")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Preliminary in vitro and in vivo assessment of a new targeted inhibitor for choroidal neovascularization in age-related macular degeneration
Authors Li W, Dong L , Ma M, Hu B, Lu Z, Liu X, Liu J , Li X
Received 25 June 2016
Accepted for publication 16 August 2016
Published 19 October 2016 Volume 2016:10 Pages 3415—3423
DOI https://doi.org/10.2147/DDDT.S115801
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Wenbo Li,1,* Lijie Dong,1,* Minwang Ma,2,* Bojie Hu,1 Zhenyu Lu,3 Xun Liu,1 Juping Liu,1 Xiaorong Li1
1Tianjin Medical University Eye Hospital, Tianjin, People’s Republic of China; 2Affiliated Hospital of Medical College of Chinese People’s Armed Police Forces (CapF), Tianjin, People’s Republic of China; 3Tianjin Precision Cell Biotechnology Co. Ltd., Tianjin, People’s Republic of China
*These authors contributed equally to this work
Abstract: Choroidal neovascularization (CNV) in age-related macular degeneration usually causes blindness. We established a novel targeted inhibitor for CNV in age-related macular degeneration. The inhibitor CR2-sFlt 1 comprises a CR2-targeting fragment and an anti-vascular endothelial growth factor (VEGF) domain (sFlt 1). The targeting of CR2-sFlt 1 was studied using the transwell assay in vitro and frozen sections in vivo using green fluorescent labeling. Transwell assay results showed that CR2-sFlt 1 migrated to the interface of complement activation products and was present in the retinal tissue of the CR2-sFlt 1-treated CNV mice. Treatment effects were assessed by investigating the VEGF concentration in retinal pigmented epithelial cell medium and the thickness of the CNV complex in the mice treated with CR2-sFlt 1. CR2-sFlt 1 significantly reduced the VEGF secretion from retinal pigmented epithelial cells in vitro and retarded CNV progress in a mouse model. Expression analysis of VEGF and VEGFRs after CR2-sFlt 1 intervention indicated the existence of feedback mechanisms in exogenous CR2-sFlt 1, endogenous VEGF, and VEGFR interaction. In summary, we demonstrated for the first time that using CR2-sFlt 1 could inhibit CNV with clear targeting and high selectivity.
Keywords: choroidal neovascularization, macular degeneration, complement activation, vascular endothelial growth factor
Introduction
Approximately 10% of patients with age-related macular degeneration (AMD) will develop choroidal neovascularization (CNV), which is the most common cause of blindness in people older than 50 years old in developed countries.1–3 CNV, which is characterized by choroidal capillary growth through the Bruch’s membrane beneath the retinal pigmented epithelial (RPE) cell layer, forms fibrovascular proliferative tissue containing vascular endothelial cells, fibroblasts, RPE cells, and various inflammatory cells.
Vascular endothelial growth factor (VEGF) plays a central role in CNV formation in AMD and usually exerts its biological effects by binding to its cell surface receptors.4 VEGF-binding receptors include VEGF receptor-1 (VEGFR-1) and VEGFR-2. Soluble VEGFR-1 (sVEGFR-1, also known as soluble fms-like tyrosine kinase-1 [sFlt 1]) is a naturally occurring protein antagonist of VEGF formed by the alternative splicing of pre-mRNA for a full-length receptor.5–7 Its angiostatic effects are exerted via two inhibitory mechanisms: VEGF sequestering and inactive heterodimer formation with two membrane-spanning isoforms of VEGFR-1 and VEGFR-2. Therefore, sFlt 1 has gained considerable attention because of its potential clinical application as an inhibitor of angiogenesis.8–11
Complement activity is a fundamental component in the pathogenesis of CNV with AMD.12–15 Drusen formation and C3 deposition in the RPE–choroid interface (primary target tissue site of AMD) were confirmed by several studies.16–19 Moreover, adequate complement activation products were identified within the CNV where iC3b/C3dg/C3d, the cell-bound cleavage fragments of C3, remain covalently attached to these sites for prolonged periods of time.20–22 In a previous study, a domain (SCR1–4 fragment of complement receptor 2 [CR2]) targeting the iC3b/C3dg/C3d ligand-binding site was employed to target the regulatory domains to complement the activation site in AMD and attenuate the progression of CNV.22–26
In the present study, we established a novel targeted inhibitor of VEGF in CNV. The inhibitor CR2-sFlt 1 comprises a CR2-targeting fragment and an anti-VEGF domain (sFlt 1). We aimed to determine the targeting property and reducing effect of CR2-sFlt 1 on CNV.
