")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Precision Medicine of Sodium Benzoate for the Treatment of Behavioral and Psychological Symptoms of Dementia (BPSD)
Authors Lin CH , Yang HT, Chen PK, Wang SH, Lane HY
Received 11 October 2019
Accepted for publication 9 February 2020
Published 20 February 2020 Volume 2020:16 Pages 509—518
DOI https://doi.org/10.2147/NDT.S234371
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yuping Ning
Chieh-Hsin Lin,1– 3 Hui-Ting Yang,4 Ping-Kun Chen,5,6 Shi-Heng Wang,7 Hsien-Yuan Lane2,8,9
1Department of Psychiatry, Kaohsiung Chang Gung Memorial Hospital, Chang Gung University College of Medicine, Kaohsiung, Taiwan; 2Graduate Institute of Biomedical Sciences, China Medical University, Taichung, Taiwan; 3School of Medicine, Chang Gung University, Taoyuan, Taiwan; 4School of Food Safety, Taipei Medical University, Taipei, Taiwan; 5School of Medicine, China Medical University, Taichung, Taiwan; 6Department of Neurology, China Medical University Hospital, Taichung, Taiwan; 7Department of Occupational Safety and Health, China Medical University, Taichung, Taiwan; 8Department of Psychiatry & Brain Disease Research Center, China Medical University Hospital, Taichung, Taiwan; 9Department of Psychology, College of Medical and Health Sciences, Asia University, Taichung, Taiwan
Correspondence: Hsien-Yuan Lane
Department of Psychiatry, China Medical University Hospital, No. 2, Yuh-Der Road, Taichung 404, Taiwan
Email [email protected]
Objective: Behavioral and psychological symptoms of dementia (BPSD) are associated with poorer prognosis of dementia. A 24-week study demonstrated that sodium benzoate, a D-amino acid oxidase (DAAO) inhibitor, surpassed placebo in improving cognitive function in early-phase Alzheimer’s disease; however, benzoate did not excel placebo in another 6-week study on BPSD. The current study examined whether the precision medicine approach was able to identify specific individuals with BPSD who could benefit from benzoate treatment.
Methods: In the randomized, double-blind, placebo-controlled, 6-week trial, 97 patients with BPSD were allocated to receive 250– 1500 mg/day of sodium benzoate or placebo. Cognitive function was measured by the Alzheimer’s disease assessment scale-cognitive subscale (ADAS-cog) and behavioral and psychological symptoms were mainly measured by Behavioral Pathology in Alzheimer’s Disease Rating Scale (BEHAVE-AD). DAAO level, amino acids (L-serine, D-serine, L-alanine, and D-alanine, glycine), and two antioxidants (catalase, superoxide dismutase) were assayed in peripheral blood.
Results: After benzoate treatment, DAAO inhibition was correlated with ADAS-cog decrease (p = 0.034), while baseline DAAO level was correlated with baseline BEHAVE-AD score. Multiple linear regression analyses showed that cognitive improvement after benzoate treatment was correlated with DAAO decrease, female gender, younger age, BMI, baseline BPSD severity, and antipsychotic use.
Conclusion: The finding suggests that sodium benzoate may have potential to benefit cognitive function in a fraction of BPSD patients after 6 weeks of treatment. Of note, the precision medicine approach may be helpful for identifying individuals who could respond to benzoate. More studies are warranted to confirm the preliminary findings.
Trial Registration: The trial was registered online (https://clinicaltrials.gov/ct2/show/NCT02103673).
Keywords: behavioral and psychological symptoms of dementia, BPSD, N-methyl-D-aspartate, D-amino acids oxidase, DAAO, inhibitor, sodium benzoate, precision medicine
Introduction
The behavioral and psychological symptoms of dementia (BPSD) develop in Alzheimer’s disease (AD), vascular dementia (VaD), and other kinds of dementia.1 Depression is common in the early stage of dementia, while psychotic symptoms are more often in later stages.2 BPSD contribute to a speedier decline of cognitive and global function and poorer prognosis.3 Psychosocial interventions may improve BPSD; however, medication is often required for more severe manifestations. To date, no pharmacological treatment has been found to have disease-modifying activity, though some medications, including several antipsychotics, may have modest effects,4–8 or approved by the US Food and Drug Administration (FDA) for treating BPSD.
Altered N-Methyl-D-aspartate receptor (NMDAR)-related neurotransmission is involved in dementia manifestations, including cognitive and behavioral domains.9 NMDAR over-activation leads to neurotoxicity, while its hypofunction results in neurodegeneration,10 suggesting that NMDAR activity needs to maintain at an optimal range.11 One of the avenues to enhance NMDAR function is via inhibiting D-amino acids oxidase (DAAO) activity.12 In a placebo-controlled trial,13 sodium benzoate, a pivotal DAAO inhibitor,14 significantly improved the cognitive function of patients with early-phase AD, without BPSD.
Precision medicine approaches have been advancing our understanding of the development and treatment of AD dementia. However, precision medicine has not yet been adequately applied by many of clinical trials for dementia.15 Increased oxidative stress also contributes to aging processes and neurodegenerative diseases, while free radicals damage cells and tissues.16 Antioxidants may help prevent and reverse cognitive deficits induced by free radicals.17 Studies indicate a link among age-related NMDAR dysfunction, oxidative stress, and senescence and related cognitive decline.18 Benzoate can inhibit reactive oxygen species19 and increase the activity of catalase (CAT), an antioxidant, in patients with schizophrenia.20 In a recent study on BPSD,21 6-week treatment of benzoate (mean doses at weeks 2, 4 and 6 were 341.8 ± 121.8 [SD], 528.4 ± 248.3 and 622.0 ± 340.6 mg/day of 622.0 mg/day, respectively) did not excel placebo in restoring cognitive function or reducing behavioral and psychological symptoms for all patients as a whole. Of note, the two treatments showed similar safety profiles.21 Therefore, the current study aimed to test whether the precision medicine approach was able to identify subgroups of BPSD individuals who could respond to benzoate treatment, by measuring antioxidants, DAAO, and NMDAR-related amino acids in peripheral blood,22 and examined their associations with treatment response of benzoate.
Methods and Materials
The methods and materials have been briefed elsewhere.21 Here we provided more details.
Standard Protocol Approvals, Registrations, and Patient Consents
Patients were recruited from the outpatient clinics at the Department of Psychiatry, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Department of Psychiatry, China Medical University Hospital, Taichung, and Department of Neurology, Lin-Shin Hospital, Taichung, which are three major medical centers in Taiwan. The study was approved by the institutional review boards of all three participating hospitals, and conducted in accordance with the current revision of the Declaration of Helsinki.
