")
Back to Journals » Journal of Inflammation Research » Volume 14
Preclinical Characterization of the Selective JAK1 Inhibitor LW402 for Treatment of Rheumatoid Arthritis
Authors Zhang N , Zhang C, Zeng Z, Zhang J, Du S, Bao C, Wang Z
Received 8 January 2021
Accepted for publication 1 April 2021
Published 21 May 2021 Volume 2021:14 Pages 2133—2147
DOI https://doi.org/10.2147/JIR.S301076
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Ning Zhang,1 Changqing Zhang,2 Zhihong Zeng,1 Jiyong Zhang,1 Shengnan Du,1 Chunde Bao,3 Zhe Wang1
1Longwood Biopharmaceuticals, Shanghai, People’s Republic of China; 2Department of Orthopaedics, Shanghai Sixth People’s Hospital, Shanghai, People’s Republic of China; 3Department of Rheumatology, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, People’s Republic of China
Correspondence: Zhe Wang Email [email protected]
Background: Research on JAK family members as therapeutic targets for autoimmune diseases has brought tofacitinib and baricitinib into clinical for the treatment of rheumatoid arthritis and other autoimmune diseases. Despite the potent efficacy of these first-generation JAK inhibitors, their broad-spectrum JAK inhibition and adverse events warrant development of a JAK1-specific inhibitor to improve their safety profile.
Methods: In this study, we characterized a JAK1-specific inhibitor, LW402, on biochemical and human whole-blood assays. We further evaluated the therapeutic efficacy of LW402 in a rat adjuvant-induced arthritis (rAIA) model and a mouse collagen-induced arthritis (mCIA) model. The safety of LW402 was evaluated in both SpragueDawley rats and cynomolgus monkeys.
Results: LW402 exhibited potent nanomolar activity against JAK1 and showed a 45-fold selectivity for inhibition of JAK1- over JAK2-dependent signaling induced by either IL6 or GM-CSF in human whole-blood assays. In the rAIA model, oral dosing of LW402 resulted in a dose-dependent improvement in disease symptoms, including reduction in paw swelling, marked reduction in the inflammatory-cell infiltration to synovial tissue, and protection of articular cartilage and bone from damage. The therapeutic efficacy of LW402 correlated well with the plasma exposure of LW402 and the extent of pSTAT3 inhibition in white blood cells. LW402 also effectively eased disease symptoms in the mCIA model. Toxicity studies in the Sprague Dawley rats and cynomolgus monkeys established a ≥ 5x therapeutic window for LW402 as drug exposures of toxicity study NOAEL dose and pharmacology study ED50 dose were compared.
Conclusion: We developed a novel JAK1-specific inhibitor LW402 with potent efficacy in rAIA and mCIA models. We established a good safety profile for LW402 in toxicity studies, and the overall superiority of LW402 should translated well to the clinical setting for the treatment of RA and other autoimmune diseases.
Keywords: rheumatoid arthritis, JAK1 inhibitor, rat-AIA model, mouse CIA
Introduction
Rheumatoid arthritis (RA) is a systemic inflammatory condition with synovitis as a major pathological symptom, which can deteriorate to joint damage and severe disability. The prevalence of RA is 0.5%–1.0% in most populations. RA occurs in a wide age range, although incidence generally peaks at late middle-age and is more common in women than men. Conventional RA therapy relies on synthetic disease-modifying antirheumatic drugs (sDMARDs), such as methotrexate, and a short period of glucocorticoids. Although the etiology of RA remains poorly understood, involvement of proinflammatory cytokines, such as TNFα and IL6 have been indicated for their contribution to disease progression. Biological DMARDs (bDMARDs), including TNFα and IL6 antibodies, have been developed for clinical therapy of RA, particularly to address the poor prognosis of sDMARD-refractory patients.1 Although a majority of bDMARD-treated RA patients can attain partial remission of the disease, certain troublesome symptoms, such as pain, fatigue, and morning joint stiffness, can accompany the treatment.2 Loss of response to existing therapies and intolerance/adverse effects causing drug discontinuation warrant investigation to pursue alternative therapies.
Involvement of proinflammatory cytokines in the disease progression of RA has attracted interest in modulating the downstream JAK–STAT pathway as an alternative RA-treatment strategy. The JAK family, which contains four members — JAK1, JAK2, JAK3, and TYK2 — are cytoplasmic tyrosine kinases critical for intracellular signal transduction of >50 cytokines and growth factors.3 JAKs bind to the intracellular moieties of type I and type II receptors and become activated as either homodimers or heterodimers upon ligand binding to outer-membrane receptors. Activation of JAKs involves cross-phosphorylation followed by phosphorylation of tyrosine residues on the intracellular domains of the receptors. These phosphorylated residues serve as docking sites for STAT factors, which are phosphorylated and detached from the docking sites to translocate into the nucleus, where they initiate the transcriptional activity of IFN-stimulated genes.4–6 Ubiquitous expression of JAK1 and JAK2 in virtually all cell types and specific expression of JAK3 in immune-system cells are important roles of JAK family members in the functioning of the immune system. Mouse and human genetic studies have linked deficiencies in JAK1 and JAK3 to severe combined immunodeficiency and TYK2 to increased susceptibility to infections.7,8 JAK2 mediates signal transduction of inflammatory cytokines, such as IFNγ, IL12, IL23, and GM-CSF.5,9
While the JAK inhibitors baricitinib,10 tofacitinib,11,12 upadacitinib13 have been approved by the FDA for treatment of RA, management of other autoimmune disease conditions, such as ankylosing spondylitis, psoriasis, dry-eye syndrome, ulcerative colitis, Crohn’s disease, and alopecia areata9,11,14–17 with JAK inhibitors have been actively explored in clinical trials. Although the first generation of JAK inhibitors provide therapeutic benefit in RA patients in comparison to conventional therapies, occurrence of serious adverse events, such as infection, herpes zoster, and thrombosis, raises safety concerns regarding their use in long-term care. With the broad involvement of JAKs in the functioning of the immune system, selective inhibition of JAKs may hold the key for minimization of adverse events in the treatment of immunoinflammatory disorders. Studies have established that JAK1 dominates JAK1/JAK3/γc signaling.18–20 It is expected that specific inhibition of JAK1 signaling without disturbance of other immunity-essential signaling via other JAK family members should retain most of the in vivo efficacy, but avoid or minimize some of the adverse effects.
In this paper, we characterized a novel JAK1-specific inhibitor, LW402,21 in preclinical studies and demonstrated a favorable efficacy and safety profile that warrants further clinical investigation.
Methods
Small-Molecule Kinase Inhibitors
LW402 was synthesized at Longwood Biopharmaceuticals.
Biochemical Assays for IC50 Determination
Recombinant JAK1, JAK2, JAK3 and TYK2 (Invitrogen) were used to develop activity assays in 50 mM HEPES (pH 7.5), 10 mM MgCl2, 2 mM DTT, 1 mM EDTA, and 0.01% BRIJ 35. The amount of JAK protein was determined per aliquot, maintaining initial velocity and linearity over time. ATP concentration corresponded with the experimentally determined Km value, and substrate concentration (ULight-conjugated JAK1 [Tyr1023] peptide; PerkinElmer) was set according to the manufacturer’s instruction. After 90 minutes’ incubation at room temperature, the amount of phosphorylated substrate was measured with the addition of 2 nM europium–anti-phosphotyrosine Ab (PerkinElmer) and 10 mM EDTA in Lance detection buffer (PerkinElmer). Compound IC50 values were determined by incubation with enzymes and ATP at room temperature for 90 minutes.
Human Whole-Blood Assays to Assess JAK1 Signaling and JAK2 Homodimer Signaling
Human whole-blood assays (WBAs) were conducted in accordance with previously published methods.25 Blood from healthy volunteers who had given informed consent was collected into sodium heparin tubes after preincubation with compounds at 37°C for 30 minutes, triggered with recombinant human IL6 (100 ng/mL; R&D Systems), human GM-CSF (20 ng/mL; PeproTech), or vehicle (PBS plus 0.1% BSA) for 20 minutes at 37°C, then treated with lysis/fix buffer (BD) for 10 minutes to lyse red blood cells and fix leukocytes. Cells were then permeabilized with precold Perm buffer III (BD) for 60 minutes, followed by incubation with anti-pSTAT1 and anti-CD4 (IL6–triggered samples) to assess JAK1 signaling or anti-pSTAT5 and anti-CD33 Abs (GM-CSF–triggered samples) to assess JAK2 homodimer signaling. Cells were analyzed on a Thermo Attune NxT cytometer.