Materials and methods
Animals
C57BL/6 mice (National Institute for Food and Drug Control, Beijing, People’s Republic of China) were bred under a 12 hour light/12 hour dark cycle. For CNV lesion preparation, 3-month-old mice were anesthetized (10% chloral hydrate), and then the pupils of the mice were dilated (using compound tropicamide eye drops). All procedures were performed in strict accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research, and approved by the Local Commission of Ethics for the Care and Use of Laboratory Animals in Tianjin, People’s Republic of China. Human RPE cells (D407) were cultured as described previously.27
Plasmid construction, expression, and purification of CR2-sFlt 1
To generate the CR2-sFlt 1-green fluorescent protein (GFP) fusion protein, we cloned complementary DNA (cDNA) sequences consisting of four N-terminal SCRs of human CR2 (NM_001006658.2) linked to human sFlt 1-His tag (RefSeq: NM_001159920.1) with a signal peptide fragment at the N-terminus of sFlt 1 sequence into eukaryotic green fluorescent protein recombinant vector (pEGFP-N2) (Figure 1). The signal peptide fragment within the CR2-sFlt 1-GFP fusion protein facilitated CR2-sFlt 1-GFP to localize outside the cells. The polyhistidine (His)-tags were used for the affinity purification of genetically modified proteins. The final plasmid constructs were transfected into HeLa cells to express the CR2-sFlt 1-GFP fusion protein, and the GFP gene was used as a reporter and directly tracked the fusion expression under a fluorescence microscope. HIS-Select Spin Columns (H7787; Sigma-Aldrich Co., St Louis, MO, USA) were employed to purify the CR2-sFlt 1-GFP fusion protein in accordance with the manufacturer’s instructions, and the concentration of the purification protein was determined using the bicinchoninic acid (BCA) Protein Assay Kit (23225; Thermo Fisher Scientific, Waltham, MA, USA).
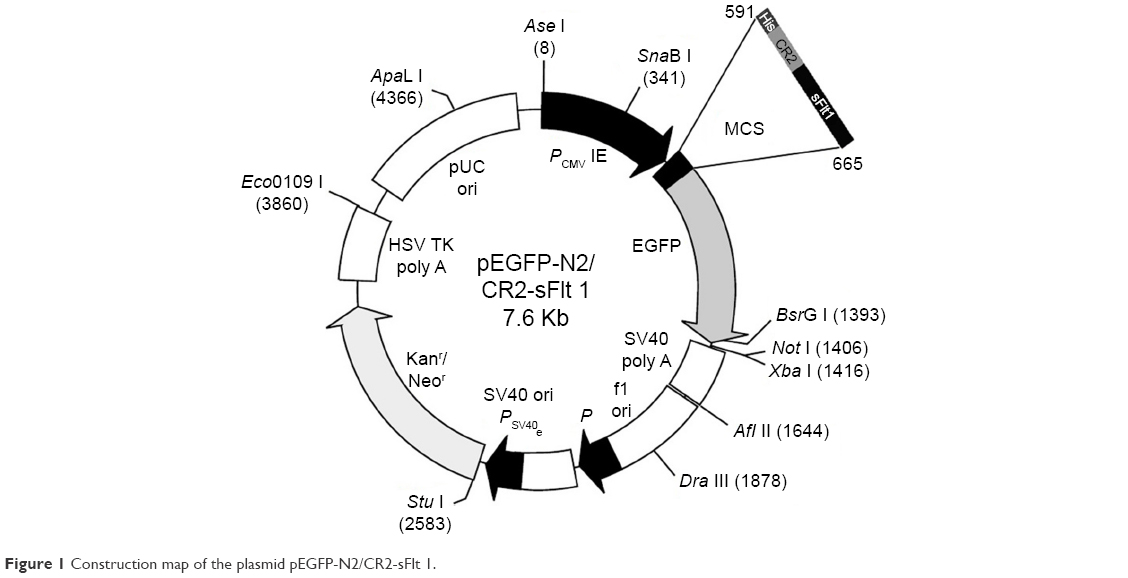 | Figure 1 Construction map of the plasmid pEGFP-N2/CR2-sFlt 1. |
Transwell chamber assay
To assess the targeting of the CR2-sFlt 1-GFP fusion protein, we analyzed the migratory ability of CR2-sFlt 1-GFP toward complement activation products. Complement activation products were prepared by incubating fresh rat sera with rabbit erythrocytes, as described in a previous work.27 Complement activation products were then detected in the lower compartment of a two-chamber Costar, which is a migratory well with a 0.4 μm pore size (3413; Corning Incorporated, Corning, NY, USA). By contrast, 200 μL of CR2-sFlt 1 (84 μg/mL) was loaded in the upper compartment. After 1 hour of incubation, images were obtained from the upper chamber, interface membrane, and lower chamber to analyze the GFP signal under a fluorescence microscope.
CNV model and CR2-sFlt 1 administration
Argon laser photocoagulation (532 nm wavelength, 100 μm spot size, 0.05 seconds duration, and 100 mW) was employed to generate four to six laser spots in each eye surrounding the optic nerve by a slit-lamp delivery system and a handheld cover slide as contact lens. The production of a vaporization bubble at the laser spot indicated the rupture of the Bruch’s membrane, which is an important factor in successful CNV. Fundus image and optical coherence tomography (OCT; Spectralis; Heidelberg Engineering, Inc., Carlsbad, CA, USA) of CNV mice were performed at the seventh day after laser injury (Figure 2A and B). For tail injections, a 20 μL volume CR2-sFlt 1 (84 μg/mL) in phosphate-buffered saline (PBS) buffer was injected at the seventh day after laser injury in 30 mice. A total of 30 mice in the control group were then subjected to a similar intensity of photocoagulation and injections of the same amounts of PBS only.