Patients were evaluated by research psychiatrists and neurologists after a thorough medical and neurological workup. Written informed consent was obtained from all participants or guardians of participants in this study. There was no disclosure of any recognizable persons in photographs, videos, or other information in this report.
The trial was registered on the ClinicalTrials.gov website (NCT02103673): https://clinicaltrials.gov/ct2/show/NCT02103673.
Study Design
The inclusion criteria, exclusion criteria, concurrent allowable medications (anti-dementia medications and antipsychotics), and doses of study medications have been described elsewhere.19 We now provided more details on the dose-escalation strategies. The dose of the study medications (sodium benzoate or placebo) was started at 250–500 mg/day (250 mg q.d. or b.i.d.), according to clinical judgement on patients’ severity and tolerability. If the patients showed poor response and fine tolerability, the dose was increased by 250–500 mg/day from week 3; and, if still poor response and fine tolerability, the dose was further increased by another 250–500 mg/day from week 5.
Patient’s medical adherence and safety were closely monitored and ensured by caregivers and research physicians and pill-counting by the study staff.
Assessments
The cognitive function was measured by the Alzheimer’s disease assessment scale-cognitive subscale (ADAS-cog)23 at baseline and endpoint, and behavioral and psychological symptoms were measured by BEHAVE-AD at weeks 0, 2, 4, and 6. ADAS-cog is the most popular cognitive assessment instrument used in AD and VaD clinical trials. Its scores range from 0 (best) to 70 (worst). BEHAVE-AD, a commonly used tool for the assessment of BPSD,24 consists of global evaluation and seven symptoms domains, including paranoid and delusional ideation, hallucinations, activity disturbances, aggressiveness, diurnal rhythm disturbances, affective disturbances, anxieties, and phobias; and higher score indicates severer symptoms.25
Clinical ratings were performed by the research psychiatrists and neurologists who were trained and experienced in the rating scales. Inter-rater reliability was analyzed with the ANOVA test. Only raters reaching the intra-class correlation coefficients of ≥0.90 during pre-study training were allowed to rate the patients. To minimize inter-rater variability, each individual patient was assessed by the same rater throughout the trial.
Laboratory Measurements
Laboratory measurements, including DAAO, amino acids, and antioxidants, were measured at baseline and endpoint.
DAAO protein concentrations were measured using commercially available enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer's recommended protocol (Cloud-Clone Corp, Houston, TX, USA). Briefly, 100 μL serum samples and the standard were added to each well of a 96-well plate. The solutions were incubated for 1 hr at 37°C. The liquid was then removed. One hundred-microliter Detection Reagent A was added to each well and incubated for 1 hr at 37°C. Each well was washed for 3 times, 100 µL of Detection Reagent B was added to each well, and the solutions were incubated for 30 mins at 37°C. Each well was washed for 5 times and then incubated with 90 µL substrate solution for 20 min at 37°C with the protection from light, and 50 µL stop solution was added to each well. A Benchmark Plus Microplate Reader (Bio-Rad) was used to read the optical density at 450 nm. The concentrations of DAAO in the samples were determined according to a standard curve.
For measuring concentrations of amino acids, plasma was firstly extracted by methanol (1:3, by volume), then filtered after 15 min centrifugation (1500 ×g) with nylon membranes (0.45mM, Minisart SRP4, Sartorius, Germany). The filtrate was diluted with proper amount of 20% methanol, derivatized with N-isobutyl-L-cysteine (IBC) and O-phthaldialdehyde mixture for 5 mins, and then injected into high-performance liquid chromatography (HPLC, L-7100 Pump, L7250 Autosampler, L-7250, with L7480 fluorescence Detector, Hitachi, Japan) for analysis. Analytical column (Grom-Sil OPA-2, 5 µm, 250 mm*4 mm, Part No: GSOP 20512S2504, SAP No: 5113679, Grace, US) with guard column (Grom-Sil OPA-2, 5 µm, 10 mm×4 mm, Part No: GSOP20512v0104V, Grace, US) was used for the determination. Isocratic elution of mobile phase A (23mM sodium acetate, pH 6.0) and B (50 mL acetonitrile in 600 mL methanol) were performed under fluorescence detection (excitation 260 nm, emission 455 nm), respectively. Retention time of each amino acid was L-serine, 33.6 mins; D-serine, 35.8 mins; glycine, 41.5 mins; L-alanine, 47.2 mins; and D-alanine, 50.3 mins, respectively.
Superoxide dismutase (SOD) levels were measured using commercially available kits according to the manufacturer's recommended protocol (Merck, Kenilworth, NJ, USA). Briefly, 200 μL of the diluted radical detector and 10 μL of standard or plasma sample were added to each well of a 96-well plate. Then, 20 μL of diluted xanthine oxidase was added to each well and then incubated on a shaker for 20 mins at room temperature. The absorbance at 450 nm was assessed with the Benchmark Plus Microplate Reader (Bio-Rad). The SOD level in the sample was determined using a standard curve and the formula.
CAT activity was measured using commercially available kits according to the manufacturer's recommended protocol (Cayman, Ann Arbor, MI, USA). Briefly, 100 μL diluted assay buffer, 30 μL methanol, 20μL plasma samples, standard and diluted Catalase (Control) were added to each well of a 96-well plate, and 20 μL diluted Hydrogen Peroxide was added to each well and incubated for 20 mins at room temperature. Next, 30 μL Potassium Hydroxide and 30 μL Catalase Purpald were added to each well and incubated for 10 mins at room temperature, and, then, 10 μL Catalase Potassium was added to each well and incubated for 5 mins at room temperature. The absorbance at 540 nm was assessed with the Benchmark Plus Microplate Reader (Bio-Rad). The CAT activity in the samples was determined using a standard curve and the formula.
Data Analysis
To ensure a sufficient sample size, a power analysis was conducted26 and a power of 80% was obtained. Under the assumption of the medium effect (Cohen’s f = 0.33), the sample size required per group was 37 to achieve a group-difference of ADAS-cog score at 4, with S.D. estimated at 6. For further confirmation, we increased the sample size per group to about 48, which was larger than that (n = 30) of the previous benzoate trial in early-phase AD13 and that (n = 20) in treatment-resistant schizophrenia.20
Chi-square test (or Fisher’s exact test) was used to compare differences of categorical variables and Student’s two-sample t-test (or Mann–Whitney U-test if the distribution was not normal) for continuous variables (including some demographic characteristics, ADAS-cog, and laboratory measurements) between two groups. Mean changes from baseline in repeated-measure assessments (weeks 2, 4, and 6) (BEHAVE-AD and GDS) were assessed using the generalized estimating equation (GEE) method with treatment, visit, and treatment-visit interaction as fixed effects and intercept as the only random effect; baseline value as the covariance. No imputation for the incomplete data was used for the GEE analysis. The working correlation matrix was specified as autoregressive of order 1, named AR(1).
