z, Xie Y. Two novel TSC2 mutations in pediatric patients with tuberous sclerosis complex: case report. Medicine. 2018;97(29):e11533. doi:10.1097/MD.0000000000011533
11. Treichel AM, Hamieh L, Nathan NR, et al. Phenotypic distinctions between mosaic forms of tuberous sclerosis complex. Genet Med. 2019;21(11):2594–2604. doi:10.1038/s41436-019-0520-3
12. Giannikou K, Lasseter KD, Grevelink JM, et al. Low-level mosaicism in tuberous sclerosis complex: prevalence, clinical features, and risk of disease transmission [published correction appears in Genet Med. 2021 Oct;23(10):2022]. Genet Med. 2019;21(11):2639–2643. doi:10.1038/s41436-019-0562-6
13. Northrup H, Aronow ME, Bebin EM, et al. Updated international Tuberous Sclerosis Complex diagnostic criteria and surveillance and management recommendations. Pediatr Neurol. 2021;123:50–66. doi:10.1016/j.pediatrneurol.2021.07.011
14. Mayer K, Ballhausen W, Leistner W, Rott HD. Three novel types of splicing aberrations in the tuberous sclerosis TSC2 gene caused by mutations apart from splice consensus sequences. BBA-MOL BASIS DIS. 2000;1502:495–507. doi:10.1016/s0925-4439(00)00072-7
15. Abdelwahed M, Touraine R, Ben-Rhouma B, et al. A novel de novo splicing mutation c.1444-2A>T in the TSC2 gene causes exon skipping and premature termination in a patient with tuberous sclerosis syndrome. IUBMB Life. 2019;71(12):1937–1945. doi:10.1002/iub.2134
16. Jiangyi W, Gang G, Guohai S, et al. Germline mutation of TSC1 or TSC2 gene in Chinese patients with bilateral renal angiomyolipomas and mutation spectrum of Chinese TSC patients. Aging. 2020;12(1):756–766. doi:10.18632/aging.102654
17. Niu Y, Huang S, Xu P, et al. A case of tuberous sclerosis complex due to a novel splicing variant of TSC2 gene. Chin j med genet. 2021;38(6):553–556. doi:10.3760/cma.j.cn511374-20200211-00072
18. Qiu C, Li C, Tong X, et al. A novel TSC1 frameshift mutation c.1550_1551del causes tuberous sclerosis complex by aberrant splicing and nonsense-mediated mRNA degradation (NMD) simultaneously in a Chinese family. Mol Genet Genomic Med. 2020;8(10):e1410. doi:10.1002/mgg3.1410
19. Cariola F, Disciglio V, Valentini AM, et al. Characterization of a rare variant (c.2635-2A>G) of the MSH2 gene in a family with Lynch syndrome [published online ahead of print, 2018 Apr 24]. Int J Biol Markers. 2018:1724600818766496. doi:10.1177/1724600818766496
20. Ye Y, Zeng Y. Whole exome sequencing identifies a novel intron heterozygous mutation in TSC2 responsible for tuberous sclerosis complex. Sci Rep. 2019;9(1):4456. doi:10.1038/s41598-019-38898-9
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.