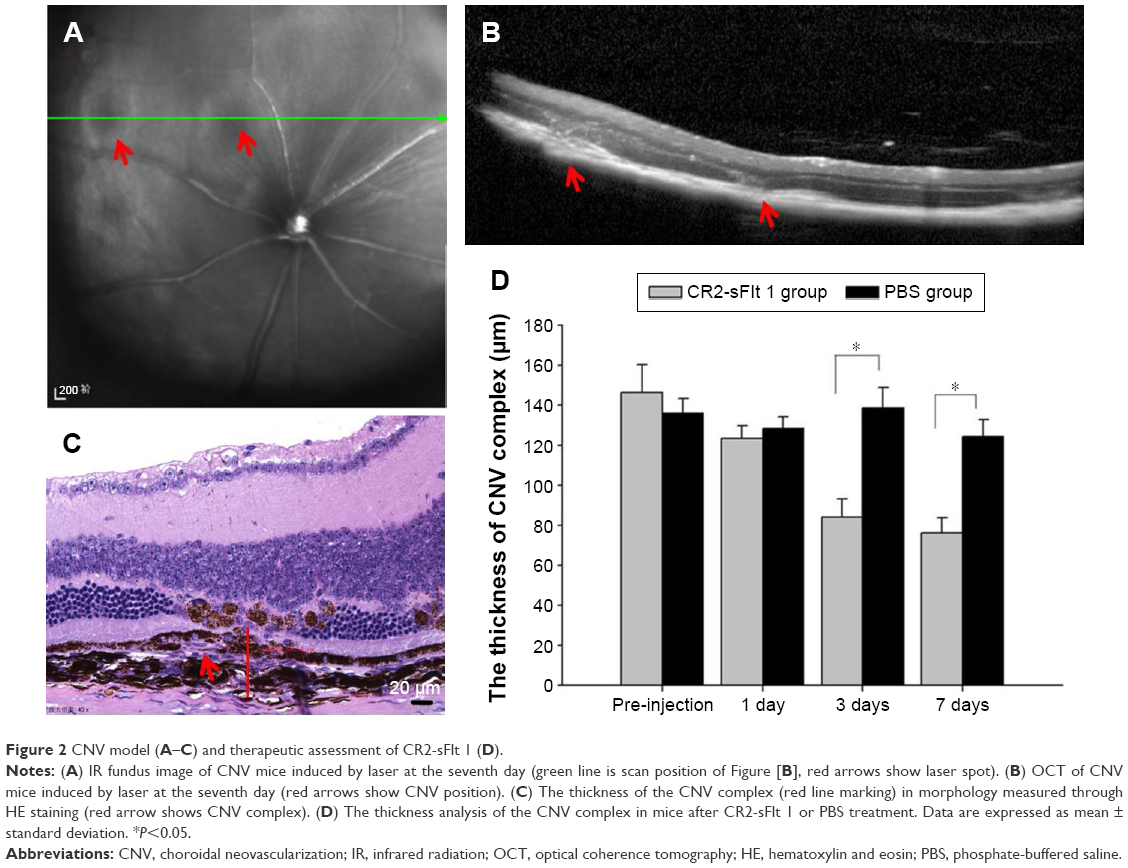 | Figure 2 CNV model (A–C) and therapeutic assessment of CR2-sFlt 1 (D). |
Histological examination
The in vivo localization of CR2-sFlt 1-GFP was performed in fresh frozen sections postfixed in ice-cold acetone. At the first day after tail vein injection, the eyes and main tissues (bladder, brain, colon, heart, kidney, liver, lung, skeletal muscle, spleen, and thyroid) of the mice were enucleated. The posterior eyecups and main tissues were fixed in 4% formaldehyde for 1 hour. After washing with PBS, the eyecups were cryoprotected in 30% sucrose overnight at 4°C and then embedded in optimal cutting temperature compound at −20°C. Sections were cut on a cryostat, placed on glass slides, and allowed to dry overnight. The GFP fluorescence image was obtained using a fluorescence microscope. To assess the CNV lesions, the mice were sacrificed on the first, third, and seventh days after injections; paraffin sections were then prepared for hematoxylin and eosin staining, and the thickness of the CNV complex was measured using the Image software (CellSens, version 1.6; Olympus America Inc., Center Valley, PA, USA) (Figure 2C).
Enzyme-linked immunosorbent assay
The supernatant of RPE cells with CR2-sFlt 1 (84, 420, and 840 μg/mL) was collected to measure VEGF levels using enzyme-linked immunosorbent assay (ELISA). Groups cultured in both normal media and bevacizumab (0.25 mg/mL) were used as control. VEGF ELISA was performed in accordance with the manufacturer’s instructions (VEGF ELISA; R&D systems, Inc., Minneapolis, MN, USA). D407 cells were cultured in a serum-free medium at a density of 1×106 cells/mL with or without VEGF for 24 hours. The conditioned media of the D407 cells were placed on a plate coated with anti-VEGF. After 2 hours, the plate was washed, and an antibody against VEGF conjugated to horseradish peroxidase was added. After another 2 hours, the plate was again washed and the substrate solution added for 30 minutes. The reaction was stopped and the fluorescence read on an ELISA reader (Thermo Max; Molecular Devices LLC, Sunnyvale, CA, USA) at 450 nm with a correction set to 540 nm. The results were plotted against the standard curve to determine the actual concentrations. Bevacizumab (0.25 mg/mL) was administered as positive control.
