")
Back to Journals » Cancer Management and Research » Volume 11
Precision oncology: lessons learned and challenges for the future
Authors Yang HT, Shah RH , Tegay D, Onel K
Received 12 January 2019
Accepted for publication 8 July 2019
Published 7 August 2019 Volume 2019:11 Pages 7525—7536
DOI https://doi.org/10.2147/CMAR.S201326
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Alexandra R. Fernandes
Hsih-Te Yang,1 Ronak H Shah,1,2 David Tegay,1 Kenan Onel3
1Medical Genetics and Human Genomics, Department of Pediatrics, Northwell Health, New York, NY, USA; 2Center for Research Informatics and Innovation, The Feinstein Institute for Medical Research, Northwell Health, New York, NY, USA; 3The Icahn School of Medicine at Mount Sinai, Department of Genetics and Genomic Sciences, New York, NY, USA
Abstract: The decreasing cost of and increasing capacity of DNA sequencing has led to vastly increased opportunities for population-level genomic studies to discover novel genomic alterations associated with both Mendelian and complex phenotypes. To translate genomic findings clinically, a number of health care institutions have worked collaboratively or individually to initiate precision medicine programs. These precision medicine programs involve designing patient enrollment systems, tracking electronic health records, building biobank repositories, and returning results with actionable matched therapies. As cancer is a paradigm for genetic diseases and new therapies are increasingly tailored to attack genetic susceptibilities in tumors, these precision medicine programs are largely driven by the urgent need to perform genetic profiling on cancer patients in real time. Here, we review the current landscape of precision oncology and highlight challenges to be overcome and examples of benefits to patients. Furthermore, we make suggestions to optimize future precision oncology programs based upon the lessons learned from these “first generation” early adopters.
Keywords: next-generation sequencing, pathogenic variant, driver mutation, actionable mutation, cancer disparities
Introduction
Driven by the precipitous drop in the cost of next-generation sequencing (NGS),1 it has become possible to perform genetic studies on a population scale to identify rare2 and common genetic variants3 associated with Mendelian disease and complex traits.4–9 This “omics” revolution has yielded a wealth of information that has catalyzed efforts in precision medicine, allowing for characterization of patients at the genomic level for more precise diagnosis and treatment.
In cancer research, whole exome sequencing (WES) and whole genome sequencing (WGS) have been successfully used to identify germline and somatic variants that drive cancer initiation or cancer progression,10–12 as well as copy number variations (CNVs) and other structural variations (SV) important in cancer progression or chemoresistance.13–18 Large-scale sequencing studies have defined clinically relevant cancer subtypes in a variety of oncology studies, such as for pancreatic cancer,19 ovarian cancer,20 breast cancer,21 and hematopoietic malignancy.22 Furthermore, by sequencing a single cancer patient at multiple time points, it is possible to study clonal evolution and tumor heterogeneity in relation to cancer etiology,23 metastatic potential,24 and drug resistance.25
Genomic aberrations in cancer are either germline or somatic mutations that can be detected in blood, other normal tissue, or malignant tissue from an affected patient. Among germline variants, those classified as pathogenic or likely pathogenic are associated with high-penetrance susceptibility to cancer.26–28 Among somatic (ie, acquired) variants found in a tumor but not in corresponding normal tissue, many are thought to be “passenger” mutations that have no functional effect but are the consequence of the genomic instability that characterizes cancer cells. Some somatic mutations are driver mutations that are actively involved in the evolution or progression of a tumor. A subset of these driver mutations are actionable genetic alterations that are sensitive to targeted therapies and have significant clinical implications in cancer treatment.
By allowing for the discovery of pathogenic germline variants, driver mutations, and other variants influencing, for example, drug metabolism, NGS studies in cancer have revealed numerous novel insights with significant clinical implications for cancer prevention, prognosis, and treatment. These discoveries, however, open new questions regarding the relationship between genetics, tumor biology, and clinical outcomes. Does an actionable mutation in one tumor type, for example, imply that the same mutation is actionable in another tumor type? To answer questions of this sort, it is necessary to integrate genomic data with clinical and family history data and follow individual patients longitudinally. Prior to new initiatives in precision medicine led by the United States government in 2015,29 integration of genomic medicine programs with electronic health records (EHR) was done largely through independent efforts. Currently, some research-oriented health care systems are collaborating on a large scale to develop revolutionary clinical precision medicine practices. In this review, we highlight the lessons learned, the challenges of running these programs, and future directions where precision oncology may lead.
Current precision oncology programs
Overall goals
Population genomics studies (Figure 1A) aim to identify germline or somatic genetic variants that confer an increased risk of disease or other phenotypes, such as an improved response to therapy.10,11 However, while the results of these studies may achieve statistical significance, they often do not translate clinically, as the effect sizes of associated variants are either small or limited to specific patient subsets which are rarely readily discernable because of incomplete or missing clinical data. To move population genomics towards precision oncology, three additional components are required (Figure 1B). The first is a biobank comprised both identified germline and multiple fresh-frozen tumor samples (ie, pre-treatment diagnostic samples, relapse samples, and metastatic samples) from as many patients as possible and across many ethnicities. The second is a comprehensive clinical data warehouse linking all samples to patients and including family history and longitudinal clinical data abstracted from the EHR, obtained in a standardized manner and using harmonized language. The final component is the capacity to return results to patients and their caregivers, in order to facilitate the use of targeted or other molecular therapies, clinical trials, and other follow-up studies. Importantly, an informatics infrastructure is necessary to ensure high-quality precision data.