Pharmacokinetic Study Formulations
For intravenous (IV) administration, LW402 was formulated in DMA/30% solutol HS15/30% HP-β-CD (5:10:85 v:v:v). For oral (per os [PO]) administration to rats, LW402 was formulated in 0.5% (v:v) methylcellulose plus 0.2% Tween 80 (v:v). For OS administration to cynomolgus monkeys, LW402 was formulated in 20% HP-β-CD with pH adjusted to 3.3 to form a clear solution.
Pharmacokinetic Study Animals
Sprague Dawley rats (6–10 weeks old) were obtained from Vital River (Beijing, China). Cynomolgus monkeys were supplied by Kunming Biomed International (Kunming, China). Animals were deprived of food for at least 16 hours before OS dosing 4–6 hours later. Cynomolgus monkey pharmacokinetic (PK) studies were carried out at Kunming Biomed International.
Pharmacokinetic Study Procedure
PK studies were conducted in accordance with previously published methods.17 In the rat PK study, LW402 was dosed via PO administration at 10, 30, and 100 mg/kg and via IV administration with a bolus via the caudal vein at 5 mg/kg. Each group consisted of three male and three female animals, and blood samples were collected via the jugular vein. In the cynomolgus monkey study, LW402 was dosed via PO administration at 10, 30, and 60 mg/kg and with a bolus via the cephalic vein of the forelimb at 5 mg/kg. Each group consisted of three male and three female animals, and blood samples were collected via the lesser saphenous vein of the hindlimb. EDTA-K2 was used as anticoagulant, and blood was taken at 0.083, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours (IV) and predose, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours (PO). LW402 plasma concentrations were determined by liquid chromatography–tandem mass spectrometry with a lower limit of quantification of 5 ng/mL. PK parameters were calculated by noncompartmental analysis using Phoenix WinNonlin 8.0 software.
Pharmacology-Study Animals and Reagents
Female Lewis rats (7–9 weeks old) were obtained from Vital River (Beijing, China) for adjuvant-induced arthritis (AIA) model establishment and study. Male DBA/1 mice (8–12 weeks old) were obtained from the Shanghai Laboratory Animal Center for collagen-induced arthritis (CIA) model establishment and study. Complete Freund’s adjuvant (CFA) was purchased from Sigma-Aldrich (F5881; St Louis, MO, USA). Mycobacterium tuberculosis (H37Ra) was purchased from Difco (Detroit, MI, USA). Chick type II collagen was purchased from Chondrex (20011; Redmond, WA, USA).
Rat AIA–Model Study
The rat-AIA study was conducted in accordance with previously published methods.10 CFA was prepared by mixing and grinding M. tuberculosis H37Ra with paraffin oil to obtain a CFA suspension at a concentration of 10 mg/mL. Eighty Lewis rats were intradermally immunized with 100 μL CFA suspension at the base of the tail on day 1. Rats were monitored and scored for clinical symptoms of arthritis after immunization and randomly allocated into ten groups (n=8) on day 11, when average clinical score had reached 2.9±0.1. Efficacy of LW402 was determined after OS administration twice daily (bis in die [BID]) of 1–100 mg/kg or once daily (quaque die [QD]) of 10–100 mg/kg for 14 days. On day 25, blood samples were collected by retro-orbital puncture into EDTA-K2 anticoagulant-containing tubes at predose (0 hours) and 1, 3, 6, 12, and 24 hours (n=3/time point) for PK analysis of LW402. pSTAT3 levels in blood samples were measured according to a previously published protocol.10
Mouse CIA–Model Study
The mouse-CIA study was conducted in accordance withpreviously published methods.25 Upon onset of disease induction on day 26, animals were randomized into five groups (n=10) and treated with vehicle (model group) and 50, 100, and 200 mg/kg (BID) LW402. Naïve mice were used as negative controls. Animals were monitored and scored for clinical symptoms of arthritis and foot thickness.
Clinical Assessment of Arthritis, X-Rays, and Histopathological Evaluation
Clinical scores were recorded twice a week on a 0–4 scale, as previously used.22 On day 26, all animals were killed and hind paws removed for X-ray evaluation and histological examination. Severity of bone erosion was blindly ranked using a modified version of the Larsen scoring method.23 For histopathological analysis, the hind paws were fixed in 3.7% (v:v) formaldehyde at 4°C for 48 hours and decalcified. Three series of 5 μm sections were taken at 100 μm intervals from the middle part of the paw. The sections were stained with H&E and safranine to visualize the severity of collagen damage, and histological analyses of joint damage were performed as previously described23 to investigate the effect of the testing compounds. Overall histological evaluations were combined into a summed score to assess the extent of drug treatment–mediated improvement of histopathological symptoms. Statistical analysis was performed using one-way ANOVA unpaired test versus the vehicle group.
Preclinical PK/PD Modeling
Raw data from LW402 studies in the rat-AIA model were used in this assessment. Pharmacodynamic (PD) values (severity-score improvement) were assessed by calculating the percentage of improvement in the treatment groups over the disease group at the end of the study. PD values were plotted against their PK AUC (BID and QD, respectively) to determine AUC50. Clinical scores measured on day 25 were correlated with drug doses to obtain ED50 values for BID and QD dosing using GraphPad Prism (GraphPad Software, La Jolla, CA).
GLP Toxicity Study
Sprague Dawley rats (6–10 weeks old) were obtained from Vital River. Cynomolgus monkeys (2.5–3.5 years old) were supplied by Guangxi Grand Forest Scientific Primate (Guangxi, China). For toxicity studies on the Sprague Dawley rats, LW402 was formulated in 0.5% methylcellulose/0.2% Tween 80 as a suspension and delivered to four groups (15/sex/group) via OS administration at 0, 30, 100, and 300 mg/kg daily for 28 days, followed by a 28-day recovery period. For toxicity studies on cynomolgus monkeys, LW402 was formulated in 20% HP-β-CD as a solution, and delivered to the four groups (five/sex/group) via OS administration at 0, 10, 30, and 60 mg/kg daily for 28 days, followed by a 28-day recovery period. During the study period, toxicity of drug dosing animals was monitored by clinical observation, body weight, food consumption, body temperature, electrocardiography, blood pressure, ophthalmoscopic examination, hematology, coagulation, clinical chemistry, urinalysis, toxicokinetics, organ weight (including male and female reproductive tissues), bone marrow, and macroscopic and microscopic examination.
Results
Biochemical and Cellular Activity of LW402
The biochemical potency and selectivity of LW402 over the JAK family of kinases were evaluated by assessing the inhibitory activity of this compound on all four JAK family members in enzymatic assays. As LW402 is an ATP-competitive kinase inhibitor, we characterized relative selectivity for JAKs in a biologically relevant manner by conducting these assays at ATP concentrations approximating those within cells. As shown in Table 1, LW402 functions as a potent inhibitor of JAK1, with IC50 for 7.7 nM. Selectivity forr JAK2, JAK3, and TYK2 was 1.65-, 22.85-, and 29.48-fold respectively.
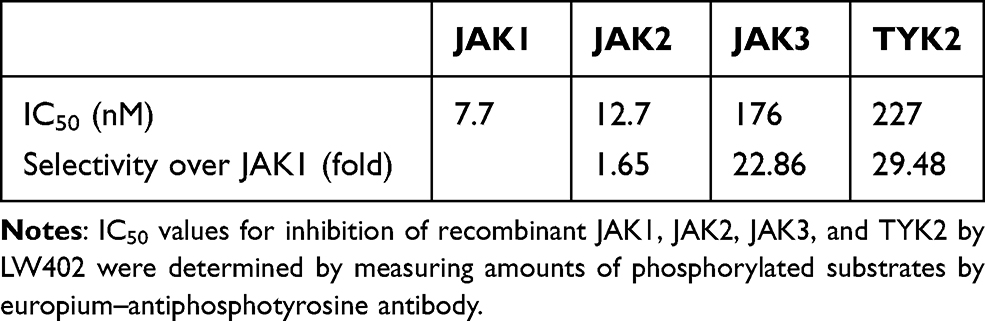 |
Table 1 LW402 Potency and Selectivity on Biochemical Assay |
Human WBAs were conducted to assess the relative inhibitory effect of JAK1 versus JAK2 following cytokine stimulation of STAT1 or STAT5 phosphorylation. IL6 stimulation triggered JAK1-mediated STAT1 phosphorylation in CD4+ lymphocytes, while GM-CSF stimulation activated JAK2-mediated STAT5 phosphorylation in myeloid cells.23,24 The effects of LW402 treatment on JAK1 vs JAK2 signaling in human WBAs are presented in Table 2. In CD4+ lymphocytes, IL6-induced STAT1 phosphorylation was inhibited by LW402, with an IC50 of 414 nM. In CD33+ myeloid cells, GM-CSF–induced STAT5 phosphorylation was inhibited by LW402, with an IC50 of 19,917 nM. In comparison, 45-fold selectivity of JAK1 over JAK2 was established for LW402 in the WBAs. In CD8+ lymphocytes, an IC50 of 844 nM was obtained for IL2-induced JAK1/3–STAT5 signaling. Together with the biochemical assays, these data indicate that LW402 is a more selective JAK1 inhibitor in a cellular environment, which is more physiologically relevant.