Quantitative real-time PCR
RNA was purified using the GeneJET RNA Purification Kit (K0731; Thermo Fisher Scientific). Total mRNA was isolated from the parental or transfected cells. RevertAid First Strand cDNA Synthesis Kit (K1622; Thermo Fisher Scientific) was utilized to generate cDNA from total mRNA. Quantitative reverse transcription polymerase chain reaction (PCR) using the SYBR Green Master Mix for VEGF, VEGFR-1, VEGFR-2, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was performed on an ABI Prism 7900 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The ratio between each respective gene and the corresponding GAPDH was calculated in accordance with the ΔΔCt threshold cycle method. The following primers were used for PCR:
VEGF, forward: 5′-GGAGGAGGCATTCGGAAAGT-3′, reverse: 5′-GCACTACCTGGGTCAGCTTCA-3′;
VEGFR-1, forward: 5′-ACAAGGCTGCCCCGACTAC-3′, reverse: 5′-CTCCTGGTATGAAATGGCAAATC-3′;
VEGFR-2, forward: 5′-GGAGGAGGCATTCGGAAAGT-3′, reverse: 5′-GCACTACCTGGGTCAGCTTCA-3′;
GAPDH, forward: 5′-TGAAACCCTAGTTCATATCTTCAAACA-3′, reverse: 5′-CTCCTTCTGTGACTCTAACTTCTCCAT-3′.
Immunoblotting and antibodies
D407 cells or retinal tissues were homogenized in radioimmunoprecipitation assay lysis buffer supplemented with protease inhibitor cocktail (P8340; Sigma-Aldrich Co.). The total lysates were collected by centrifugation at 16,000× g for 15 minutes at 4°C. The concentration of lysates was determined using BCA assay. The same amounts of proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes. After incubation with blocking buffer for 1 hour at room temperature, the membranes were probed with antibodies specific to VEGF (ab46154; Abcam, Cambridge, MA, USA), VEGFR-1 (ab32152; Abcam), and VEGFR-2 (ab2349; Abcam), with GAPDH (ab8245; Abcam) as the loading control.
Statistical analysis
All data were analyzed using a commercially available statistical software package (SPSS, version 21.0; IBM Corporation, Armonk, NY, USA). The parameters were presented as mean ± standard deviation. Expression of gene mRNA, the thicknesses of the CNV complex, and the secretion of VEGF were analyzed using analysis of variance. A P-value of <0.05 was considered significant.
Results
Targeting of CR2-sFlt 1 to complement activation sites in vitro and in vivo
The targeting performance of CR2-sFlt 1 was confirmed by performing the transwell assay to analyze the chemotaxis of GFP-labeled CR2-sFlt 1 toward complement cleavage fragments. The complement cleavage fragments were deposited in the lower chamber, whereas the purified CR2-sFlt 1-GFP protein was loaded in the upper transwell chamber (Figure 3A). After 1 hour of incubation, no GFP signal was detected in the upper chamber suspension (Figure 3B). Most increased green signals were excited within the interface membrane of the transwell chamber (Figure 3C). Some green signals passed through the membrane and were localized in the lower chamber suspension (Figure 3D). This result suggests that the CR2 domain can target CR2-sFlt 1 to complement activation sites in vitro.