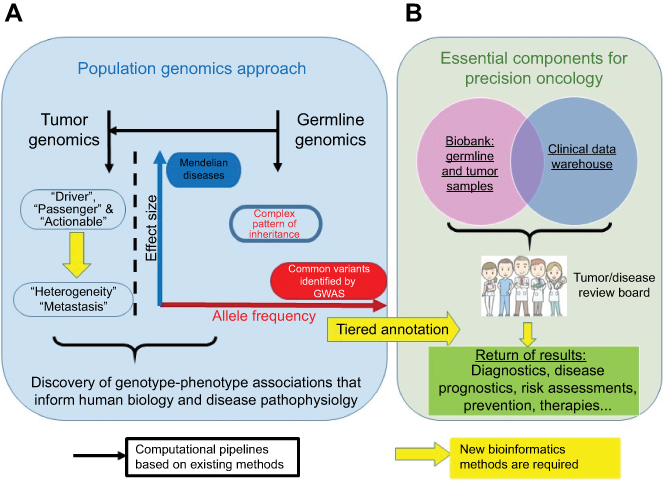 |
Figure 1 From population genomics to precision medicine. (A) Current population genomics strategies. (B) Essential components for the translation of population genomics to precision oncology. |
Many hospitals and research institutes have independent programs integrating population genetics into precision medicine. While these programs have reported similar results, there has been a large amount of redundant work, potentially resulting in suboptimal use of funding resources, clinician efforts, and patient participation.30 The availability and application of NGS technologies in clinical care have prompted the development of common standards such as the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) guidelines31 for reporting clinically actionable variants, and have led to the generation of massive datasets. Furthermore, NGS technologies have led to the development of novel algorithms and tools facilitating data mining and interpretation through interdisciplinary collaborations among clinicians, molecular pathologists, computational biologists, medical geneticists, bioinformaticians, statisticians, and laboratory technicians, producing a precision medicine ecosystem.32
Strategies and study designs
We have listed many of the current large-scale precision oncology projects in Figure 2. Investigational strategies consist largely of two study designs: 1) a disease-agnostic approach in which the participants are mostly healthy,33 not ascertained for a specific status,34 and broadly recruited from primary care and specialty clinics9 and 2) a disease-focused approach where patients are recruited with a specific disease in mind at the outset. Examples of disease-focused studies in pediatric oncology include BASIC3,35 GREAT KIDS,36 INFORM,37 iCat,38,39 Peds-MiOncoSeq,40 and PIPseq.41 In adult oncology, studies (eg, SHIVA42) seek to identify targets for molecular therapies in patients with advanced cancer. Treatment studies such as these generally have one of the two designs. A basket trial recruits participants who share the same genetic mutation across multiple cancer types, whereas an umbrella trial recruits patients with a single type of cancer and assigns them to different arms based upon specific mutations for which therapies are being tested.42
 |
Figure 2 Precision medicine programs stratified by study design and organization scale. This figure includes the 14 programs from USA, Europe, and Asia cited in this review. |
Each investigational strategy has pros and cons. Disease-agnostic studies require a larger sample size than do disease-focused studies, because the prevalence of any given cancer in the general population is generally quite low. Consequently, disease-agnostic studies generally require significantly more resources. Disease-agnostic approaches allow researchers to explore a different set of questions compared to disease-focused studies. While it can be easier to access and share information within a single-center study, a multi-center study can typically access a broader patient population and a larger pool of resources.
Beyond disease-agnostic and disease-focused studies, many research programs aim to identify disease risks and other personal health information through pre-symptomatic genomic screening (eg, The PeopleSeq Consortium).43 Instead of enrolling patients diagnosed with or suspected of having specific diseases, this consortium initiated a predispositional personal genome sequencing program to sequence the genomes of ostensibly healthy participants who can directly access their genomic data for sequencing-derived genetic findings.
Patients, samples, and clinical data
The implementation of a precision oncology program starts with efficient patient enrollment. This includes patient selection, informed consent, centralized sample collection (a biobank), and a computerized laboratory information management system (LIMS) for tracking these components. Integrating patient enrollment, family history, and clinical information into a patient-centric data warehouse is an essential requirement for precision medicine, but is only feasible within organized health systems (Figure 2), eg, the GREAT KIDS program at The University of Chicago,36 BASIC3 clinical study at Baylor College of Medicine,44 and MyCode community health initiative at the Geisinger Health System.45
After enrollment, a unique and secure identification number is assigned to each patient,36 which is then associated with EHR information and clinical specimens that are collected in either an identified or de-identified manner, depending upon the study consenting process and participant preference. However, even within a single health system, a patient’s EHR is typically fragmented and distributed in different clinical and research departments with specialized information systems, such as LIMS, radiology imaging (picture archiving and communication systems, PACS) and pharmacy (prescription systems).46 With the commitment of decision-makers in a cancer center, data exchange and integration are facilitated by common standards (Health Level Seven) and tools of ETL (Extract, Transform, Load).46 The Electronic Medical Records and Genomics (eMERGE) network was recently organized to facilitate linking EHR phenotype data to biobanked samples and genotype information across multiple health systems.47 In pharmacogenetic studies to investigate drug efficacy and safety, the eMERGE strategy has been shown to be more cost-effective than traditional single-institution study designs.48
As coupling EHR systems with biobank data for discovery in genome science has become increasingly prevalent, a major challenge has emerged in how to define clinical human traits or symptoms in EHR systems. The ICD codes, currently in their tenth iteration, were developed and are maintained by the WHO for universal diagnosis and procedure documentation in the EHR. Human Phenotype Ontology classifications allow standardization of phenotypic trait nomenclature. The adoption of standards in coding and documenting traits and the harmonization of these terms across EHRs and data warehouses have facilitated phenome-wide association studies, where many phenotypes can be compared to a single genetic variant.49–51 Using this method, the DiscovEHR study9 of the Geisinger MyCode community45 successfully identified novel rare protective variants in PCSK9 that reduce the risk of cardiovascular disease by decreasing levels of low-density lipoprotein cholesterol (LDL-C), and developed a drug to lower LDL-C in the healthy population.52
Return of information
Patient follow-up, whereby personalized genomic results are returned, is an essential component of a precision medicine clinical program or trial, and has already been implemented in the MyCode community,45 BASIC3 study,44 and the GREAT KIDS program.36
For the return of results to patients, in general, research sequencing results must be confirmed in a CLIA-certified laboratory.53 Furthermore, before genomic data can be returned, it must be subjected to expert review by a multidisciplinary medical board, which often includes oncologists, molecular pathologists, genetic counselors, laboratory technicians, and biostatisticians. Generally, germline and somatic results are returned separately, because they generally have different implications (risk and therapy, respectively). However, it is increasingly clear that germline variants can have therapeutic implications, such as in the burgeoning field of pharmacogenomics, or in BRCA status and the use of poly ADP ribose polymerase (PARP) inhibitors.54
A germline report returns one of the five pathogenicity annotations for genomic variants (pathogenic, likely pathogenic, unknown significance, likely benign, or benign).35,45 In 2013, the ACMG issued recommendations for the standardized reporting of actionable germline information, by releasing a list of 56 genes that harbor known pathogenic and/or likely pathogenic variants leading to severe diseases. This list is regularly updated as more genomic information is gathered; currently, there are 59 reportable genes (ACMG SF v2.0).55 The ACMG recommendations are only guidelines, however, as some studies (such as the DiscovEHR study) report the Geisinger-76 genes, which significantly overlap the ACMG list.9 Based on a joint consensus recommendation from the ACMG and the AMP, these standards and guidelines31 have been established for use by genetic review boards and genomic annotation tools, such as InterVar56 and CharGer,28 used to properly classify germline variants into the aforementioned five categories.
