")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 12
Precision medicine approaches for the management of Ewing sarcoma: current perspectives
Authors Rizk VT , Walko CM, Brohl AS
Received 31 October 2018
Accepted for publication 29 December 2018
Published 17 January 2019 Volume 2019:12 Pages 9—14
DOI https://doi.org/10.2147/PGPM.S170612
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin Bluth
Victoria T Rizk,1 Christine M Walko,2 Andrew S Brohl3,4
1University of South Florida Hematology/Oncology Fellowship, 2DeBartolo Family Personalized Medicine Institute, 3Sarcoma Department, 4Chemical Biology and Molecular Medicine Program, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL, USA
Abstract: Advancements in molecular and genetic techniques have significantly furthered our biological understanding of Ewing sarcoma (ES). ES is typified by a driving TET–ETS fusion with an otherwise relatively quiet genome. Detection of one of several characteristic fusions, most commonly EWSR1–FLI1, is the gold standard for diagnosis. We discuss the current role of precision medicine in the diagnosis, treatment, and monitoring of ES. Continued efforts toward molecularly guided approaches are actively being pursued in ES to better refine prognosis, identify germline markers of disease susceptibility, influence therapeutic selection, effectively monitor disease activity in real time, and identify genetic and immunotherapeutic targets for therapeutic development.
Keywords: Ewing sarcoma, next generation sequencing, genomics, liquid biopsy, targeted therapy
Introduction
Sarcomas are a heterogeneous group of mesenchymal neoplasms that account for 1% of all adult malignancies, with >50 histologic subtypes identified and a reported incidence of 13,040 cases in 2018.1 Ewing sarcoma (ES) as well as the more encompassing classification Ewing sarcoma family of tumors (ESFT) represents a significant percentage of bone sarcomas mainly effecting the pediatric and adolescent population with a mild predominance in the male gender. Despite an overall improvement in sarcoma-related deaths over the last two decades, patients presenting with more advanced ES continue to have a poor prognosis.2
The genetic profile of ESFT is dominated by the driving reciprocal translocation between EWRS1 and ETS family transcription factors. Continuing research has provided insight into the prognostic value of certain genetic alterations reported in ES, as well as the potential future targets to further individual treatment regimens.3 In this report, we review the molecular landscape of ES with a focus on translational aspects. We discuss the current role of precision medicine in the diagnosis, prognosis, and management of ESFT.
Genomic landscape
Several large next-generation sequencing (NGS) studies have helped to define the genomic landscape of ES.4–6 ES is a fusion-associated malignancy with recurrent translocations between EWSR1 and members of the ETS family of transcription factors, most commonly FLI1.7 Similar to other fusion-driven malignancies,8 ESFT is characterized by a low somatic mutational burden suggesting a primacy of the EWSR1–ETS fusion as a driver. Recurrent oncogenic mutations have been noted, however, in several tumor suppressors including STAG2 (15%–22%), TP53 (6%–7%), and CDKN2A (12%–28%).4–6 Of note, ESFT displays a stark paucity of mutations in genes involved in kinase signaling pathways. Epigenomic and transcriptomic mapping have provided genome-wide overviews of the ES cellular program.9 Indirectly targeting the fusion via associated epigenomic or transcriptomic changes has been challenging, but remains an area of active research.10
Germline genetics
Germline genetics may play a role in the development of ES and can have important implications for the patient as well as the family members. A large case–control study of 1,162 patients with a variety of sarcoma subtypes found a high burden of likely pathogenic germline alterations in sarcoma patients relative to a control population with variants in TP53, ATM, ATR, BRCA2, and ERCC2 being commonly represented.11 Focusing on the ES population, a germline sequencing study utilizing whole-genome or whole-exome sequencing in 175 patients with ES identified pathogenic or likely pathogenic mutations in 13.1% of patients, with significant enrichment in genes involved in hereditary breast cancer and DNA repair pathways.12 A separate germline analysis that included 46 pediatric and adolescent patients with ES also found 5 patients with damaging alterations in one of TP53, PMS2, or RET.13 Germline alterations in more common cancers such as ovarian, breast, and colon have led to personalized medicine strategies unique to the individual’s cancer biology. Given the overlap in genetic pathways seen in ES, these germline findings might benefit ES survivors and their families though the utilization of existing screening and surgical risk reduction strategies for genes such as BRCA1/2, APC, and TP53. Furthermore, germline variants may suggest clinical use of novel targeted therapies, ideally in the setting of a clinical trial. As examples, carriers of BRCA1 or BRCA2 deleterious germline mutations may benefit from PARP inhibitors, even outside of breast or ovarian cancer.14
Diagnosis
Morphologically, ES family tumors can closely resemble other small round blue cell tumors. While histology and immunohistochemistry can be suggestive, it is now considered standard of care to confirm the diagnosis by detection of one of the typical ESFT-associated translocations.