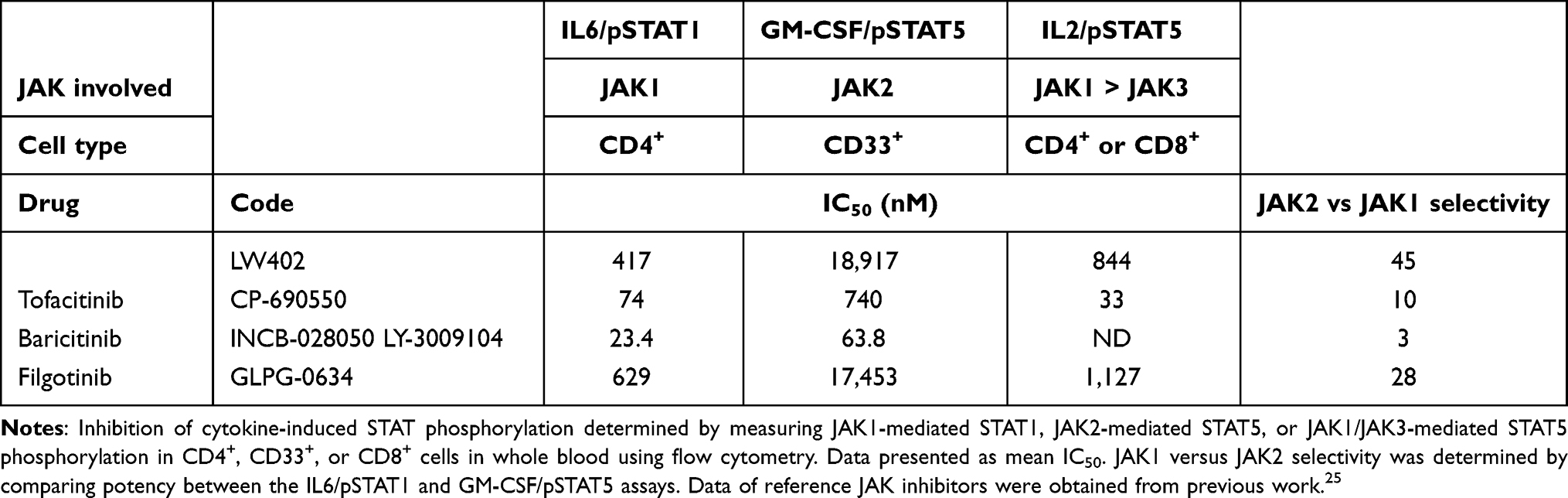 |
Table 2 Potency and Selectivity of LW402 Assayed in WBAs |
Evaluation of LW402 in the Rat-AIA Model
The therapeutic potential of LW402 for RA treatment was evaluated in a rat-AIA model. Lewis rats were intradermally immunized with a CFA suspension and monitored for clinical symptoms of arthritis. On day 11, when average clinical score had reached 2.9, animals were treated with LW402 by OS gavage BID at 1, 3, 10, 30, and 100 mg/kg or QD at 10, 30, and 100 mg/kg. Assessment of disease symptoms was conducted and clinical scores plotted over treatment time. As shown in Figure 1A and B, LW402 treatment inhibited disease progression in a dose-dependent manner in both BID or QD dosing regimen. For BID, treatment significantly improved disease symptoms compared to the model group (G2) for the 10, 30, and 100 mg/kg dosing groups from day 14 to day 25. For QD, both the 30 and 100 mg/kg dosing groups exhibited significant improvement in clinical scores from day 14 (Table 3). Correlations between normalized clinical scores on day 25 and drug dose were analyzed, which showed ED50 of 7.99 mg/kg and 24.23 mg/kg for BID and QD respectively (Figure 1C and D).
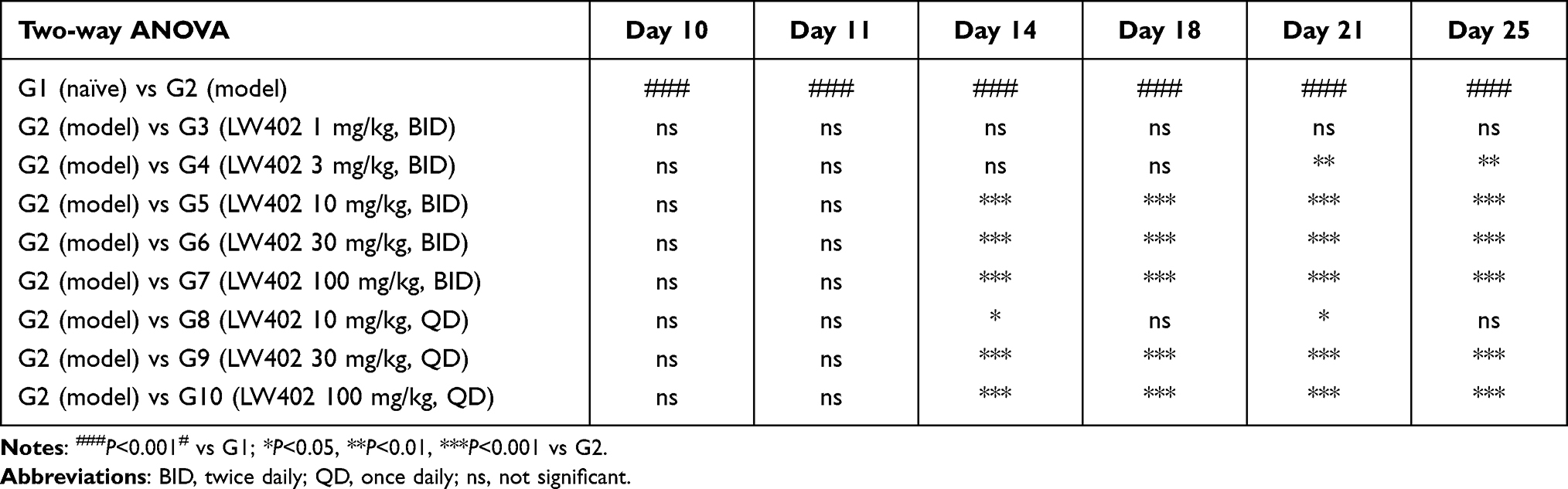 |
Table 3 Statistical Analysis of LW402 Effect on AIA Disease Progression |
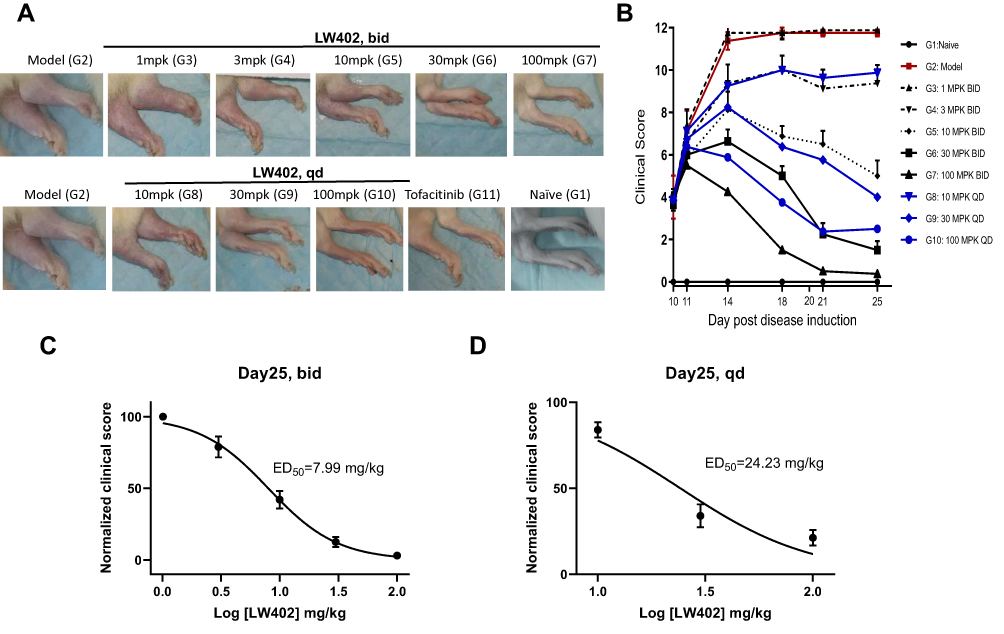 |
Figure 1 Efficacy of LW402 in rat AIA model following PO BID and QD administration. Abbreviations: PO, per os (orally); BID, bis in die (twice daily); QD, quaque die (once daily). Notes: Disease was inducedon day 0 and obvious clinical signs started to appearon day 7. Animals were then scored daily and randomizedon day 10 based on clinical score, and treatment was started on thesame day. Animals were assessed on disease development at the indicated time points. (A) Representative images of the knee joint for each treatment group on day 25. (B) Clinical scores assessed over time. Normalized clinical scores were plotted against logarithm of doses (mg/kg) to calculate ED50s for BID (C) and QD (D) dosing regimens. |
Hind paws at the end point were examined by X-ray, and representative radiographs are shown in Figure 2A. The model group (G2) exhibited severe destructive abnormalities, with all the metatarsal bones showing definite bone erosion and at least one of the tarsometatarsal joints being completely eroded, leaving some bony joint outlines partly preserved. These symptoms were almost completely cured in the100 mg/kg LW402 group (G7). Larsen scores of the X-rays are shown in Figure 2B. The model group showed an average Larsen score of 8.75. LW402 inhibited Larsen scores in a dose-dependent fashion, with the100 mg/kg BID group exhibited a Larsen score of 0 as the naïve group. When dosed on a QD regimen, LW402 also significantly reduced Larsen scores to 3.25 and 1.25 for the 30 and100 mg/kg treatment groups, respectively. These data suggested that LW402 treatment in a BID or QD regimen effectively protected the animals from CFA-induced bone damage. Histological analysis of the paw joints following H&E or safranine