A somatic report returns clinically actionable acquired variants. Currently, these are mostly identified through the panel sequencing of a predefined gene list, and are typically performed on samples obtained at diagnosis. These genetic alterations can be annotated with four tiers of actionability (as in the GREAT KIDS program64 and BASIC3 study62) or five tiers of actionability (as in the iCat study59,73) based on evidence of their clinical utility, which assists physicians with proper decision-making in patient care. Researchers are working to harmonize and establish standards and knowledge databases with respect to clinical actionability of molecular targets, and to leverage that information to facilitate the implementation of precision medicine in the clinical management of cancer patients.57–59 For example, the ESMO Scale of Clinical Actionability for molecular Targets (ESCAT)57 project defines six tiers of clinical evidence and ranks genomic alterations as molecular targets for FDA-approved or investigational drugs. Recently, these knowledge databases and rules of evidence for clinical interpretation have been integrated and coordinated to annotate cancer variants using an ontology-based modeling framework.60 One limitation of most current approaches is that most sequenced tumor samples are obtained as part of the initial diagnostic evaluation of a patient (for solid tumor patients, this is typically the only time point at which a sample is obtained), but the data obtained are used to guide therapeutic decision-making for relapsed, refractory, or metastatic tumors in patients who no longer respond to conventional therapies. This lack of re-sequencing may result in the targeting of mutations common in drug-sensitive clones that have been eradicated in relapsed or in metastatic tumors, or conversely, the masking of mutations found in the drug-resistant clones that may have comprised only a minor subpopulation within the pre-treatment tumor. Alternatively, the mutations acquired during solid tumor progression and drug therapy can be detected as biomarkers for patient prognosis using liquid biopsies, with the caveat that the noisy background that impacts data quality and utility must be defined in prospective clinical trials.61,62
Challenges to clinical implementation
Data challenges
Clinically favorable responses to therapies targeting actionable mutations are limited due to a dearth of FDA-approved companion therapies, ambiguous scientific contexts, and imperfect treatment algorithms.63
One of the first targeted therapies was an anti-EGFR monoclonal antibody for metastatic colorectal cancer (mCRC). About 75% of mCRC patients overexpress EGFR within their tumors, suggesting that targeted inhibition of EGFR may improve outcomes. However, initial response rates to anti-EGFR monotherapy in heavily pretreated mCRC patients were only 10%.64 A subsequent study demonstrated a minor improvement in response rates (cetuximab with supportive care versus supportive care alone) of patients bearing wild type KRAS, a gene downstream of EGFR.65 However, most mCRC patients carrying wild type KRAS in their tumors did not respond to anti-EGFR therapy. Other studies showed that mutations in other genes downstream of EGFR (BRAF, NRAS, and PIK3CA) are also significantly associated with a low response rate in patients with KRAS wild type tumors.66 Thus, these findings show that EGFR status alone is insufficient to assess the likelihood that a patient will benefit from anti-EGFR therapy, but rather that a broader pathway-based genomic profile is required to identify those mCRC patients most likely to respond. Besides mutations at EGFR and KRAS as therapeutic targets in mCRC or lung cancer, the anti-HER2 antibody trastuzumab, and adjuvant chemotherapy, are beneficial in cases of HER2 overexpression detected by immunohistochemistry and in situ hybridization, as well as amplified ERBB2 (HER2) copy number characterized by DNA/RNA-seq67 in breast cancer patients. A famous example of a SV as a molecular target is in genomic rearrangement at anaplastic lymphoma kinase (ALK)68 or receptor tyrosine kinase (ROS1)69 through gene fusion. Targeted therapy led to constitutive activation of tumor growth pathways in a subgroup of non-small cell lung cancer (NSCLC) patients responsive to crizotinib or ceritinib therapy. A variety of genomic variants in the relevant genes within a pathway may reveal potential matched therapies or new indications for existing approved drugs.
This finding inspired further clinical trials to determine whether pathway-based molecular profiling (MP)-guided therapy can be beneficial to advanced or refractory cancer patients.70–73 Von Hoff et al conducted a pilot study which showed that 27% of patients treated with MP-based targeted therapies exhibited longer progression-free survival than did patients treated similarly without initial MP.70 The BATTLE Phase II clinical trial extended this observation by trying to match targeted therapies based on biomarkers from needle biopsies with positive results for sorafenib treatment in NSCLC patients harboring KRAS mutations.71 These pioneer projects demonstrated the potential feasibility and predictive utility of MP for targeted therapy. Tourneau et al further investigated off-label use of molecularly targeted therapy based on tumor MP in comparison with conventional therapy for advanced cancer, by performing a feasibility trial in the SHIVA cohort randomized Phase II clinical trial.72,74 However, MP-guided targeted therapy did not significantly improve progression-free survival, and more grades 3–4 adverse events were reported in patients receiving targeted therapy than in the control group.72 Reviewing these MP clinical trials indicates that existing treatment algorithms for selecting molecularly targeted agents might be inadequate.30,63,75
Clonal diversity and biological challenges
Because of current technical limitations, only the genetics of the dominant clones within a tumor can be assessed using MP. Tumor heterogeneity and clonal diversity within cancers has been shown across sample types obtained from a single individual, including blood25,76–78 and tumor biopsy,23,79 and in primary tumors and metastases.24,80,81 Mutational shifts during treatment and in metastasis can select for minor clones and thereby reduce the efficacy of treatments chosen solely using the profile of the original tumor. NGS has been shown to reliably detect driver mutations of clones/subclones of differing variant allele frequencies (VAF) with high depth and coverage,25,82 although very deep sequencing may introduce artifacts that are difficult to distinguish from minor subclones. Introduction of artifacts in NGS can be avoided by using newer methods that implement unique molecular identifiers.83,84
Clonal heterogeneity within tumors is a roadblock to precision oncology, as treating the dominant clone could potentially spur the growth of a resistant subclone. Griffith et al tracked tumor evolution and refined the clonal architecture of acute myeloid leukemia over approximately 500 days within a single patient (treated with an induction chemotherapy and four rounds of consolidation chemotherapy) to demonstrate that: 1) all clonal populations originated from clones existing prior to therapy and no new subclones emerged following treatment and 2) dominant TP53-mutant clonal populations observed at relapse but difficult to detect at diagnosis were selected for by chemotherapy, likely because the TP53 mutations conferred a drug resistance phenotype and resultant growth advantage.