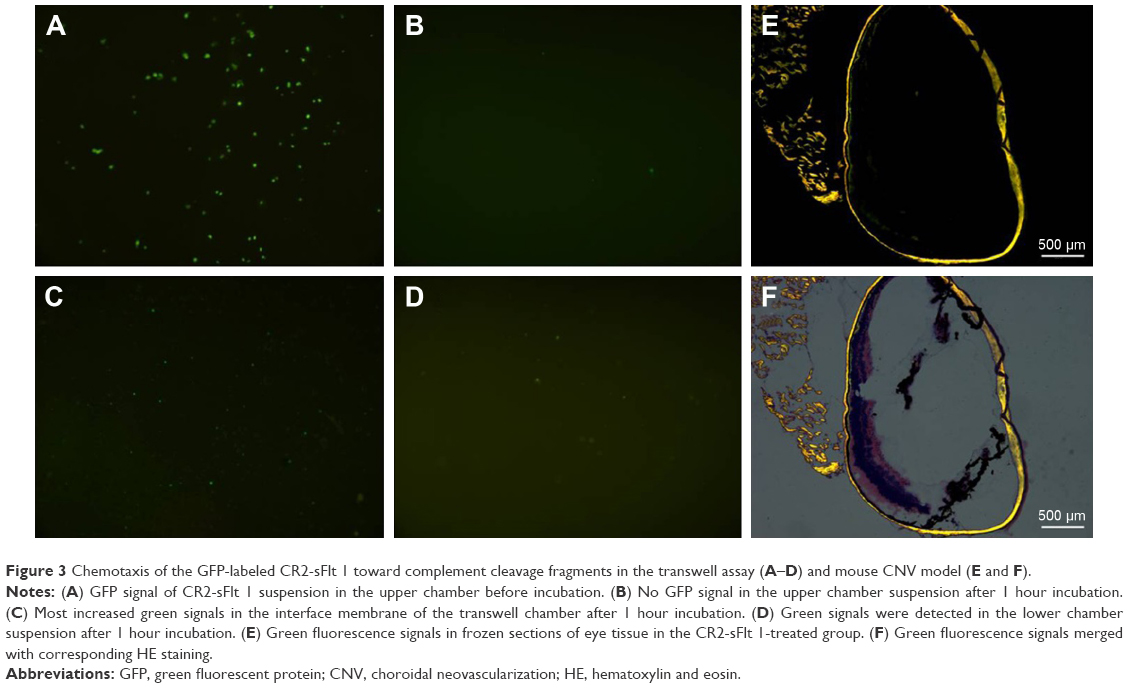 | Figure 3 Chemotaxis of the GFP-labeled CR2-sFlt 1 toward complement cleavage fragments in the transwell assay (A–D) and mouse CNV model (E and F). |
In the mouse CNV model, on the first day after tail vein injection, frozen sections of retinal tissue in the CR2-sFlt 1-treated group showed green fluorescence signals, but the signals were significantly stronger in the optic disk area and laser spots. This result suggests that CR2-sFlt 1 was mobilized and deposited in the retina of the CNV mice (Figure 3E and F). No fluorescence was observed in the main tissues, including the bladder, brain, colon, heart, kidney, liver, lung, skeletal muscle, spleen, and thyroid.
Therapeutic assessment of CR2-sFlt 1
Histological analysis was performed to investigate the development of CNV in the mice after tail vein treatment with CR2-sFlt 1 or vehicle (PBS). Animals received tail vein injections on the seventh day after laser photocoagulation. On the first, third, and seventh days after injection, the mice were sacrificed to prepare the paraffin section of retinal tissue. The thickness of the CNV complex was 146.17±14.15 μm on the seventh day after laser photocoagulation in the untreated mice. On the first, third, and seventh days after CR2-sFlt 1 treatment, the thicknesses of the CNV complex were 123.17±6.62, 73.32±9.26, and 66.36±7.52 μm, respectively. This result indicates that CR2-sFlt 1 significantly reduced CNV lesion sizes when compared with PBS treatment (*P<0.05) (Figure 2D).
ELISA results revealed the effect of CR2-sFlt 1 on the secretion of VEGF in RPE cells. CR2-sFlt 1 significantly reduced VEGF secretion from RPE cells (Figure 4). After 24 hours, the VEGF concentration in the medium reduced in the CR2-sFlt 1-treated groups compared with the normal control group. After 48 hours, the VEGF concentration in the medium with 84 μg/mL CR2-sFlt 1 was reduced. However, the VEGF concentration in the medium with 420 and 840 μg/mL CR2-sFlt 1 slightly rebounded. After 72 hours, the increase in VEGF concentration in the medium with 84 μg/mL CR2-sFlt 1 was lower than that in the control group. A total of 84 μg/mL CR2-sFlt 1 was generated with the adequate dosage of intervention. However, CR2-sFlt 1 was less effective than bevacizumab. Thus, the optimal therapeutic dosage of CR2-sFlt 1 treatment must be identified in future studies.