staining illustrated CFA-induced pathological development and infiltration of inflammatory cells in joint tissue in the model group (Figure 2C and D, G2-2-L and G2-2-R) compared to the naïve group (Figure 2C and D, G1-2-L and G1-2-R). LW402 treatment ameliorated pathological development in joint tissue in a dose-dependent fashion. In the 100 mg/kg (BID) groups, joint tissue exhibited normal histological appearance, indicating complete protection from CFA-induced damage of the articular cartilage and bone (Figure 2C and D, G7-3-L and G7-3-R). Figure 2E presents the effects of LW402 treatment on composite pathological score of all histological observations, including joint-tissue inflammation, pannus damage, cartilage damage, bone resorption, and periosteal new-bone formation. The base level of composite pathological scores in naïve mice was 1.63, and the model group exhibited a composite pathological score of 18.81. LW402 treatment dose-dependently reduced composite pathological scores to 13.53, 9.78, and 5.44 for the 10, 30, and 100 mg/kg BID groups, and 12.50, 7.41 for the 30 and 100 mg/kg QD groups, respectively, and these reductions were significantly different from the model group. Histological evaluation data were in good agreement with the assessments of clinical symptoms (clinical score) and X-ray radiographs (Larsen score).
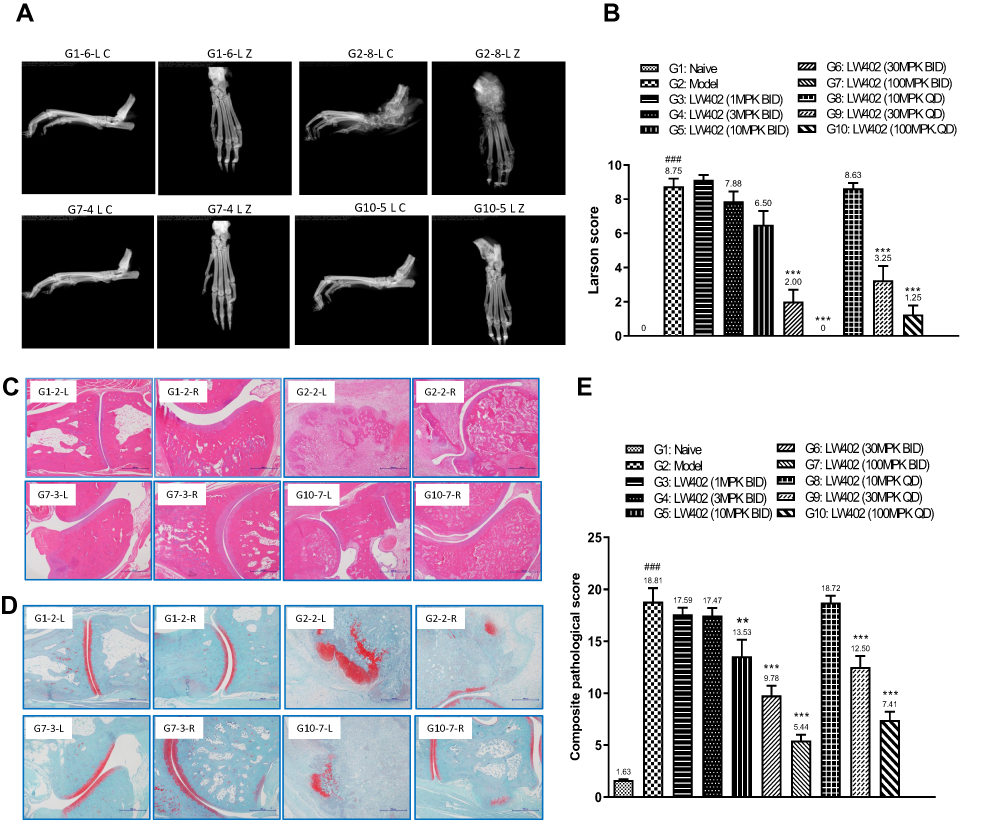 |
Figure 2 Pathological evaluation of the effect of LW402 treatment. Notes: (A) X-ray radiographs taken on hind paws. (B) Larsen scores for the radiographs. (C) Paw joint stained with H&E. (D) Paw joint stained with safranine. (E) Composite pathological scores for the hind paws. For B and E, mean values are shown above the columns. ###P<0.001 vs G1; **P<0.01, ***P<0.001 vs G2. |
PK/PD Correlation of LW402 in the Rat-AIA Model
Correlations between drug effects on clinical scores and drug exposure after LW402 dosing were investigated in the rat-AIA model. Following the completion of the efficacy study, animals were administered a final dose on day 25 and bled at various time points to assess plasma concentrations of LW402.
Figure 3A shows the LW402 plasma concentrations of various BID-dosing groups for Cmax to 1–12 hours postdosing. In the 100 mg/kg group, plasma concentrations of unbound LW402 were above the IC50 of JAK1 inhibition at all time points, and thus a BID-dosing regimen with a 12-hour interval kept the LW402 concentration constantly above the IC50 of JAK1 inhibition in the 100 mg/kg group, which agreed with the potency of LW402 in ameliorating CFA-induced disease symptoms at this dose. At 30 mg/kg, LW402 plasma concentration was above the IC50 of JAK1 inhibition at 3 hours, but slightly below IC50 at 6 hours. At 10 mg/kg, LW402 plasma concentration was above the IC50 of JAK1 inhibition for <3 hours (Figure 3A), but nevertheless still significantly improved CFA-induced increase in clinical scores (Table 3). Lower doses of 1–3 mg/kg did not have sufficient LW402 exposure to be above the IC50 of JAK1 inhibition, which correlated with the minimal efficacy in these treatment groups.