A similar story emerges in NSCLC, where tumors become resistant to first-generation EGFR tyrosine kinase inhibitors (TKIs) gefitinib and erlotinib by acquiring an EGFR T790M mutation.85 Third-generation EGFR TKIs, such as rociletinib, were developed to treat patients with these EGFR T790M resistance mutations.86 However, rociletinib-resistant biopsies show both wild type and T790M clones coexisting prior to treatment, suggesting that clonal heterogeneity can impart resistance.87 Furthermore, tracking and profiling circulating tumor DNA, using liquid biopsy technology, and incorporating this data into personalized polygenetic trees reveal early-stage cancer evolution by showing the contribution of clonal heterogeneity to adjuvant chemotherapy resistance and metastasis in high-risk patients.88
These studies imply that overcoming clonal diversity is a major hurdle to the successful adoption of precision oncology.
Cancer disparities and population diversity challenges
Cancer disparities are differences in cancer measures—incidence, prevalence, mortality, and others—among specific populations. The actions of and interactions between nongenetic factors (eg, socioeconomic status, culture, diet, stress, geography, environment) and genetic factors contribute to cancer phenotypes. Precision oncology programs that do not account for population heterogeneity are likely to propagate health disparities by mistakenly ascribing a genetic foundation to outcomes or other clinical measures that actually result from nongenetic factors. The reference genome was built from a global reference population where 86% of genomic variants were restricted to a single continental group.89 When performing sequence alignment and variant calling, population-specific genetic variation may be ignored, resulting in a reference genome biased toward the European population and against non-European populations.
Despite the critical modifying role of nongenetic factors in explaining population differences in cancer etiology and response to therapy, genetic factors also contribute to disparities. For example, a cohort study using Children’s Oncology Group outcomes data reported that children of African ancestry have a higher prevalence of high-risk neuroblastoma and poorer outcomes than do patients of European ancestry.90 Later, Gamazon et al demonstrated that differences in the VAF of a risk variant in SPAG16 between African and European populations contributed significantly to this observed disparity. Similar findings have been reported in populations of African ancestry with prostate cancer.91
Moreover, differences in the genomic architecture of different populations can be a useful tool for fine-mapping studies of disease-associated loci. For example, using data obtained through the 1000 Genomes Project, it was found that individuals of African ancestry have smaller haplotype blocks and higher genetic heterogeneity than European and Asian populations at the 11q13 breast cancer susceptibility.92 Asimit et al performed a transethnic fixed-effects meta-analysis on simulated data of varying ancestral composition, allelic heterogeneity, and minor allele frequency.93 They concluded that inclusion of African genomes yields a significant improvement in fine-mapping resolution. Hungate et al employed a similar polyethnic strategy to discover a novel locus associated with pediatric B-lineage ALL and the risk variant defining the region, which they then functionally analyzed.94
Currently, non-European populations are underrepresented in virtually all population health studies, thereby limiting our capacity to deconvolute the relative roles of genetic and nongenetic factors in human health and disease. This discrepancy remains as one of the most important barriers to overcome in the path towards precision oncology. Only through inclusiveness will it be possible to learn from and benefit everyone.
Opportunities and future developments
Translating genetic variants into actionable therapies
The results of clinical trials to test the feasibility of MP-guided targeted therapies have frequently been unsatisfactory. The intuitive hypothesis in these trials is that patients with alterations in genes and pathways that render them sensitive to targeted therapies should be treatable with these therapies. However, the lack of success may indicate that our current knowledge of molecular disease etiologies and drug mechanisms of action has not kept pace with the technological advances in genomics.75
Typically, driver mutations are identified in patient samples by comparing paired germline and somatic samples from the same patient following review by an expert panel.44 However, manual variant refinement by a tumor board is labor-intensive and non-reproducible. To scale up and enhance reproducibility, Ainscough et al developed a deep learning approach to automate and increase the efficiency of review processes for refinement of somatic variants.95 Prediction of therapeutic response from diagnostics and prognostics in breast cancer is demonstrated by machine learning that combines genomic data including copy number, mutations, and isoform expression.67 By incorporating tumor heterogeneity, we propose a bioinformatics framework (Figure 3) to identify actionable mutations for targeted therapy using genomic profiling. The first part of this framework uses ultra-deep sequencing of tumor tissue to characterize driver mutations/genes within each tumor subclone (Figure 3A). These driver mutations are annotated and filtered by functional impact (using ANNOVAR96 or VEP97) and clinical interpretation (using OncoKB58 or CIViC59). Once mutations have been characterized in the tumor subclones, a multi-pronged precision oncology approach includes (Figure 3B–D): 1) finding genes that harbor driver mutation(s) that might be directly druggable or exclusive to drug targeting by repositioning drugs described in the literature98; 2) finding genetic defects where inhibition of another gene would cause death via synthetic lethality, as in the use of PARP inhibitors in breast cancer patients with BRCA1/2 mutations54; 3) using mutations as biomarkers to monitor drug response; 4) identifying residues mutated within multiple clones, which imply shared etiology99; and 5) modeling drug–protein interactions in order to predict potential mechanisms of resistance (eg, the structural changes in the EGFR ectodomain in tumors resistant to anti-EGFR therapy in NSCLC were accurately modeled before crystal structures were available).100 Notably, these five approaches are not mutually exclusive and indeed should be utilized in parallel. Also, therapies that attack targets shared among clones should be used. Furthermore, this approach should be repeated for each relapse or metastatic sample in order to identify mutations enriched at relapse or in metastasis, as these mutations are likely to point towards the drivers of these processes.