Therapeutic effect sizes (Cohen’s d) were used to determine the magnitude of improvement for the continuous variables resulting from benzoate treatment vs placebo. Multiple linear regression analyses were used to generate predictive models for treatment response.
Fisher’s exact test was used to compare the between-group differences in the dropout rates. All data were analyzed by IBM SPSS Statistics (version 22.0; SPSS Inc.). All p values were based on two-tailed tests with a significance level of 0.05.
Data Availability Statement
The protocol of this study is online (https://clinicaltrials.gov/ct2/show/NCT02103673). The individual de-identified participant data are available to qualified investigators on request to the corresponding author after the request is approved by the Institutional Review Board and Data Safety Monitoring Board of Kaohsiung Chang Gung Memorial Hospital, Institutional Review Board and Data Safety Monitoring Board of China Medical University Hospital, and Institutional Review Board and Data Safety Monitoring Board of Lin-Shin Hospital.
Results
One hundred and twenty-one patients were screened, and 97 of them were eligible and randomized; all the randomized patients completed at least one follow-up (at week 2), and 84 (86.6%) of them completed the 6-week trial (Figure 1). The dropout rate (12.2%) of the benzoate group was similar to that (14.6%) of the placebo group (p = 0.20) (Figure 1).
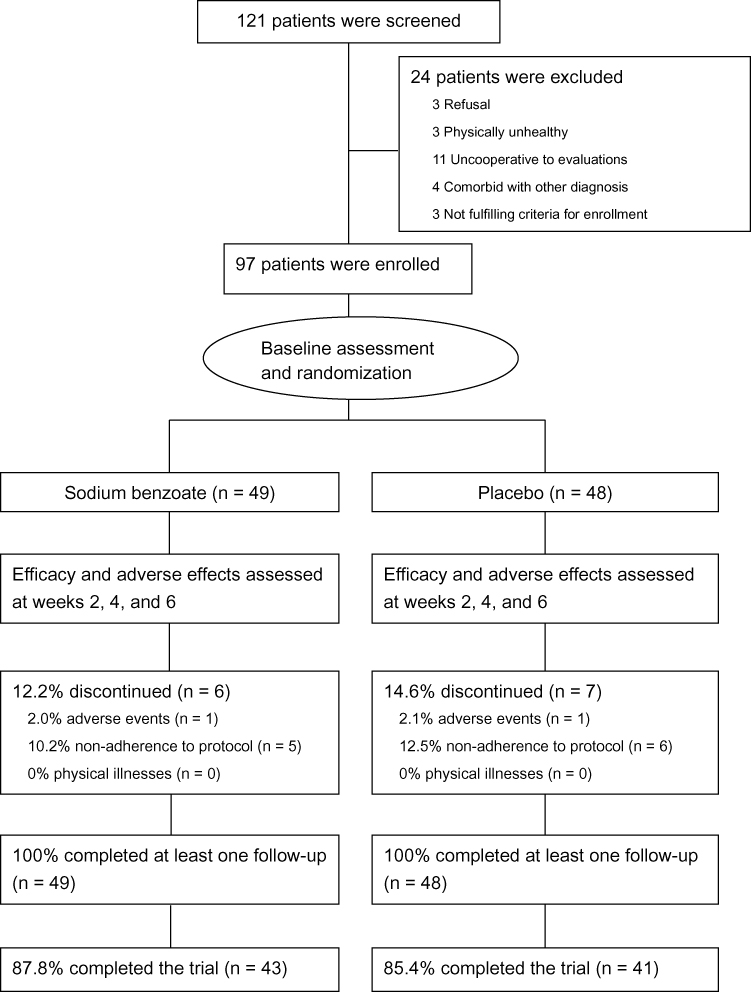 |
Figure 1 Flow diagram and disposition of the two treatment groups. |
Demographic data (including gender, age, diagnosis, age at illness onset, CDR), education level, body mass index (BMI), anti-dementia medication use, and antipsychotics use at baseline were similar between the benzoate group (n = 49) and the placebo group (n = 48). For details, refer to Table 1 in our previous article.21
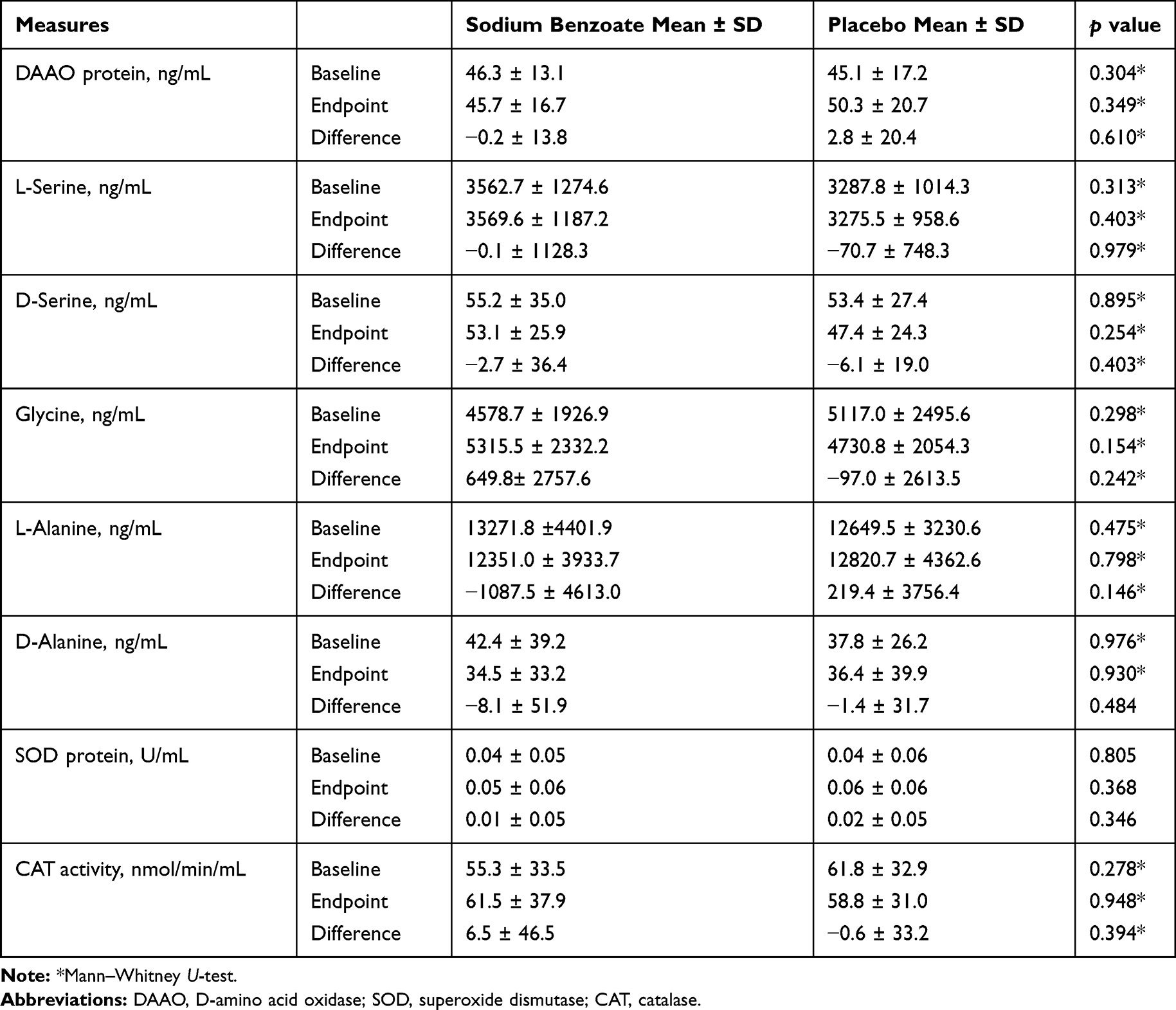 |
Table 1 Results of Measures of Serum DAAO, Plasma Amino Acids, and Plasma Antioxidants Over the 6-Week Treatment Between Sodium Benzoate and Placebo Groups |
Blood Levels of DAAO, Amino Acids, and Antioxidants
All laboratory parameters were measured at baseline and endpoint. The DAAO concentration showed a minimal but insignificant decrease in the benzoate group, when compared to the placebo group (Table 1).