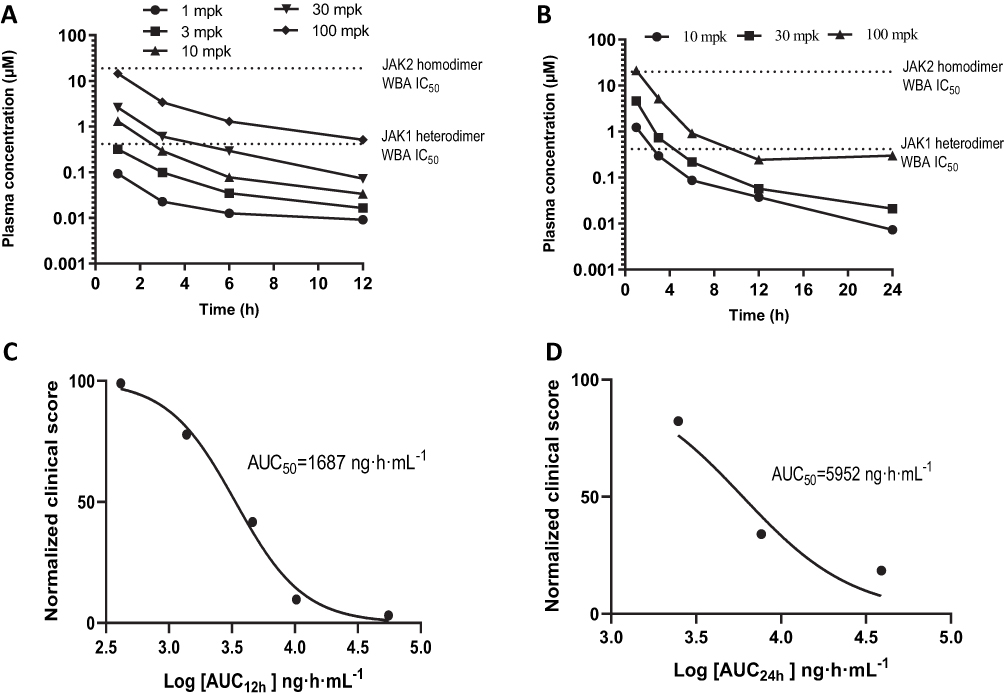 |
Figure 3 PK/PD correlations for LW402. Abbreviations: BID, bis in die (twice daily); QD, quaque die (once daily). Notes: Plasma concentrations of LW402 at various time points were plotted for BID (A) and QD (B) dosing regimens. IC50 values obtained in WBAs for inhibition of JAK1 or JAK2 were added to the plots. Normalized clinical scores were plotted against logarithm of drug exposure (AUC24) to calculate AUC50 for BID (C) and QD (D) dosing regimens. |
Figure 3B shows LW402 plasma drug concentrations of various QD-dosing groups from their peak levels at 1 –24 hours postdosing. At 100 mg/kg, LW402 maintained a plasma concentration of unbound drug above or close to the IC50 of JAK1 inhibition for 1–24 hours. At 30 mg/kg, LW402 plasma concentrations stayed above the IC50 of JAK1 inhibition for <6 hours, but this exposure was sufficient to produce significant improvement in disease symptoms (Figure 2A and B). It is noteworthy that plasma levels of unbound LW402 were below the IC50 of JAK2 inhibition for all dosing levels at all time points. Therefore, the treatment minimally affected the JAK2-signaling pathway at the dosage levels studied.
Based on the plasma concentrations, LW402 exposure (AUC0–t) were plotted against the clinical scores to estimate the AUC50 of LW402 for the rat-AIA treatment. The estimated AUC50(0-12) was 1,687 h/ng/mL (or AUC50(0-24) of 3,373 h/ng/mL for 2 bid doses) for BID dosing and AUC50(0-24)was 5,952 h/ng/mL for QD dosing.(Figure 3C and D).
Inhibition of pSTAT3 in White Blood Cells Following LW402 Treatment
Blood samples were collected at various time points following the last dosing of LW402 on day 25 of the efficacy study. Following removal of the red blood cells by lysis, the remaining white blood cells were assayed for STAT3 phosphorylation (Figure 4A). In comparison to the naïve animals that exhibited a baseline pSTAT3 level of 382 pg/mL, the model group and all treatment groups showed starting pSTAT3 levels of 2,062–3,871 pg/mL, indicating continuous induction of the STAT3-signaling pathway in the white blood cells in response to CFA induction. Variation of the pSTAT3 levels at T0 reflected differences in LW402 levels due to drug treatment. Following dosing, LW402 reduced pSTAT3 levels in a dose-dependent fashion. Inhibition plateaued at 1 hour, which correlated with the maximum plasma concentration of LW402 at 1 hour. In comparison to the model group, the decrease in pSTAT3 was statistically significant in the 10, 30, and 100 mg/kg groups. At 3 hours, pSTAT3 levels had started to recover, but the decrease was still significant in the 100 mg/kg group. By 12 hours, pSTAT3 levels had returned to their predosing levels in all treatment groups. The measured pSTAT3 at 1 hour post dosing and the logarithm of doses correlated linearly in the 1–100 mg/kg dose range (Figure 4B).
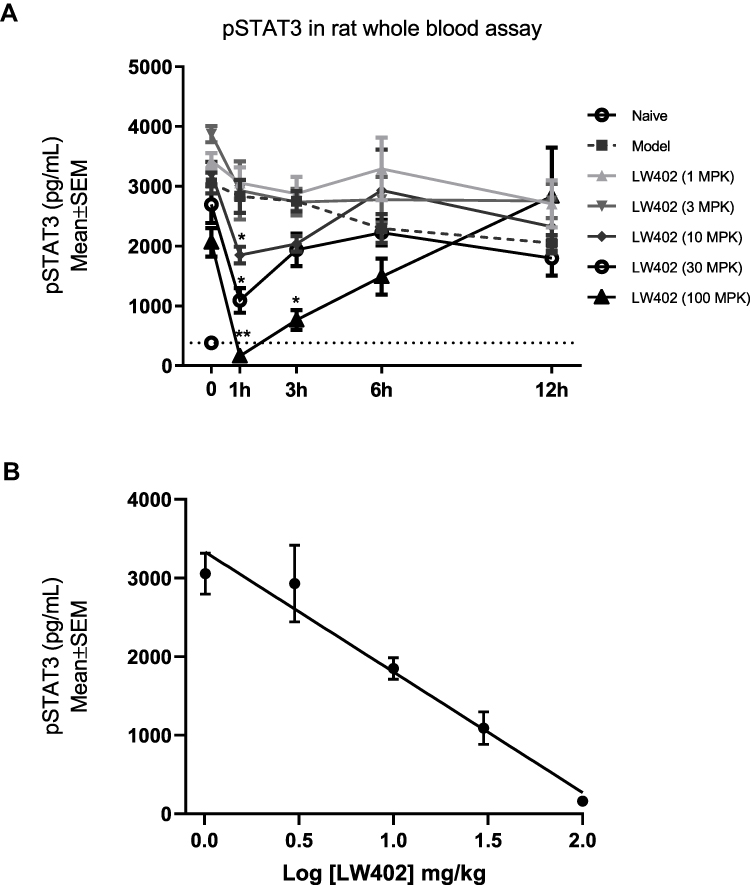 |
Figure 4 Effect of LW402 treatment on STAT3 phosphorylation in whole blood. Notes: Animals (n=8) were orally dosed with LW402 or vehicle (model) and blood collected at 0 hours (predosing) and various time points. (A) STAT3 phosphorylation was quantified as described in the Methods section. Baseline level of STAT3 phosphorylation was established with blood from naïve animals. *P<0.05, **P<0.01. (B) Correlation between logarithm of doses and pSTAT3 levels measured at 1 hour postdosing. |
Evaluation of LW402 in the Mouse-CIA Model
The therapeutic potential of LW402 for treatment of RA was further evaluated in a mouse-CIA model. Male DBA/1 mice were intradermally injected with chick type II collagen emulsion and monitored for clinical symptoms of arthritis. On day 26, when disease symptoms started to appear, animals were randomized and treated with LW402 at 50, 100, and 200 mg/kg PO BID. The effect of drug treatment on disease symptoms and foot thickness is shown in Figure 5A and B, respectively. LW402 treatment at 50 mg/kg significantly inhibited clinical scores from day 29. Higher doses of LW402 only marginally improved these disease symptoms. These results had good correlation with treatment effects on foot thickness (Figure 5B).