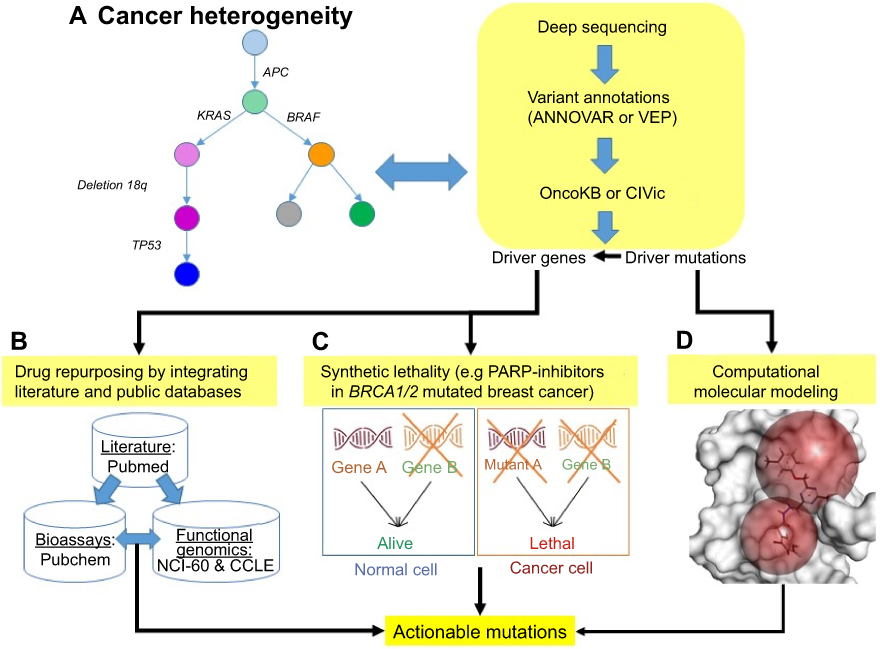 |
Figure 3 Translating pathogenic variants into matched molecularly targeted therapy. (A) Upon ultra-deep sequencing data, driver mutations/genes can be discovered by modeling tumor evolution, and further annotated by tools and databases. (B) Literature-based drug repurposing98 is used to target the driver genes by integrating drug and compound bioassays (PubChem: https://pubchem.ncbi.nlm.nih.gov/) and function genomics (NCI-60: https://dtp.cancer.gov/discovery_development/nci-60/, and CCLE: https://portals.broadinstitute.org/ccle) databases. (C) Synthetic lethality is a novel anticancer strategy to increase the specificity of a drug target in cancer cells harboring actionable mutations while decreasing off-target effects on normal tissues (eg, inhibiting PARP in breast cancer with BRCA1/2 mutations).54 (D) Structural modeling is used to evaluate whether drug–target interactions are directly mediated by actionable mutation(s) or other mutated residue(s). |
One consideration is the cost of ultra-deep sequencing. To address the need for compromise between cost and exploration of tumor heterogeneity, we suggest performing ultra-deep targeted sequencing using a gene panel—a focused set of target genes derived from tiered annotations of actionable genetic alterations.36,101 By implementing these considerations, it may be possible to use genomic profiles to develop matched therapies with greater efficacy and safety.
Genomic and clinical data aggregation
The Exome Aggregation Consortium (ExAC) is an alliance of genomic scientists accumulating and harmonizing WES data sets from a multitude of sequencing projects.102 With the addition of WGS data, this association is referred to as the Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org/). Data from ExAC have been used in the discovery of naturally occurring knockout variants in human protein-coding genes,102 the characterization of rates and properties of genetic intolerance to rare CNV,103 and the discovery of individuals with pathogenic variants implicated in a number of Mendelian disorders.104 Although the goal of ExAC and gnomAD is to make reference data sets of diverse populations available for study by the wider scientific community, this is largely limited to genomics data. To facilitate collaborations and consortia to catalyze precision oncology, clinical information (EHR and disease phenotype) with long-term follow up is required in addition to genomic data. This will allow for the identification of genotype-phenotype associations. The technical barriers to the sharing of clinical information include a disuse of common standardized disease terms, definitions, and ontologies across health care systems and EHRs. Consequently, it is often unclear if a term used by one provider in reference to one patient has the same meaning when used by a second provider in reference to a second patient. Moreover, it is often impossible to discern whether unrecorded clinical data are negative or simply missing.105 To promote sharing of high-quality and standardized genomic and clinical data, the Global Alliance for Genomics and Health—an international coalition of over 470 stakeholder organizations from more than 60 countries (http://genomicsandhealth.org/)—was founded to develop interoperable solutions. Instead of building centralized repositories, the Global Alliance suggests that shared data could be stored by their originating institutions and assessed and analyzed by members of the global research community through secure cloud-embedded network solutions.105 Besides integrating clinical information with genomic data across each health system, this international alliance points towards a path for publicly accessible clinical and genomic data resources.
Concluding thoughts
The goal of precision oncology is to use genetics to guide cancer prevention and treatment, in order to maximize positive outcomes and minimize adverse events. Many lessons, both expected and unexpected, have emerged from the early forays into large-scale precision oncology programs. Here, we have discussed some of the major findings and barriers to success resulting from these massive efforts. We avoided including topics such as pharmacogenomics,106 immuno-oncology,107 and epigenomics108 in this review, as each necessitates a separate review article.