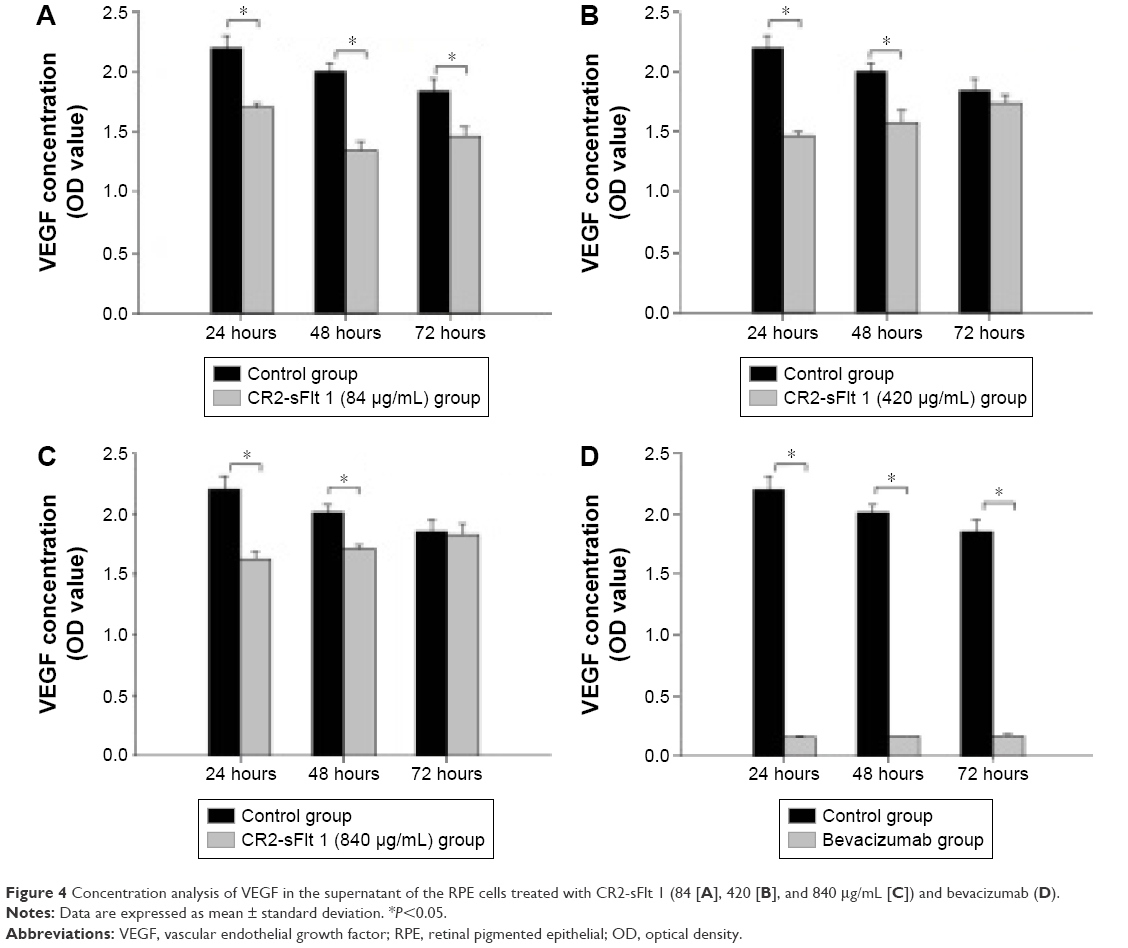 | Figure 4 Concentration analysis of VEGF in the supernatant of the RPE cells treated with CR2-sFlt 1 (84 [A], 420 [B], and 840 μg/mL [C]) and bevacizumab (D). |
Effects of CR2-sFlt 1 on the expression of VEGF and VEGFRs
VEGF, VEGFR-1, and VEGFR-2 were significantly upregulated at 24 hours and then dramatically decreased at 48 hours in the RPE cells treated with 84 μg/mL CR2-sFlt 1. However, VEGF and VEGFRs slightly increased again at 72 hours (Figure 5A). VEGF expression significantly increased in the CNV mice on the seventh day compared with that on the first day after PBS injection. After CR2-sFlt 1 intervention, VEGF expression significantly decreased on the seventh day unlike that in the PBS-treated group at the same time point (Figure 5B).
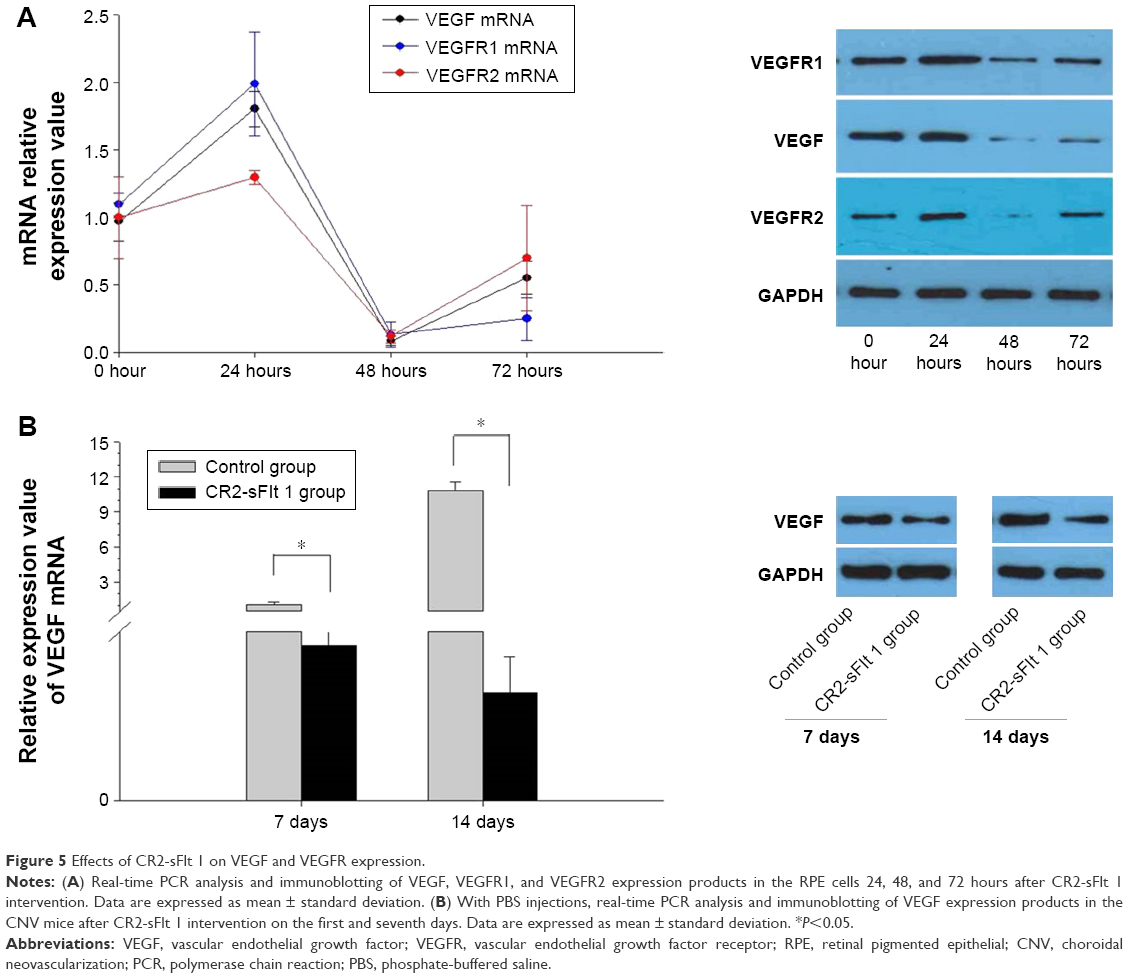 | Figure 5 Effects of CR2-sFlt 1 on VEGF and VEGFR expression. |
Discussion
Various antiangiogenic therapies, such as bevacizumab, ranibizumab, conbercept, and aflibercept, have been developed. These therapies target the VEGF–VEGFR system to inhibit CNV. An inhibitor of the alternative pathway of complement activation linked to a CR2-targeting fragment also exists; this inhibitor has been proven effective in mouse CNV, but has not yet entered clinical practice.22 The novel CR2-sFlt 1 developed in this study can be “targeted” to sites of complement activation lesions and bind with VEGF with high affinity. This occurrence then inhibits the progression of mouse CNV.
The main results of the current study are as follows.
- The CR2-sFlt 1 fusion protein mobilizes from remote tissues to the C3-deposited RPE/choroid interface in a target organ and requires mobilization signals provided primarily by the CR2 domain. The targeting of CR2-sFlt 1 was demonstrated both in vitro and in vivo. In vitro, the transwell assay showed that the CR2-sFlt 1 in the upper chamber could migrate to the interface incubated with a solution containing complement activation products. In vivo, CR2-sFlt 1 transported from remote tissues to the C3-deposited RPE/choroid interface in a target organ visualized using imaging technology.
- CR2-sFlt 1, a soluble VEGF inhibitor, significantly reduced CNV. We confirmed that administering CR2-sFlt 1 by tail vein injection reduces the thickness of the CNV complex.
- CR2-sFlt 1 exerted its action on the secreted VEGF in RPE cells by binding VEGF with high affinity, thus preventing VEGF from interacting with VEGFR-1 and VEGFR-2. The formation of the VEGF-CR2-sFlt 1 complex failed to activate the VEGF–VEGFR signaling pathway within RPE cells, which provide protection against CNV.