For the five amino acids (L-serine, D-serine, glycine, L-alanine, D-alanine), there were also no significant differences in terms of baseline levels, endpoint levels, and changes from baseline to endpoint between the two groups (Table 1). In addition, no significant correlations were found between DAAO levels and amino acid levels in both benzoate and placebo groups (data not shown).
For the antioxidants, the SOD changes from baseline to endpoint were similar between the two groups, while CAT tended to increase after benzoate treatment, albeit insignificantly (Table 1).
Blood Correlates of Clinical Outcomes
We further analyzed blood assays of DAAO, amino acids, and antioxidants vs clinical outcomes of ADAS-cog and BEHAVE-AD (the details of clinical outcomes have been described elsewhere)19 in the two groups (Table 2). In the benzoate group, baseline DAAO level was correlated with baseline BEHAVE-AD score. The DAAO change was correlated with ADAS-cog change (p = 0.034).
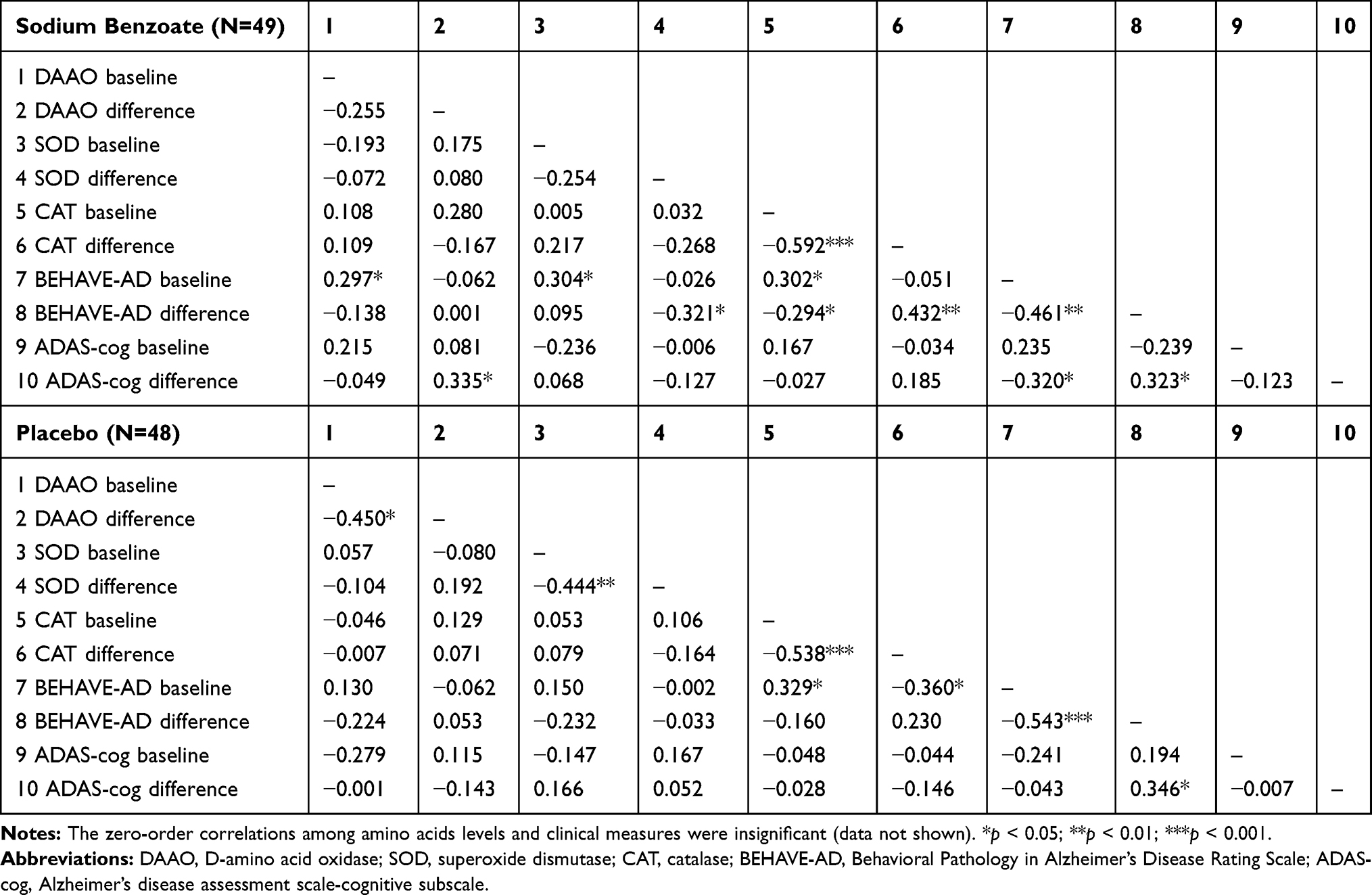 |
Table 2 Zero-Order Correlations Among Laboratory and Clinical Measures at Baseline and Their Difference Values (Endpoint – Baseline) |
In the placebo group, the baseline BEHAVE-AD score was significantly correlated with the baseline CAT level (p = 0.024) and negatively correlated with the CAT change (p = 0.021). There was no significant correlation between any other two items among DAAO level or change, SOD level or change, BEHAVE-AD score or change, and ADAS-cog score or change in the placebo group (Table 2). The correlations among amino acids levels and clinical measures were insignificant in both groups (data not shown).