The first generation of precision oncology programs has demonstrated significant benefits to patients, showing that >30% of cancer patients have at least one actionable variant,109 and the use of WGS or WES data can influence management for >50% of patients.9,35,37,39–41 By further using circulating tumor DNA sequencing (as in the TARGET study),110 drug combination for targeting multiple actionable variants (as in the I-PREDICT study),111 or tumor microenvironment with RNA sequencing (as in the WINTHER trial),112 three recent innovative precision oncology studies demonstrate not only high rates of actionability (41%, 49%, and 52%, respectively) but also relatively high efficacy of identifying matched therapies (4%, 11%, and 4%, respectively).113
Despite the evidence of success of precision oncology as outlined in this article, significant barriers remain before precision oncology can become a standard of care. Paramount among these obstacles are the significant cost and infrastructure requirements of genomics, and the urgent need for inclusivity to overcome biases and limitations inherent in studies comprised largely individuals of European ancestry. Furthermore, even after clinical and genomic data have been collected, there remain the difficulties of data standardization and harmonization.
Despite these challenges, there is considerable cause for optimism. The costs of NGS technologies and assays continue to decline, and clinicians and scientists continually form consortia which use common terminology in the prospective collection of clinical data. Moreover, there is increasing recognition of the importance of inclusivity in studies to overcome disparities in cancer treatment. This is an exciting time in the development of precision oncology, as new technologies make it possible to explore the genomic landscape of cancer at a resolution and scale unimaginable just a few years ago. The challenge is to translate these discoveries into clinical practice.
Acknowledgment
The authors thank Andrew Shih (Feinstein Institutes for Medical Research (FIMR), Northwell, NY) for his comments on an earlier version of this manuscript, and Robert P Adelson (FIMR, Northwell, NY) for editing of and commenting on the revised manuscript.
Author contributions
This manuscript was drafted by H-TY and KO, with significant contributions from RHS and DT. KO oversaw the project. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
All authors have no conflicts of interest in this work.
References
1. Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456(7218):53–59. doi:10.1038/nature07517
2. Tennessen JA, Bigham AW, O’Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337(6090):64–69. doi:10.1126/science.1219240
3. Genomes Project Consortium, Abecasis GR, Altshuler D, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi:10.1038/nature09534
4. Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–1879. doi:10.1001/jama.2014.14601
5. Chong JX, Buckingham KJ, Jhangiani SN, et al. The genetic basis of mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet. 2015;97(2):199–215. doi:10.1016/j.ajhg.2015.06.009
6. Holm H, Gudbjartsson DF, Sulem P, et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet. 2011;43(4):316–320. doi:10.1038/ng.781
7. Do R, Stitziel NO, Won HH, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518(7537):102–106. doi:10.1038/nature13917
8. Steinberg S, Stefansson H, Jonsson T, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet. 2015;47(5):445–447. doi:10.1038/ng.3246
9. Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354:6319. doi:10.1126/science.aal1794
10. Gundem G, Perez-Llamas C, Jene-Sanz A, et al. IntOGen: integration and data mining of multidimensional oncogenomic data. Nat Methods. 2010;7(2):92–93. doi:10.1038/nmeth0210-92
11. Dees ND, Zhang Q, Kandoth C, et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res. 2012;22(8):1589–1598. doi:10.1101/gr.134635.111
12. Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. doi:10.1038/nature12213
13. Handsaker RE, Korn JM, Nemesh J, McCarroll SA. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet. 2011;43(3):269–276. doi:10.1038/ng.768
14. Zack TI, Schumacher SE, Carter SL, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45(10):1134–1140. doi:10.1038/ng.2760
15. Carter L, Rothwell DG, Mesquita B, et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med. 2017;23(1):114–119. doi:10.1038/nm.4239
16. Nik-Zainal S, Alexandrov LB, Wedge DC, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149(5):979–993. doi:10.1016/j.cell.2012.04.024
17. Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3(1):246–259. doi:10.1016/j.celrep.2012.12.008
18. Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi:10.1038/nature12477
19. Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. doi:10.1038/nature14169
20. Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521(7553):489–494. doi:10.1038/nature14410
21. Nik-Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534(7605):47–54. doi:10.1038/nature17676
22. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221. doi:10.1056/NEJMoa1516192
23. Nik-Zainal S, Van Loo P, Wedge DC, et al. The life history of 21 breast cancers. Cell. 2012;149(5):994–1007. doi:10.1016/j.cell.2012.04.023
24. Gibson WJ, Hoivik EA, Halle MK, et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat Genet. 2016. doi:10.1038/ng.3602
25. Griffith M, Miller CA, Griffith OL, et al. Optimizing cancer genome sequencing and analysis. Cell Syst. 2015;1(3):210–223. doi:10.1016/j.cels.2015.08.015
26. Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373(24):2336–2346. doi:10.1056/NEJMoa1508054
27. Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA. 2017;318(9):825–835. doi:10.1001/jama.2017.11137
28. Huang K-L, Mashl RJ, Wu Y, et al. Pathogenic germline variants in 10,389 adult cancers. Cell. 2018;173(2):355–370.e14. doi:10.1016/j.cell.2018.03.039
29. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793–795. doi:10.1056/NEJMp1500523
30. Tannock IF, Hickman JA. Limits to personalized cancer medicine. N Engl J Med. 2016;375(13):1289–1294. doi:10.1056/NEJMsb1607705
31. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
32. Aronson SJ, Rehm HL. Building the foundation for genomics in precision medicine. Nature. 2015;526(7573):336–342. doi:10.1038/nature15816