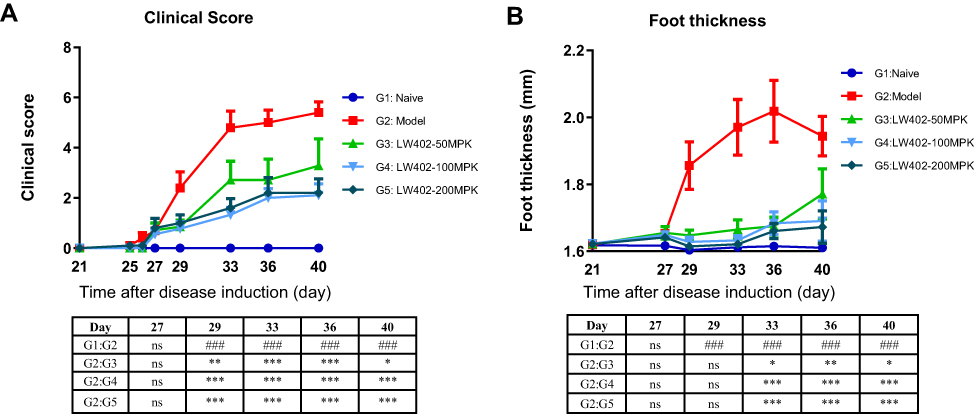 |
Figure 5 Efficacy of LW402 in mCIA model. Notes: Animals were immunized on day 0 and randomized for treatment on day 26, when obvious clinical signs started to appear. (A) Clinical scores assessed over time. (B) Foot thickness assessed over time. ###P<0.001 vs G1; *P<0.05*, P<0.01**, ***P<0.001 vs G2. |
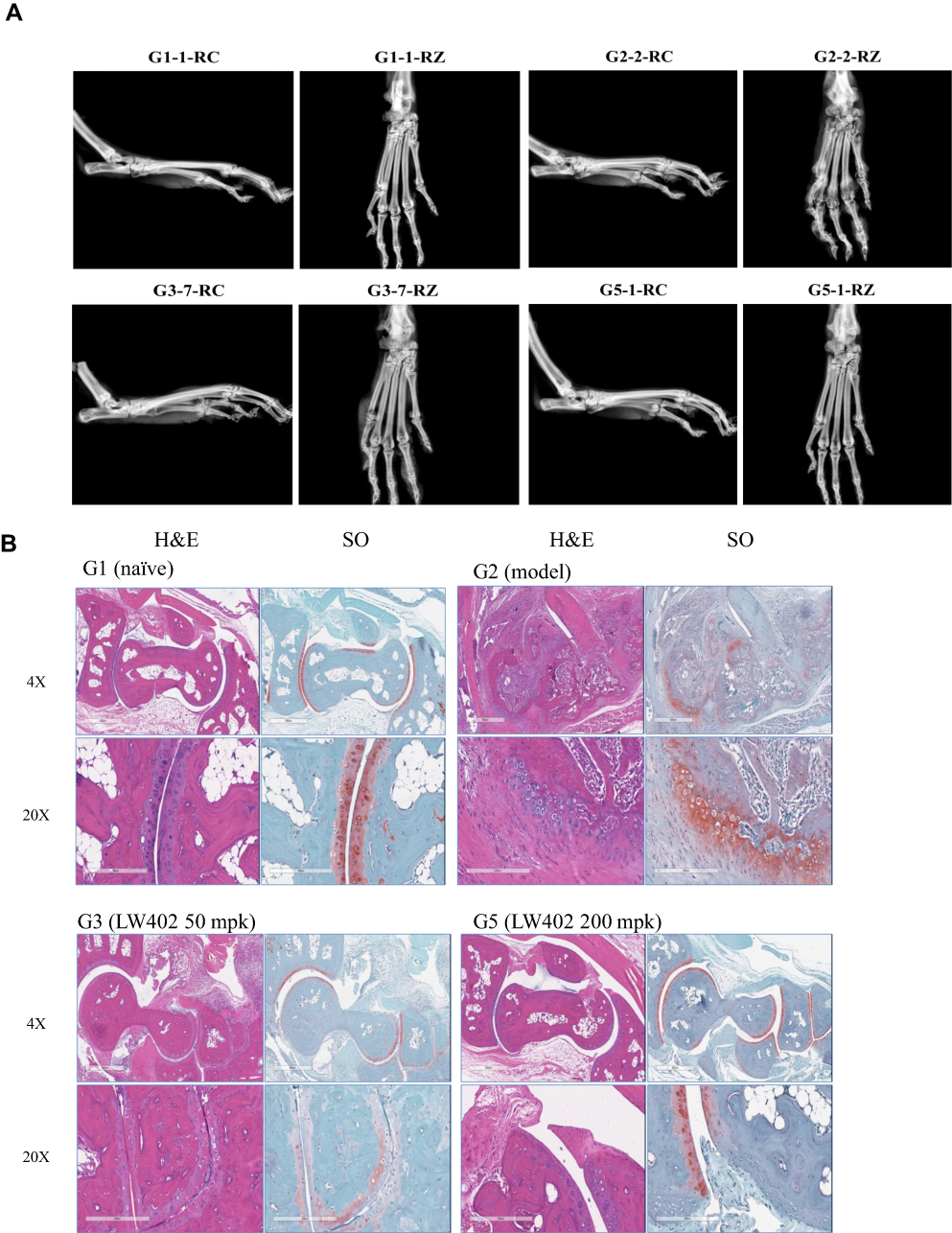 |
Figure 6 Continued. |
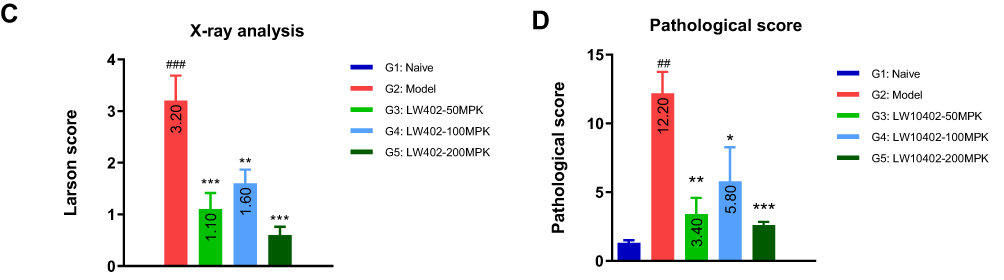 |
Figure 6 X-ray and histological analyses. Notes: (A) Representative X-ray radiographs. (B) Representative H&E and safranine images. (C) Larsen scores for the X-ray radiographs. (D) Pathological scores of histological analyses. ##P<0.01, ###P<0.001 vs G1; *P<0.05, **P<0.01, ***P<0.001 vs G2. |
At the termination of the study, the hind paws were subjected to X-ray analyses (Figure 6A). The X-rays of the model group showed that most of the animals exhibited moderate to severe destructive abnormalities in the metatarsal bones and erosion of the tarsal bones. These pathological symptoms were ameliorated by LW402 treatment at all doses. Larsen scores for X-rays are presented in Figure 6C. The model group exhibited an average Larsen score of 3.2. LW402 treatment significantly reduced Larsen scores to 1.1, 1.6, and 0.6 for the 50, 100, and 200 mg/kg groups, respectively. Histological analyses are shown in Figure 6B. In the model group, animals exhibited moderate to marked infiltration of inflammatory cells in periarticular tissue with moderate to marked edema. Cartilage damage was evident from mild marginal-zone proteoglycan loss, with small areas of surface erosion. Bone absorption was evident from small definite areas of resorption in distal tibial trabecular, cortical, or tarsal bones on low magnification. Average scores for inflammation, pannus, cartilage damage, and bone reabsorption were 3.2, 3.2, 3.0 and 2.8, respectively, constituting a composite score of 12.2 for the model group. LW402 treatment reduced composite pathological scores to 3.4, 5.8, and 2.6 for the 50, 100, and 200 mg/kg treatment groups, respectively. All these reductions were statistically significant in comparison to the model group (Figure 6D).
GLP Toxicity Evaluation of LW402
The preclinical safety of LW402 was evaluated in 28-day GLP toxicity studies in both Sprague Dawley rats and cynomolgus monkeys.
Sprague Dawley rats were dosed with 30, 100, and 300 mg/kg of LW402 via OS gavage daily for 28 days. At 30 mg/kg, treatment slightly decreased the spleen weight, but recovered after a 4-week recovery period. At 100 mg/kg, treatment had decreased white blood cells, lymphocytes, and thymus and spleen weights by day 29, but all these had recovered by day 57. Decreased lymphocytic cellularity in the thymus, spleen, inguinal lymph nodes and mesenteric lymph nodes were noted, but all had recovered after a 4-week recovery period. At 300 mg/kg, decreases in white blood cells and lymphocytic cellularity in the thymus, spleen, mandibular lymph nodes, inguinal lymph nodes, and mesenteric lymph nodes were noted, but all had recovered by the end of recovery period. No test-related abnormality was noted on ophthalmoscopic examinations, coagulation, clinical chemistry, or urinalysis. Compared to vehicle-treated animals, male animals showed 10.5% slower growth in body weight at day 28, while female animals exhibited 8.1% slower growth in body weight at day 35. Based on these findings, the no observed adverse-effect level (NOAEL) was taken as 100 mg/kg. At the NOAEL dose, the AUClast of LW402 on day 28 was 18,740 h/ng/mL and 35,390 h/ng/mL for males and females, respectively. In comparison to AUC50 measured in the BID dosing regimen, LW402 exhibited a therapeutic window of five- to tenfold.
In the cynomolgus monkey study, animals were dosed with 10, 30, and 60 mg/kg of LW402 via OS gavage daily for 28 days. At 10 mg/kg, treatment had slightly decreased thymus weight, but this had recovered after 4-weeks’ recovery. At 30 mg/kg, treatment had decreased red blood cells, hemoglobin, hematocrit, cellularity in cortex/medulla of thymus, and in white pulp of spleen in males on day 29, but these changes had recovered by the end of the recovery period. At 60 mg/kg, decreases in hematopoietic cells and cellularity in thymus, spleen, lymph nodes (mesenteric, mandibular, and inguinal), and Peyer’s patches were noted on day 28. In addition, decreases in blood pressure and increases in heart rate were noted, but these changes were reversible as the plasma concentration of the drug declined. No test-related abnormalities were noted for body weight, food consumption, ophthalmoscopic examinations, coagulation, urinalysis, or bone marrow. Based on these observations, 30 mg/kg was regarded as the NOAEL dose. At the NOAEL dose, the AUClast of LW402 on day 28 was 17,570 h/ng/mL and 15,770 h/ng/mL in males and females, respectively. In comparison to AUC50 measured on the BID-dosing regimen, the toxicity studies in cynomolgus monkeys supported a therapeutic window of fivefold for LW402.