With multiple linear regressions (stepwise method) for the benzoate group (Table 3), ADAS-cog score decrease was significantly associated with higher baseline BEHAVE-AD score, greater DAAO level decrease, higher BMI, female, antipsychotics use, and younger age (adjusted R square = 0.464); and BEHAVE-AD score decrease was significant with higher baseline BEHAVE-AD score, greater CAT decrease, and higher BMI (adjusted R square = 0.446).
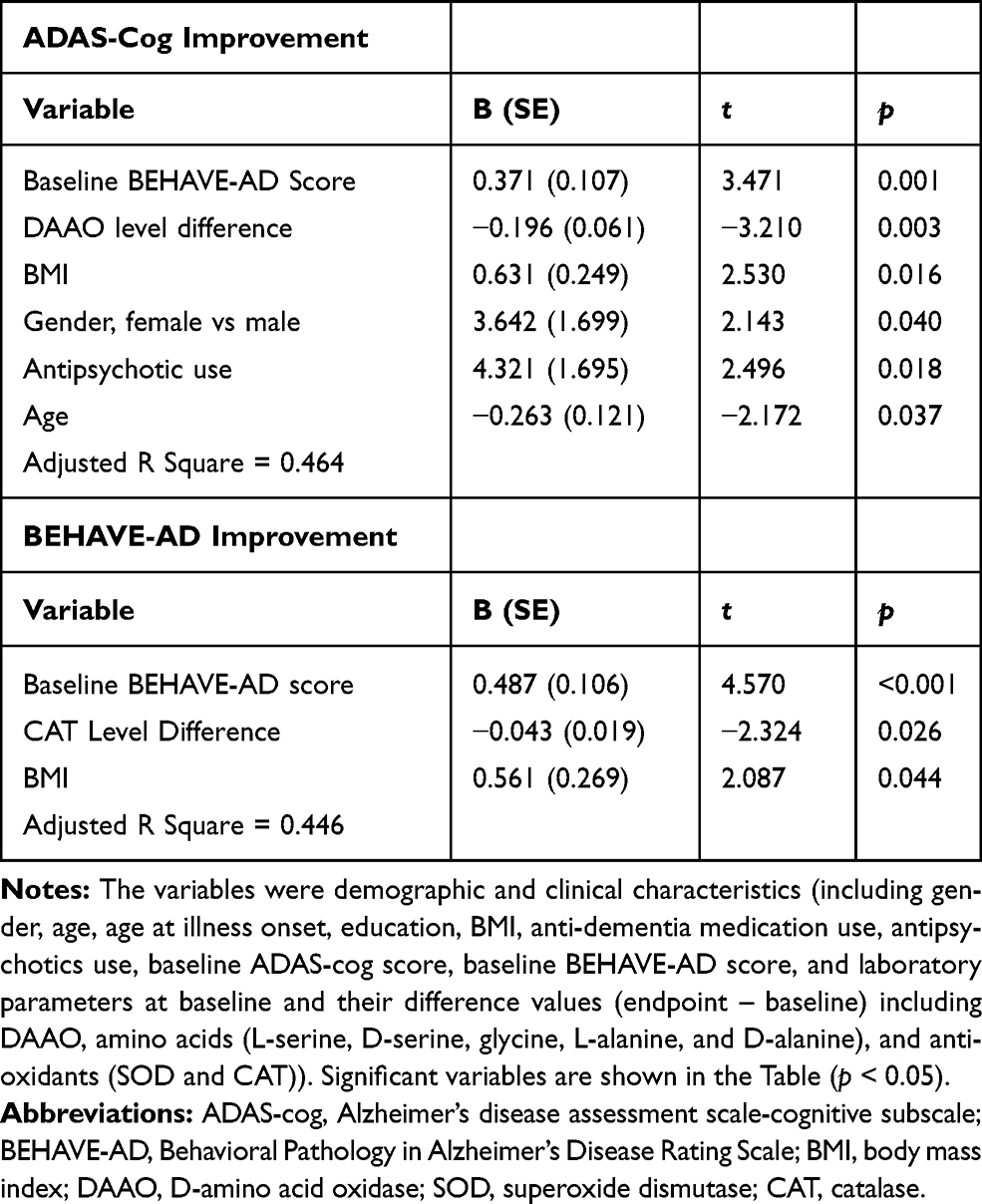 |
Table 3 Multiple Linear Regression Analyses of Independent Factors Associated with ADAS-Cog and BEHAVE-AD Improvements (Score Reduction from Baseline to Endpoint) in Sodium Benzoate Group (Stepwise) |
In comparison, with multiple linear regressions (stepwise method) for the placebo group (Table 4), ADAS-cog score decrease was significantly associated with male and lower baseline D-alanine level (adjusted R square = 0.228); and BEHAVE-AD score decrease was significant with higher baseline BEHAVE-AD score and more glycine increase (adjusted R square = 0.490).