33. Chen R, Shi L, Hakenberg J, et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotechnol. 2016;34(5):531–538. doi:10.1038/nbt.3514
34. Telenti A, Pierce LC, Biggs WH, et al. Deep sequencing of 10,000 human genomes. Proc Natl Acad Sci U S A. 2016;113(42):11901–11906. doi:10.1073/pnas.1613365113
35. Parsons DW, Roy A, Yang Y, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016. doi:10.1001/jamaoncol.2015.5699
36. Pinto N, Volchenboum SL, Skol AD, et al. Establishing a translational genomics infrastructure in pediatric cancer: the GREAT KIDS experience. Per Med. 2015;12(3):221–229. doi:10.2217/pme.14.90
37. Worst BC, van Tilburg CM, Balasubramanian GP, et al. Next-generation personalised medicine for high-risk paediatric cancer patients – the INFORM pilot study. Eur J Cancer. 2016;65:91–101. doi:10.1016/j.ejca.2016.06.009
38. Marron JM, DuBois SG, Glade Bender J, et al. Patient/parent perspectives on genomic tumor profiling of pediatric solid tumors: the Individualized Cancer Therapy (iCat) experience. Pediatr Blood Cancer. 2016;63(11):1974–1982. doi:10.1002/pbc.26137
39. Harris MH, DuBois SG, Glade Bender JL, et al. Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: the Individualized Cancer Therapy (iCat) Study. JAMA Oncol. 2016. doi:10.1001/jamaoncol.2015.5689
40. Mody RJ, Wu YM, Lonigro RJ, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. 2015;314(9):913–925. doi:10.1001/jama.2015.10080
41. Oberg JA, Glade Bender JL, Sulis ML, et al. Implementation of next generation sequencing into pediatric hematology-oncology practice: moving beyond actionable alterations. Genome Med. 2016;8(1):133. doi:10.1186/s13073-016-0389-6
42. West HJ. Novel precision medicine trial designs: umbrellas and baskets. JAMA Oncol. 2017;3(3):423. doi:10.1001/jamaoncol.2016.5299
43. Linderman MD, Nielsen DE, Green RC. Personal genome sequencing in ostensibly healthy individuals and the PeopleSeq consortium. J Pers Med. 2016;6(2):14. doi:10.3390/jpm6020014
44. Scollon S, Bergstrom K, Kerstein RA, et al. Obtaining informed consent for clinical tumor and germline exome sequencing of newly diagnosed childhood cancer patients. Genome Med. 2014;6(9):69. doi:10.1186/s13073-014-0069-3
45. Carey DJ, Fetterolf SN, Davis FD, et al. The Geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet Med. 2016;18(9):906–913. doi:10.1038/gim.2015.187
46. Maissenhaelter BE, Woolmore AL, Schlag PM. Real-world evidence research based on big data: motivation-challenges-success factors. Onkologe (Berl). 2018;24(Suppl 2):91–98. doi:10.1007/s00761-018-0358-3
47. Gottesman O, Kuivaniemi H, Tromp G, et al. The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genet Med. 2013;15(10):761–771. doi:10.1038/gim.2013.72
48. Bowton E, Field JR, Wang S, et al. Biobanks and electronic medical records: enabling cost-effective research. Sci Transl Med. 2014;6(234):234cm233. doi:10.1126/scitranslmed.3008604
49. Denny JC, Ritchie MD, Basford MA, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26(9):1205–1210. doi:10.1093/bioinformatics/btq126
50. Roden DM, Denny JC. Integrating electronic health record genotype and phenotype datasets to transform patient care. Clin Pharmacol Ther. 2016;99(3):298–305. doi:10.1002/cpt.321
51. Verma A, Leader JB, Verma SS, et al. Integrating clinical laboratory measures and Icd-9 code diagnoses in phenome-wide association studies. Pac Symp Biocomput. 2016;21:168–179.
52. Schwartz ML, Williams MS, Murray MF. Adding protective genetic variants to clinical reporting of genomic screening results: restoring balance. JAMA. 2017;317:1527. doi:10.1001/jama.2017.1533
53. Shevchenko Y, Bale S. Clinical versus research sequencing. Cold Spring Harb Perspect Med. 2016;6(11):a025809. doi:10.1101/cshperspect.a025809
54. Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi:10.1056/NEJMoa0900212
55. Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249–255. doi:10.1038/gim.2016.190
56. Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017;100(2):267–280. doi:10.1016/j.ajhg.2017.01.004
57. Mateo J, Chakravarty D, Dienstmann R, et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann Oncol. 2018;29:1895–1902. doi:10.1093/annonc/mdy263
58. Chakravarty D, Gao J, Phillips SM, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017;1:1–16. doi:10.1200/PO.17.00011
59. Griffith M, Spies NC, Krysiak K, et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet. 2017;49(2):170–174. doi:10.1038/ng.3774
60. Wagner AH, Walsh B, Mayfield G, et al. A harmonized meta-knowledgebase of clinical interpretations of cancer genomic variants. bioRxiv. 2018. doi:10.1101/366856
61. Emamekhoo H, Lang JM. Are liquid biopsies ready for primetime? Cancer. 2019;125(6):834–837. doi:10.1002/cncr.31644
62. Campos CDM, Jackson JM, Witek MA, Soper SA. Molecular profiling of liquid biopsy samples for precision medicine. Cancer J. 2018;24(2):93–103. doi:10.1097/PPO.0000000000000311
63. Voest EE, Bernards R. DNA-guided precision medicine for cancer: a case of irrational exuberance? Cancer Discov. 2016;6(2):130–132. doi:10.1158/2159-8290.CD-15-1321
64. Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337–345. doi:10.1056/NEJMoa033025
65. Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–1765. doi:10.1056/NEJMoa0804385
66. De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–762. doi:10.1016/S1470-2045(10)70130-3
67. Lesurf R, Griffith OL, Griffith M, et al. Genomic characterization of HER2-positive breast cancer and response to neoadjuvant trastuzumab and chemotherapy-results from the ACOSOG Z1041 (Alliance) trial. Ann Oncol. 2017;28(5):1070–1077. doi:10.1093/annonc/mdx048
68. Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. doi:10.1056/NEJMoa1408440
69. Shaw AT, Solomon BJ. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2015;372(7):683–684. doi:10.1056/NEJMc1415359
70. Von Hoff DD, Stephenson JJ
71. Kim ES, Herbst RS, Wistuba II, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1(1):44–53. doi:10.1158/2159-8274.CD-10-0010
72. Le Tourneau C, Delord JP, Goncalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16(13):1324–1334. doi:10.1016/S1470-2045(15)00188-6
73. Meric-Bernstam F, Brusco L, Shaw K, et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol. 2015;33(25):2753–2762. doi:10.1200/JCO.2014.60.4165