- The therapeutic effect of CR2-sFlt 1 was achieved with tail vein injections and did not require intraocular applications. Current inhibitors of VEGF could inhibit the ocular neovascularization efficiently, but require repeated intraocular injections that carry a risk of injection-related adverse events, including ocular inflammation, retinal injury or detachment, and endophthalmitis. Moreover, commercial VEGF inhibitors, such as VEGF monoclonal antibody or VEGF-trap-inactivated VEGF, are potent without selectivity. In other words, these inhibitors failed to distinguish pathological VEGF from physiological VEGF. Normal VEGF plays an essential role in maintaining vascular structure and function. The overinhibition of VEGF inevitably causes future complications. Therefore, these disadvantages prompt the development of efficient VEGF inhibitors that can potentially be administered less frequently and act more selectively and potently. Herein, we proposed the use of CR2-sFlt 1, a unique VEGF blocker, as a systemic approach for ocular delivery with clear-cut targeting that successfully circumvents adverse events.
Our data also indicated the existence of feedback mechanisms among exogenous CR2-sFlt 1, endogenous VEGF, and VEGFR interaction. The expression of VEGF in RPE cells was upregulated shortly after CR2-sFlt 1 intervention, indicating that negative feedback regulations may be immediately triggered by the interaction between endogenous VEGF and CR2-sFlt 1. The high VEGF expression in RPE cells was temporary, and the amount of VEGF secreted by RPE cells always decreased with CR2-sFlt 1 intervention. VEGFR expression was consistent with the VEGF changes in RPE cells. The effectiveness of CR2-sFlt 1 remains unsatisfactory and needs to be improved. An optimal therapeutic dosage of CR2-sFlt 1 treatment must be determined in the future.
Conclusion
The CR2 domain within CR2-sFlt 1 can target CR2-sFlt 1 to sites of complement deposition within the CNV area. The targeted inhibitor specific for VEGF significantly retarded CNV progress in a mouse model. We demonstrated for the first time that using CR2-sFlt 1 could inhibit CNV with clear targeting and high selectivity.
Acknowledgment
This research was supported by grants from the National Natural Science Foundation of China (Nos 81400425 and 81570872) and Tianjin Municipal Science and Technology Commission (15JCYBJC24900).