Discussion
In recent years, significant advances have been made in understanding the links between JAK-family members and their involvement in autoimmune diseases. The first generation of small-molecule inhibitors substantiated the therapeutic potential of JAK inhibitors in these diseases. In this study, a JAK1 inhibitor of the purine analogue-compound class LW402 was evaluated. On biochemical assays, LW402 demonstrated high selectivity for JAK1 over JAK3 and TYK2, but exhibited less than twofold selectivity over JAK2. We compared JAK1 selectivity with biochemical assays of several of the approved JAK inhibitors (Table 4). All these exhibited less than threefold selectivity for JAK1 against JAK2. Given the high sequence and structural identity of the protein-tyrosine kinase domains between JAK1 and JAK2,26 specific inhibition of JAK1 at the biochemical level may represent a challenge. However, when evaluated on WBAs, LW402 exhibited improved JAK1 selectivity of 45-fold, higher than that of other JAK inhibitors (Table 2). At present, no uneqivocal explanation for the discrepancy between the biochemical and whole-blood JAK-inhibition profiles can be provided. Such a discrepancy was also observed in a study using filgotinib and likely contributing factors extensively discussed,25 including difference in biochemical assay using only the C-terminal quarter of the JAK proteins vs the whole enzyme in WBA, and that the JAKs in the cellular environment are subject to posttranslational modifications, such as phosphorylation, which affect their IC50 values. In addition, LW402 exhibited more selectivity in inhibition of IL2-induced JAK1/3–STAT5 signaling in CD8+ T cells than GM-CSF–induced JAK2–STAT5 signaling in CD33+ myeloid cells.
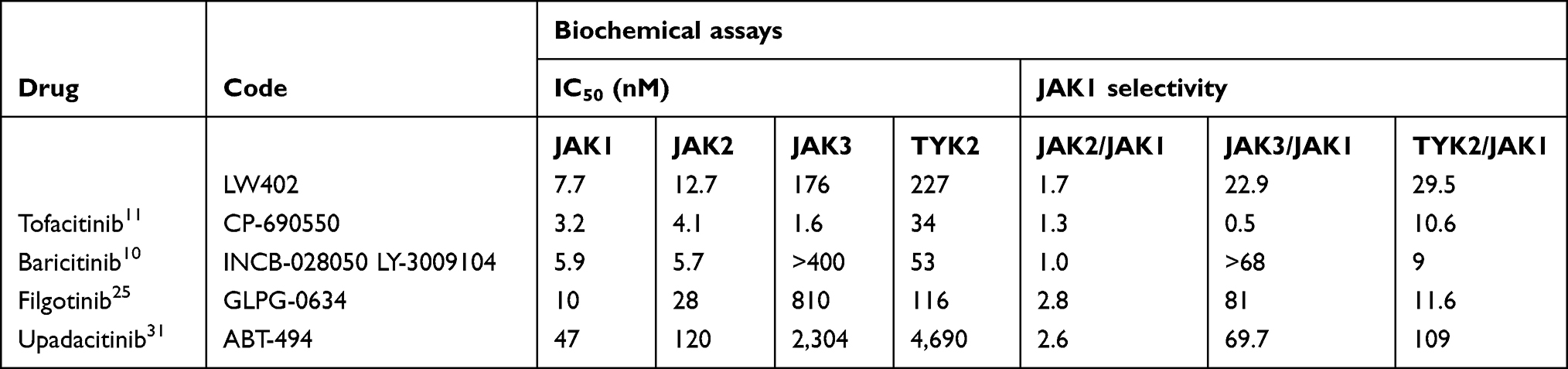 |
Table 4 Comparison of in Vitro Activity and Selectivity of JAK Inhibitors |
JAK1 selectivity against JAK3 on the biochemical assay for LW402 was less than that of filgotinib, upadacitinib, and baricitinib, but JAK1 selectivity against TYK2 was more than all of these except upadacitinib. Clinical translation of the JAK1 selectivity of LW402 balancing therapeutic potency and adverse events will be elucidated upon further investigation. In line with the in vitro data, LW402 exhibited potent inhibition of JAK1-mediated STAT3 phosphorylation in the rat-AIA model, and the extent of inhibition of JAK1 activity correlated well with the reduction in CFA-induced inflammation and damage to joint cartilage and bone. ED50 was estimated to be 24.23 mg/kg (QD) and 7.99 mg/kg (BID). At ED50 doses for both BID and QD, LW402 plasma concentrations were below the IC50 of JAK2, but stayed above the IC50 of JAK1 for 3–6 hours (Figure 3A and B). These data indicate that <6 hours’ exposure above the IC50 of JAK1 was sufficient to produce effective therapeutic efficacy in the AIA model. Continuous inhibition of the JAK-signaling pathway not being a prerequisite for achieving therapeutic efficacy in the AIA model was also demonstrated in another study, where inhibition of STAT3 for <4 hours by baricitinib was sufficient to produce therapeutic efficacy.10 Taken together, these preclinical PK/PD results provide guidance for effective dose determination in future clinical trials.
Some studies have tended to compare drugs by virtue of their in vitro/in vivo potency and judged superiority of a drug without taking into consideration NOAEL doses in animal toxicity. Such comparisons would erroneously reckon superiority of a drug based solely on its IC50 or ED50. LW402 exhibited IC50 of 417 nM for inhibition of IL6-stimulated JAK1/STAT1 signaling in CD4+ cells (Table 2). In the in vivo pharmacology studies, LW402 exhibited an ED50 of 7.99 mg/kg (human equivalent dose [HED] 80 mg) and 24.23 mg/kg (HED 240 mg) on BID and QD dosing, respectively, in the rat-AIA model and an efficacious dose of 50 mg/kg (HED 250 mg) in the mouse-CIA model on BID . In the toxicology study, LW402 had an NOAEL dose of 30 mg/kg (HED 600 mg QD) in Cynomolgus monkey. Based on the AUC50 of 3,373 h/ng/mL of LW402 established in the rat-AIA model, and an average AUC of 16,670 h/ng/mL at the 30 mg/kg NOAEL dose in the cynomolgus monkey GLP study, we estimated a therapeutic window of ≥5× for LW402, which is much higher than a maximum of double the therapeutic window established for all approved JAK inhibitors. In future clinical studies, we expect LW402 to reach its therapeutic effective dose in a range of 75–150 mg, which is much lower than the equivalent human NOAEL dose of 600 mg, as established in the toxicity study.
While JAK1 inhibition will inevitably decrease immune cells, certain side effects observed in clinical trials, such as anemia, neutropenia, and thrombocytopenia, appear to be attributed mainly to JAK2 inhibition.27–30 Selective JAK1 inhibition could potentially reduce or eliminate some JAK2 inhibition–mediated adverse events. Several JAK1-specific inhibitors including filgotinib25 and upadacitinib (ABT-494),31,32 have been developed for RA treatment. A recent study compared the efficacy and safety profiles of tofacitinib, baricitinib, upadacitinib, peficitinib, and filgotinib in RA treatment following meta-analysis of multiple-arm randomized clinical trials,33 reporting treatment outcome of SUCRA (surface under the cumulative ranking curve) 100% for the highest-ranked treatment and 0 for the lowest-ranked treatment. In terms of efficacy, peficitinib (150 mg), filgotinib (200 mg), tofacitinib (5 mg), upadacitinib (15 mg), and baricitinib (4 mg) achieved SUCRA values of 0.97, 0.70, 0.48, 0.32, and 0.17 respectively, for American College of Rheumatology 20% response. As for clinical safety, the JAK1-selective inhibitors upadacitinib and filgotinib exhibited superior safety profiles with SUCRA values of 0.29 and 0.15, respectively, for serious adverse events, while tofacitinib, peficitinib and baricitinib exhibited SUCRA values of 0.98, 0.66, and 0.65, respectively.
Conclusion
We characterized a JAK1-specific inhibitor, LW402, in preclinical studies and demonstrated excellent therapeutic efficacy on the rat-AIA and mouse-CIA models and a superior therapeutic window in GLP toxicity studies. The preclinical efficacy and safety profiles of LW402 validate further efforts to develop this drug. We hope the overall advantage of LW402 over other JAK inhibitors will be translated to the clinical setting in the treatment of RA and other autoimmune diseases.