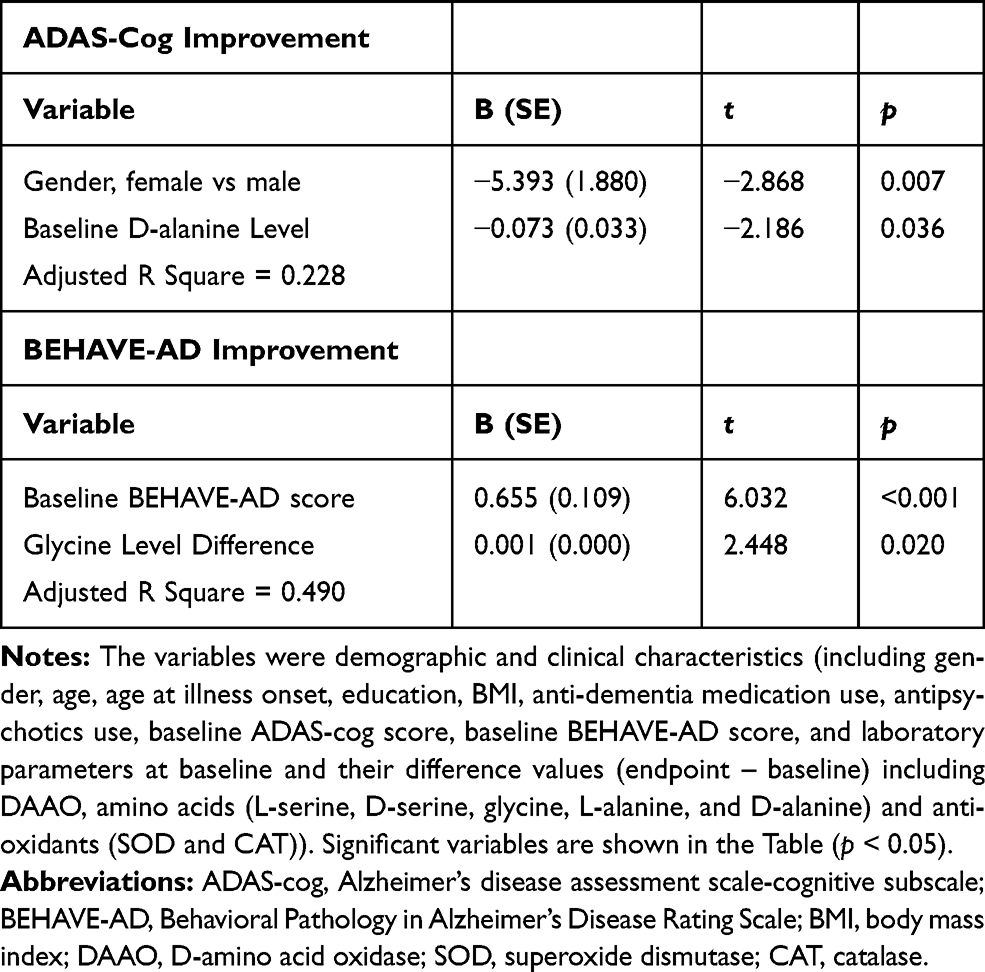 |
Table 4 Multiple Linear Regression Analyses of Independent Factors Associated with ADAS-Cog and BEHAVE-AD Improvements (Score Reduction from Baseline to Endpoint) in Placebo Group (Stepwise) |
Discussion
The study results showed that cognitive improvement after benzoate treatment was correlated with DAAO decrease, female gender, younger age, BMI, baseline BPSD severity, and antipsychotic use (Tables 2 and 3). More studies are warranted to confirm the findings and to identify target populations using precision medicine approach.
It is important but difficult to treat BPSD of all types of dementia.15 In the current trial, whose clinical outcome has been published elsewhere,21 with a lower mean dose (622.0 mg/day) of sodium benzoate than that (716.7 mg/day) of the previous trial on AD,13 DAAO levels only marginally and insignificantly declined; and all five amino acids (D-serine, L-serine, D-alanine, L-alanine, glycine) generally remained unaltered. Regarding the dose–response relationship, 2 g/day, rather than 1 g/day, of sodium benzoate has been demonstrated to improve overall symptomatology of clozapine-resistant schizophrenia.20 Therefore, whether higher-dose benzoate treatment can benefit clinical performances and change levels of DAAO and amino acids requires studies.
The current finding that there was no change on D-serine and D-alanine with treatment supports the antecedent finding that sodium benzoate at reported clinical therapeutic concentration did not affect D-alanine metabolism in dogs.27 Therefore, clinical efficacy of sodium benzoate for treatment of dementia maybe not mediated through changes of D-serine or D-alanine. However, its effects on other amino acids such as D-aspartate28 remain unknown. Further studies are needed to elucidate how sodium benzoate exert its therapeutic activity.29
DAAO is implicated in oxidative stress.30 Studies indicated that the DAAO level in peripheral blood increased with the severity of cognitive deficits in the elderly31 and decreased after 6-week treatment of benzoate in patients with schizophrenia.20 The insignificant DAAO decrease after benzoate therapy in this study may have been due to the relatively lower mean dose (as aforementioned) of benzoate compared to that (2000 mg/day) in the previous study for schizophrenia.20
The current study showed that precision medicine approach also helped identify individuals who benefited from benzoate treatment in reducing behavioral and psychological symptoms, although benzoate did not excel placebo in this domain for all patients as a whole.21 At baseline, higher CAT activity was positively correlated with higher BEHAVE-AD score in both treatment groups (Table 2). And, higher baseline CAT and BMI predicted more decrease in BPSD (more BEHAVE-AD score reduction) after benzoate treatment (Tables 2 and 3). In addition, less CAT increase (even after controlling for other variables) was correlated with more decrease in BPSD after benzoate treatment (Tables 2 and 3). Benzoate also increased the CAT activity in a cell model of neurotoxicity.32 However, after adding benzoate to clozapine for treatment of schizophrenia, the increase of CAT was correlated with the decrease of psychotic symptoms.20 One possible explanation for the finding of the current study is that excessively increase of CAT made patients with BPSD over-vigorous and active, and the caregiver consequently regarded the emergent behaviors disturbing.
It is always crucial to treat dementia as early as possible to stop or at least slow down its progression; compared to treatment for early-phase dementia, treatment for late-phase dementia is much more difficult.33 In addition, the role of NMDAR varies with dementia phases.11 NMDAR enhancement may be beneficial for early-phase AD.13 However, NMDAR over-excitation leads to excitotoxicity at late-stage AD.11,34 Since BPSD are more common in the late-phase dementia, it is expectable that the result of the current trial turned to be negative as a whole.21
Psychosis is associated with worse cognitive decline in patients with dementia.35 Though no medications have been approved for treatment of BPSD, antipsychotics have been commonly used for treatment of BPSD.4–8,36,37 Adverse effects of antipsychotics may counterpoise their benefits for the treatment of psychosis and agitation;38 on the other hand, patients with stable BPSD symptoms under antipsychotics treatment may have better competence and more potential to improve in cognitive function.39 In the current trial, around half of subjects (25/49 [51%] in the benzoate group; 22/48 [46%] in the placebo group) were receiving antipsychotics,21 thereby perhaps reducing the potential of benzoate efficacy for not only psychotic symptoms but also cognitive impairment. Benzoate exerted antipsychotic properties in a mouse model40 and in patients with schizophrenia.41 Future studies in antipsychotics-free patients are needed to elucidate the potential efficacy of benzoate for BPSD.
Low BMI and BMI decline are known risk factors for increased incidence and mortality of dementia.42 In this study, higher BMI was associated with better treatment response of benzoate, lending support to the importance of adequate nutrition in patients with BPSD.