74. Le Tourneau C, Paoletti X, Servant N, et al. Randomised proof-of-concept phase II trial comparing targeted therapy based on tumour molecular profiling vs conventional therapy in patients with refractory cancer: results of the feasibility part of the SHIVA trial. Br J Cancer. 2014;111(1):17–24. doi:10.1038/bjc.2014.211
75. Hyman DM, Taylor BS, Baselga J. Implementing genome-driven oncology. Cell. 2017;168(4):584–599. doi:10.1016/j.cell.2016.12.015
76. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–510. doi:10.1038/nature10738
77. Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. doi:10.1016/j.cell.2012.06.023
78. Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–726. doi:10.1016/j.cell.2013.01.019
79. Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47(5):458–468. doi:10.1038/ng.3273
80. Gundem G, Van Loo P, Kremeyer B, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520(7547):353–357. doi:10.1038/nature14347
81. McPherson A, Roth A, Laks E, et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat Genet. 2016;48(7):758–767. doi:10.1038/ng.3573
82. Gerstung M, Beisel C, Rechsteiner M, et al. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun. 2012;3:811. doi:10.1038/ncomms1814
83. Kivioja T, Vaharautio A, Karlsson K, et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods. 2011;9(1):72–74. doi:10.1038/nmeth.1778
84. Kennedy SR, Schmitt MW, Fox EJ, et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat Protoc. 2014;9(11):2586–2606. doi:10.1038/nprot.2014.170
85. Cortot AB, Janne PA. Molecular mechanisms of resistance in epidermal growth factor receptor-mutant lung adenocarcinomas. Eur Respir Rev. 2014;23(133):356–366. doi:10.1183/09059180.00004614
86. Sequist LV, Soria JC, Goldman JW, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015;372(18):1700–1709. doi:10.1056/NEJMoa1413654
87. Piotrowska Z, Niederst MJ, Karlovich CA, et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov. 2015;5(7):713–722. doi:10.1158/2159-8290.CD-15-0399
88. Abbosh C, Birkbak NJ, Wilson GA, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545(7655):446–451. doi:10.1038/nature22364
89. Genomes Project Consortium, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi:10.1038/nature15393
90. Henderson TO, Bhatia S, Pinto N, et al. Racial and ethnic disparities in risk and survival in children with neuroblastoma: a Children’s Oncology Group study. J Clin Oncol. 2011;29(1):76–82. doi:10.1200/JCO.2010.29.6103
91. Gamazon ER, Pinto N, Konkashbaev A, et al. Trans-population analysis of genetic mechanisms of ethnic disparities in neuroblastoma survival. J Natl Cancer Inst. 2013;105(4):302–309. doi:10.1093/jnci/djs503
92. Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93(5):779–797. doi:10.1016/j.ajhg.2013.10.012
93. Asimit JL, Hatzikotoulas K, McCarthy M, Morris AP, Zeggini E. Trans-ethnic study design approaches for fine-mapping. Eur J Hum Genet. 2016;24(9):1330–1336. doi:10.1038/ejhg.2016.1
94. Hungate EA, Vora SR, Gamazon ER, et al. A variant at 9p21.3 functionally implicates CDKN2B in paediatric B-cell precursor acute lymphoblastic leukaemia aetiology. Nat Commun. 2016;7:10635. doi:10.1038/ncomms10635
95. Ainscough BJ, Barnell EK, Ronning P, et al. A deep learning approach to automate refinement of somatic variant calling from cancer sequencing data. Nat Genet. 2018;50(12):1735–1743. doi:10.1038/s41588-018-0257-y
96. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi:10.1093/nar/gkq603
97. McLaren W, Gil L, Hunt SE, et al. The ensembl variant effect predictor. Genome Biol. 2016;17(1):122. doi:10.1186/s13059-016-0974-4
98. Yang HT, Ju JH, Wong YT, Shmulevich I, Chiang JH. Literature-based discovery of new candidates for drug repurposing. Brief Bioinform. 2017;18(3):488–497. doi:10.1093/bib/bbw030
99. Chang MT, Asthana S, Gao SP, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34(2):155–163. doi:10.1038/nbt.3391
100. Bertotti A, Papp E, Jones S, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526(7572):263–267. doi:10.1038/nature14969
101. Uzilov AV, Ding W, Fink MY, et al. Development and clinical application of an integrative genomic approach to personalized cancer therapy. Genome Med. 2016;8(1):62. doi:10.1186/s13073-016-0313-0
102. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi:10.1038/nature19057
103. Ruderfer DM, Hamamsy T, Lek M, et al. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat Genet. 2016;48(10):1107–1111. doi:10.1038/ng.3638
104. Tarailo-Graovac M, Zhu JYA, Matthews A, van Karnebeek CDM, Wasserman WW. Assessment of the ExAC data set for the presence of individuals with pathogenic genotypes implicated in severe Mendelian pediatric disorders. Genet Med. 2017. doi:10.1038/gim.2017.50
105. The Clinical Cancer Genome Task Team of the Global Alliance for Genomics and Health. Sharing clinical and genomic data on cancer – the need for global solutions. N Engl J Med. 2017;376(21):2006–2009. doi:10.1056/NEJMp1612254
106. Weinshilboum RM, Wang L. Pharmacogenomics: precision medicine and drug response. Mayo Clin Proc. 2017;92(11):1711–1722. doi:10.1016/j.mayocp.2017.09.001
107. Steuer CE, Ramalingam SS. Tumor mutation burden: leading immunotherapy to the Era of precision medicine? J Clin Oncol. 2018;36(7):631–632. doi:10.1200/JCO.2017.76.8770
108. Moran S, Martinez-Cardus A, Boussios S, Esteller M. Precision medicine based on epigenomics: the paradigm of carcinoma of unknown primary. Nat Rev Clin Oncol. 2017;14(11):682–694. doi:10.1038/nrclinonc.2017.97
109. AACR Project GENIE Consortium. AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. 2017;7(8):818–831. doi:10.1158/2159-8290.CD-17-0151
110. Rothwell DG, Ayub M, Cook N, et al. Utility of ctDNA to support patient selection for early phase clinical trials: the TARGET study. Nat Med. 2019;25(5):738–743. doi:10.1038/s41591-019-0380-z
111. Sicklick JK, Kato S, Okamura R, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. 2019;25(5):744–750. doi:10.1038/s41591-019-0407-5
112. Rodon J, Soria JC, Berger R, et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med. 2019;25(5):751–758. doi:10.1038/s41591-019-0424-4
113. Le Tourneau C, Borcoman E, Kamal M. Molecular profiling in precision medicine oncology. Nat Med. 2019;25(5):711–712. doi:10.1038/s41591-019-0442-2
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.