Disclosure
The authors alone were responsible for the content and writing of the paper. The authors report no conflicts of interest in this work.
References
Semeraro F, Morescalchi F, Duse S, et al. Aflibercept in wet AMD: specific role and optimal use. Drug Des Devel Ther. 2013;7:711–722. | ||
Panos GD, Gatzioufas Z, Petropoulos IK, et al. Effect of ranibizumab on serous and vascular pigment epithelial detachments associated with exudative age-related macular degeneration. Drug Des Devel Ther. 2013;7:565–569. | ||
Tranos P, Vacalis A, Asteriadis S, et al. Resistance to antivascular endothelial growth factor treatment in age-related macular degeneration. Drug Des Devel Ther. 2013;7:485–490. | ||
Kwak N, Okamoto N, Wood JM, Campochiaro PA. VEGF is major stimulator in model of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2000;41(10):3158–3164. | ||
Gehlbach P, Demetriades AM, Yamamoto S, et al. Periocular gene transfer of sFlt-1 suppresses ocular neovascularization and vascular endothelial growth factor-induced breakdown of the blood-retinal barrier. Hum Gene Ther. 2003;14(2):129–141. | ||
He Y, Smith SK, Day KA, et al. Alternative splicing of vascular endothelial growth factor (VEGF)-R1 (FLT-1) pre-mRNA is important for the regulation of VEGF activity. Mol Endocrinol. 1999;13(4):537–545. | ||
Kendall RL, Wang G, Thomas KA. Identification of a natural soluble form of the vascular endothelial growth factor receptor, FLT-1, and its heterodimerization with KDR. Biochem Biophys Res Commun. 1996;226(2):324–328. | ||
Bainbridge JW, Mistry A, De Alwis M, et al. Inhibition of retinal neovascularisation by gene transfer of soluble VEGF receptor sFlt-1. Gene Ther. 2002;9(5):320–326. | ||
Honda M, Sakamoto T, Ishibashi T, Inomata H, Ueno H. Experimental subretinal neovascularization is inhibited by adenovirus-mediated soluble VEGF/flt-1 receptor gene transfection: a role of VEGF and possible treatment for SRN in age-related macular degeneration. Gene Ther. 2000;7(11):978–985. | ||
Lai CM, Brankov M, Zaknich T, et al. Inhibition of angiogenesis by adenovirus-mediated sFlt-1 expression in a rat model of corneal neovascularization. Hum Gene Ther. 2001;12(10):1299–1310. | ||
Lai YK, Shen WY, Brankov M, et al. Potential long-term inhibition of ocular neovascularisation by recombinant adeno-associated virus-mediated secretion gene therapy. Gene Ther. 2002;9(12):804–813. | ||
Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103(7):2328–2333. | ||
Bora NS, Kaliappan S, Jha P, et al. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: role of factor B and factor H. J Immunol. 2006;177(3):1872–1878. | ||
Bora NS, Kaliappan S, Jha P, et al. CD59, a complement regulatory protein, controls choroidal neovascularization in a mouse model of wet-type age-related macular degeneration. J Immunol. 2007;178(3):1783–1790. | ||
Bora NS, Jha P, Lyzogubov VV, et al. Recombinant membrane-targeted form of CD59 inhibits the growth of choroidal neovascular complex in mice. J Biol Chem. 2010;285(44):33826–33833. | ||
Hageman GS, Luthert PJ, Victor Chong NH, et al. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20(6):705–732. | ||
Yates JR, Sepp T, Matharu BK, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357(6):553–561. | ||
Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73(6):887–896. | ||
Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134(3):411–431. | ||
Huang Y, Qiao F, Atkinson C, et al. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. J Immunol. 2008;181(11):8068–8076. | ||
Fridkis-Hareli M, Storek M, Mazsaroff I, et al. Design and development of TT30, a novel C3d-targeted C3/C5 convertase inhibitor for treatment of human complement alternative pathway-mediated diseases. Blood. 2011;118(17):4705–4713. | ||
Rohrer B, Long Q, Coughlin B, et al. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50(7):3056–3064. | ||
Rohrer B, Coughlin B, Bandyopadhyay M, Holers VM. Systemic human CR2-targeted complement alternative pathway inhibitor ameliorates mouse laser-induced choroidal neovascularization. J Ocul Pharmacol Ther. 2012;28(4):402–409. | ||
Banda NK, Levitt B, Glogowska MJ, et al. Targeted inhibition of the complement alternative pathway with complement receptor 2 and factor H attenuates collagen antibody-induced arthritis in mice. J Immunol. 2009;183(9):5928–5937. | ||
Hu X, Tomlinson S, Barnum SR. Targeted inhibition of complement using complement receptor 2-conjugated inhibitors attenuates EAE. Neurosci Lett. 2012;531(1):35–39. | ||
Song H, He C, Knaak C, et al. Complement receptor 2-mediated targeting of complement inhibitors to sites of complement activation. J Clin Invest. 2003;111(12):1875–1885. | ||
Li W, Chen S, Ma M, Qian J, Ma X. Complement 5b-9 complex-induced alterations in human RPE cell physiology. Med Sci Monit. 2010;16(1):BR17–BR23. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.