Limitation
This study is limited by the short duration of treatment. Usually, it needs 6–12 months to ameliorate the cognitive decline of dementia.43 In comparison to the 24-week benzoate treatment for early-phase AD,13 its efficacy appeared similar to that of placebo in this 6-week study. A longer-duration trial is deserved to further clarify its efficacy. Secondly, repeated measures after short duration (6 weeks) may enhance the learning effect of cognitive tests, therefore improving the ADAS-cog performance (from 28.3 [mean] at baseline to 25.8 at endpoint) even in the placebo group21 and perhaps outshining the true drug effect. Thirdly, as aforementioned, the average dose of benzoate in this study was lower than that of the previous study in early-phase AD (716.7 mg/day),13 mainly due to the older patients in the current trial compared to the previous study (mean age 75.5 years vs 70.2 years). Finally, we measured only serum DAAO level. The correlation of serum DAAO level with brain DAAO level and activity deserves study in the future. We add this as a limitation on page.
Conclusions
The finding suggests that sodium benzoate may have potential to benefit cognitive function in a fraction of BPSD patients after only 6 weeks of treatment.
BPSD are very heterogeneous and usually unstable, compared to cognitive deficit per se. Diverse manifestations and courses of BPSD may represent numerous pathogeneses.15 Whether sodium benzoate can advantage at least a portion of patients with BPSD, particularly at their younger age and earlier-phase of illness deserves further studies. Consequently, applying precision molecular medicine may be capable of predicting benzoate effects on dementia and related BPSD.
In the future, longer-duration, higher-doses (and perhaps also lower doses) clinical trials are necessary to determine the efficacy and safety of sodium benzoate for the treatment of BPSD, with the aid of precision medicine strategies.
Acknowledgments
This work was supported by National Science Council and Ministry of Science and Technology, Taiwan (MOST 105-2325-B-039-005; MOST 107-2632-B-039-001; MOST 107-2628-B-182A-002; MOST 108-2314-B-039-002), National Health Research Institutes (NHRI-EX108-10731NI, NHRI-EX108-10816NC), China Medical University Hospital, Taiwan (DMR-108-096), and Taiwan Ministry of Health and Welfare Clinical Trial and Research Center of Excellence (MOHW 108-TDU-B-212-133004). The sponsors were not involved in study design; collection, analysis and interpretation of data; writing of the report; and the decision to submit the article for publication. ClinicalTrials.gov: DAOIB for the treatment of cognitive function and behavioral and psychological symptoms of dementia; https://clinicaltrials.gov/ct2/show/NCT02103673; NCT02103673.
Disclosure
All authors report no conflicts of interest in this work.
References
1. Steinberg M, Shao H, Zandi P, et al. Point and 5-year period prevalence of neuropsychiatric symptoms in dementia: the Cache County Study. Int J Geriatr Psychiatry. 2008;23(2):170–177. doi:10.1002/(ISSN)1099-1166
2. Paulsen JS, Salmon DP, Thal LJ, et al. Incidence of and risk factors for hallucinations and delusions in patients with probable AD. Neurology. 2000;54(10):1965–1971. doi:10.1212/WNL.54.10.1965
3. Rabins PV, Mace NL, Lucas MJ. The impact of dementia on the family. JAMA. 1982;248(3):333–335. doi:10.1001/jama.1982.03330030039022
4. Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA. 2005;293(5):596–608. doi:10.1001/jama.293.5.596
5. Davies SJ, Burhan AM, Kim D, et al. Sequential drug treatment algorithm for agitation and aggression in Alzheimer’s and mixed dementia. J Psychopharmacol. 2018;32(5):509–523. doi:10.1177/0269881117744996
6. Dyer SM, Harrison SL, Laver K, Whitehead C, Crotty M. An overview of systematic reviews of pharmacological and non-pharmacological interventions for the treatment of behavioral and psychological symptoms of dementia. Int Psychogeriatr. 2018;30(3):295–309. doi:10.1017/S1041610217002344
7. Masopust J, Protopopová D, Vališ M, Pavelek Z, Klímováet B. Treatment of behavioral and psychological symptoms of dementias with psychopharmaceuticals: a review. Neuropsychiatr Dis Treat. 2018;14:1211–1220. doi:10.2147/NDT
8. Ohno Y, Kunisawa N, Shimizu S. Antipsychotic treatment of behavioral and psychological symptoms of dementia (BPSD): management of extrapyramidal side effects. Front Pharmacol. 2019;10:1045. doi:10.3389/fphar.2019.01045
9. Francis PT. Altered glutamate neurotransmission and behaviour in dementia: evidence from studies of memantine. Curr Mol Pharmacol. 2009;2(1):77–82. doi:10.2174/1874467210902010077
10. Lin CH, Huang YJ, Lin CJ, Lane HY, Tsai GE. NMDA neurotransmission dysfunction in mild cognitive impairment and Alzheimer’s disease. Curr Pharm Des. 2014;20(32):5169–5179. doi:10.2174/1381612819666140110115603
11. Huang YJ, Lin CH, Lane HY, Tsai GE. NMDA neurotransmission dysfunction in behavioral and psychological symptoms of Alzheimer’s disease. Curr Neuropharmacol. 2012;10(3):272–285. doi:10.2174/157015912803217288
12. Sasabe J, Miyoshi Y, Suzuki M, et al. D-amino acid oxidase controls motoneuron degeneration through D-serine. Proc Natl Acad Sci U S A. 2012;109(2):627–632. doi:10.1073/pnas.1114639109
13. Lin CH, Chen PK, Chang YC, et al. Benzoate, a D-amino acid oxidase inhibitor, for the treatment of early-phase Alzheimer disease: a randomized, double-blind, placebo-controlled trial. Biol Psychiatry. 2014;75(9):678–685. doi:10.1016/j.biopsych.2013.08.010
14. Van den Berghe-snorek S, Stankovich MT. Thermodynamic control of D-amino acid oxidase by benzoate binding. J Biol Chem. 1985;260(6):3373–3379.