Abbreviations
WBA, whole-blood assay; AIA, adjuvant-induced arthritis; DMARD, disease-modifying antirheumatic drug; BID, bis in die (twice daily); QD, quaque die (once daily); PO, per os (orally); HED, human equivalent dose; NOAEL, no observed adverse-effect level.
Data Sharing Statement
All other data generated or analyzed during this study are included in this published article (and its Additional files).
Ethics Approval and Consent to Participate
The 3D BioOptima IACUC Committee approved the rat PK study, and the study was conducted following the 3D BioOptima Animal Care and Use Guidelines. The Kunming Biomed International IACUC Committee approved the monkey PK study, and the study was conducted following the Kunming Biomed International Animal Care and Use Guidelines. The ChemPartner IACUC Committee approved the pharmacology studies in rat and mice and the studies were conducted following the ChemPartner Animal Care and Use Guidelines. The Joinn Laboratories IACUC Committee approved the GLP toxicity studies in rats and monkeys and the studies were conducted following the Joinn Laboratories Animal Care and Use Guidelines. The human WBA studies were approved by the Institutional Ethics Committee of Shanghai Longwood Biopharmaceuticals,and adhered to the Declaration of Helsinki.
Acknowledgments
We acknowledge Dr Lixin Wang, Dr Wei Wang, Chunli Li, Sen Yao, and Jie Sun (ChemPartner, Shanghai) for compound testing.
Author Contributions
NZ, CZ, ZZ, JZ, SD, CB, and ZW made significant contributions to the work reported, drafted, wrote, substantially revised, or critically reviewed the article; agreed on the journal to which the article has been submitted; reviewed and agreed on all versions of the article before submission, during revision, the final version accepted for publication, and any significant changes introduced at the proofing stage, and agree to take responsibility and be accountable for the contents of the article. NZ and ZW were the principal investigators of the in vitro assays and animal studies. CZ, ZZ, JZ, SD, and CB were involved in study design and data interpretation.
Funding
The study was funded by Longwood Biopharma, Shanghai, China.
Disclosure
Ning Zhang, Zhihong Zeng, Jiyong Zhang, Shengnan Du, and Zhe Wang are employees at Longwood Biopharmaceuticals, Shanghai, China. Ning Zhang and Zhe Wang report patent WO 2018/133875 being issued. The authors report no other conflicts of interest in this work.
References
1. Smolen JS, Aletaha D. Rheumatoid arthritis therapy reappraisal: strategies, opportunities and challenges. Nat Rev Rheumatol. 2015;11:276–289. doi:10.1038/nrrheum.2015.8
2. Taylor PC, Moore A, Vasilescu R, et al. A structued literature review of the burden of illness and unmet needs in patients with rheumatoid arthritis: a current perspective. Rheumatol Int. 2016;36:685–695. doi:10.1007/s00296-015-3415-x
3. Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signalling via the JAK/STAT pathway. Protein Sci. 2018;27:1984–2009.
4. O’Sullivan LA, Liongue C, Lewis RS, Stephenson SE, Ward AC. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol Immunol. 2007;44:2497–2506. doi:10.1016/j.molimm.2006.11.025
5. Vainchenker W, Dusa A, Constantinescu SN. JAKs in pathology: role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Semin Cell Dev Biol. 2008;19:385–393. doi:10.1016/j.semcdb.2008.07.002
6. Michalska A, Blaszczyk K, Wesoly J, Bluyssen HR. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Front Immunol. 2018;9:1135. doi:10.3389/fimmu.2018.01135
7. Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–287. doi:10.1111/j.1600-065X.2008.00754.x
8. O’Shea JJ, Pesu M, Borie DC, Changelian PS. A new modality for immunosuppression: targeting the JAK/STAT pathway. Nat Rev Drug Discov. 2004;3:555–564. doi:10.1038/nrd1441
9. O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36:542–550. doi:10.1016/j.immuni.2012.03.014
10. Fridman JS, Scherle PA, Collins R, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184:5298–5307. doi:10.4049/jimmunol.0902819
11. Meyer DM, Jesson MI, Li X, et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J Inflamm. 2010;7:41. doi:10.1186/1476-9255-7-41
12. Fleischmann R, Kremer J, Cush J, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. New Engl J Med. 2012;367:495–507. doi:10.1056/NEJMoa1109071
13. Van Vollenhoven R, Takeuchi T, Pangan AL, et al. A phase 3, randomized, controlled trial comparing upadacitinib monotherapy to MTX monotherapy in MTX-naive patients with active rheumatoid arthritis. Arthritis Rheumatol. 2018;70:990–992.
14. Milici AJ, Kudlacz EM, Audoly L, Zwillich S, Changelian P. Cartilage preservation by inhibition of Janus kinase 3 in two rodent models of rheumatoid arthritis. Arthritis Res Ther. 2008;10:R14. doi:10.1186/ar2365
15. Kudlacz E, Conklyn M, Andresen C, Whitney-Pickett C, Changelian P. The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur J Pharmacol. 2008;582:154–161. doi:10.1016/j.ejphar.2007.12.024
16. Changelian PS, Flanagan ME, Ball DJ, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003;302:875–878. doi:10.1126/science.1087061
17. Dowty ME, Jesson MI, Ghosh S, et al. Preclinical to clinical translation of tofacitinib, a Janus kinase inhibitor, in rheumatoid arthritis. J Pharmacol Exp Ther. 2014;348:165–173. doi:10.1124/jpet.113.209304
18. Cox L, Cools J. JAK3 specific kinase inhibitors: when specificity is not enough. Chem Biol. 2011;18:277–278. doi:10.1016/j.chembiol.2011.03.002
19. Haan C, Rolvering C, Raulf F, et al. Jak1 has a dominant role over Jak3 in signal transduction through γc-containing cytokine receptors. Chem Biol. 2011;18:314–323. doi:10.1016/j.chembiol.2011.01.012
20. Thoma G, Nuninger F, Falchetto R, et al. Identification of a potent Janus kinase 3 inhibitor with high selectivity within the Janus kinase family. J Med Chem. 2011;54:284–288. doi:10.1021/jm101157q
21. Wang Z, Fan GQ, Yang S, Zeng ZH Preparation of 1H-pyrrolo[2,3-d] pyrimidine derivatives as Jak kinase inhibitors. 2019. US-2019-0382408-A1.
22. Brand DD, Latham KA, Rosloniec EF. Collagen-induced arthritis. Nat Protoc. 2007;2:1269–1275. doi:10.1038/nprot.2007.173
23. Lin HS, Hu CY, Chan HY, et al. Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents. Br J Pharmacol. 2007;150:862–872. doi:10.1038/sj.bjp.0707165
24. Ghoreschi K, Jesson MI, Li X, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol. 2011;186:4234–4243. doi:10.4049/jimmunol.1003668
25. van Rompaey L, Galien R, van der Aar EM, et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol. 2013;191:3568–3577. doi:10.4049/jimmunol.1201348
26. Williams NK, Bamert RS, Lucet IS. Dissecting specificity in the Janus kinases: the structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J Mol Biol. 2009;387:219–232. doi:10.1016/j.jmb.2009.01.041
27. Lee YH, Song GG. Comparison of the efficacy and safety of tofacitinib and filgotinib in patients with active rheumatoid arthritis: a Bayesian network meta-analysis of randomized controlled trials. Z Rheumatol. 2019;79:590–603. doi:10.1007/s00393-019-00733-x
28. Greenwald MW, Fidelus-Grot R, Levy R, Liang J, Vaddi K, Williams WV. A randomized dose-ranging, placebo-controlled study of INCB028050, a selective JAK1 and JAK2 inhibitor in subjects with active rheumatoid arthritis. Arthritis Rheum. 2010;62:S911.
29. O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368:161–170. doi:10.1056/NEJMra1202117
30. Kremer JM, Bloom BJ, Breedveld FC, Zwillich SH. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009;60:1895–1905. doi:10.1002/art.24567
31. Fleischmann R, Pangan AL, Song I, et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase 3, double‐blind, randomized controlled trial. Arthritis Rheumatol. 2019;71:1788–1800. doi:10.1002/art.41032
32. Parmentier JM, Voss J, Graff C, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. 2018;2:23. doi:10.1186/s41927-018-0031-x
33. Lee YH, Song GG. Comparative efficacy and safety of tofacitinib, baricitinib, upadacitinib, filgotinib and peficitinib as monotherapy for active rheumatoid arthritis. J Clin Pharm Ther. 2020;45:674–681. doi:10.1111/jcpt.13142
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.