15. Lin CH, Lane HY. The role of N-Methyl-D-Aspartate receptor neurotransmission and precision medicine in behavioral and psychological symptoms of dementia. Front Pharmacol. 2019;10:540. doi:10.3389/fphar.2019.00540
16. Serrano F, Klann E. Reactive oxygen species and synaptic plasticity in the aging hippocampus. Ageing Res Rev. 2004;3(4):431–443. doi:10.1016/j.arr.2004.05.002
17. Bickford PC, Gould T, Briederick L, et al. Antioxidant-rich diets improve cerebellar physiology and motor learning in aged rats. Brain Res. 2000;866(1–2):211–217. doi:10.1016/S0006-8993(00)02280-0
18. Guidi M, Kumar A, Foster TC. Impaired attention and synaptic senescence of the prefrontal cortex involves redox regulation of NMDA receptors. J Neurosci. 2015;35(9):3966–3977. doi:10.1523/JNEUROSCI.3523-14.2015
19. Modi KK, Roy A, Brahmachari S, Rangasamy SB, Pahan K. Cinnamon and its metabolite sodium benzoate attenuate the activation of p21rac and protect memory and learning in an animal model of Alzheimer’s disease. PLoS One. 2015;10(6):e0130398. doi:10.1371/journal.pone.0130398
20. Lin CH, Lin CH, Chang YC, et al. Sodium benzoate, a D-amino acid oxidase inhibitor, added to clozapine for the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Biol Psychiatry. 2018;84(6):422–432. doi:10.1016/j.biopsych.2017.12.006
21. Lin CH, Chen PK, Wang SH, Lane HY. Sodium benzoate for the treatment of behavioral and psychological symptoms of dementia (BPSD): a randomized, double-blind, placebo-controlled, 6-week trial. J Psychopharmacol. 2019;33(8):1030–1033. doi:10.1177/0269881119849815
22. Tremolizzo L, Beretta S, Ferrarese C. Peripheral markers of glutamatergic dysfunction in neurological diseases: focus on ex vivo tools. Crit Rev Neurobiol. 2004;16(1–2):141–146. doi:10.1615/CritRevNeurobiol.v16.i12.150
23. Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry. 1984;141(11):1356–1364.
24. Reisberg B, Borenstein J, Salob SP, Ferris SH, Franssen E, Georgotas A. Behavioral symptoms in Alzheimer’s disease: phenomenology and treatment. J Clin Psychiatry. 1987;48 Suppl:9–15.
25. Reisberg B, Borenstein J, Franssen E, Shulman E, Steinberg G, Ferris SH. Remediable behavioral symptomatology in Alzheimer’s disease. Hosp Community Psychiatry. 1986;37(12):1199–1201.
26. Faul F, Erdfelder E, Buchner A, Lang AG. Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behav Res Methods. 2009;41(4):1149–1160. doi:10.3758/BRM.41.4.1149
27. Popiolek M, Tierney B, Steyn SJ, DeVivo M. Lack of effect of sodium benzoate at reported clinical therapeutic concentration on d-alanine metabolism in dogs. ACS Chem Neurosci. 2018;9(11):2832–2837. doi:10.1021/acschemneuro.8b00229
28. Errico F, Nisticò R, Napolitano F, et al. Increased D-aspartate brain content rescues hippocampal age-related synaptic plasticity deterioration of mice. Neurobiol Aging. 2011;32(12):2229–2243. doi:10.1016/j.neurobiolaging.2010.01.002
29. Harrison PJ. D-Amino acid oxidase inhibition: a new glutamate twist for clozapine augmentation in schizophrenia? Biol Psychiatry. 2018;84(6):396–398. doi:10.1016/j.biopsych.2018.06.001
30. Lu JM, Gong N, Wang YC, Wang YX. D-Amino acid oxidase-mediated increase in spinal hydrogen peroxide is mainly responsible for formalin-induced tonic pain. Br J Pharmacol. 2012;165(6):1941–1955. doi:10.1111/j.1476-5381.2011.01680.x
31. Lin CH, Yang HT, Chiu CC, Lane HY. Blood levels of D-amino acid oxidase vs. D-amino acids in reflecting cognitive aging. Sci Rep. 2017;7(1):14849. doi:10.1038/s41598-017-13951-7
32. Arabsolghar R, Saberzadeh J, Khodaei F, Borojeni RA, Khorsand M, Rashedinia M. The protective effect of sodium benzoate on aluminum toxicity in PC12 cell line. Res Pharm Sci. 2017;12(5):391–400. doi:10.4103/1735-5362.213984
33. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–292. doi:10.1016/j.jalz.2011.03.003
34. Hsu WY, Lane HY, Lin CH. Medications used for cognitive enhancement in patients with schizophrenia, bipolar disorder, Alzheimer’s disease, and Parkinson’s disease. Front Psychiatry. 2018;9:91. doi:10.3389/fpsyt.2018.00091
35. Zahodne LB, Ornstein K, Cosentino S, Devanand DP, Stern Y. Longitudinal relationships between Alzheimer disease progression and psychosis, depressed mood, and agitation/aggression. Am J Geriatr Psychiatry. 2015;23(2):130–140. doi:10.1016/j.jagp.2013.03.014
36. Lane HY, Chang YC, Su MH, Chiu CC, Huang MC, Chang WH. Shifting from haloperidol to risperidone for behavioral disturbances in dementia: safety, response predictors, and mood effects. J Clin Psychopharmacol. 2002;22(1):4–10. doi:10.1097/00004714-200202000-00002
37. Devanand DP, Mintzer J, Schultz SK, et al. Relapse risk after discontinuation of risperidone in Alzheimer’s disease. N Engl J Med. 2012;367(16):1497–1507. doi:10.1056/NEJMoa1114058
38. Schneider LS, Tariot PN, Dagerman KS, et al. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355(15):1525–1538. doi:10.1056/NEJMoa061240
39. Bertrand E, van Duinkerken E, Landeira-Fernandez J, et al. Behavioral and psychological symptoms impact clinical competence in Alzheimer’s disease. Front Aging Neurosci. 2017;9:182. doi:10.3389/fnagi.2017.00182
40. Matsuura A, Fujita Y, Iyo M, Hashimoto K. Effects of sodium benzoate on pre-pulse inhibition deficits and hyperlocomotion in mice after administration of phencyclidine. Acta Neuropsychiatr. 2015;27(3):159–167.
41. Lane HY, Lin CH, Green MF, et al. Add-on treatment of benzoate for schizophrenia: a randomized, double-blind, placebo-controlled trial of D-amino acid oxidase inhibitor. JAMA Psychiatry. 2013;70(12):1267–1275. doi:10.1001/jamapsychiatry.2013.2159
42. Garcia-Ptacek S, Kareholt I, Farahmand B, Cuadrado ML, Religa D, Eriksdotter M. Body-mass index and mortality in incident dementia: a cohort study on 11,398 patients from SveDem, the Swedish Dementia registry. J Am Med Dir Assoc. 2014;15(6):447e441–447. doi:10.1016/j.jamda.2014.03.001
43. Steele LS, Glazier RH. Is donepezil effective for treating Alzheimer’s disease? Can Fam Physician. 1999;45:917–919.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.