More than 85% of Ewing’s sarcomas are defined by the t(11;22)(q24;q12) translocation that fuses the EWSR1 gene on chromosome 22 with the FLI1 gene on chromosome 11.15 The remaining 10%–15% of cases are characterized by alternate translocations resulting in the EWSR1 gene instead being fused with other ETS transcription factors including ERG, ETV1, ETV4, or FEV or rarely by EWSR1 being replaced by another member of the TET family of transcription factors, FUS16 (Table 1).
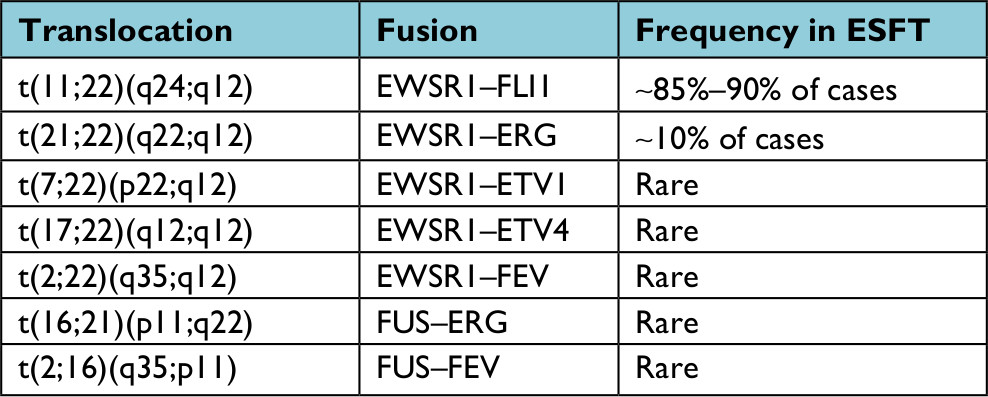 | Table 1 Current TET–ETS fusion pairings identified in Ewing sarcoma Abbreviation: ESFT, Ewing sarcoma family of tumors. |
Clinical molecular testing for ESFT translocations is most commonly performed using fluorescence in-situ hybridization (FISH) or reverse transcription-PCR (RT-PCR). Due to the limitations of these technologies, most molecular pathology laboratories do not test for all of the less common TET–ETS fusion pairings. Therefore, a negative FISH or RT-PCR does not rule out the diagnosis of ESFT and more extensive molecular evaluation (eg, NGS) may be necessary to solidify the diagnosis. An increasing number of alternate fusion drivers have been characterized in “Ewing-like” tumors, including CIC–DUX4 or CIC–FOXO4 and BCOR–CCNB3.17–20 Though sometimes designated as “Ewing family” or “Ewing-like” tumors, gene expression profiling of these non-TET–ETS fusion sarcomas indicate they are molecularly distinct from ESFT and should probably be considered a different entity.4,19
Treatment
Primary treatment for localized ES includes multi-agent neoadjuvant chemotherapy, most commonly vincristine, doxorubicin, and cyclophosphamide alternating with ifosfamide and etoposide, given every 2 weeks. This is then followed by local therapy (surgery and/or radiation) followed by adjuvant chemotherapy. Following this rigorous regimen, patients with localized disease have a survival rate of ~70%.21,22 Unfortunately, patients who develop recurrence in addition to the 20%–30% of patients who present with metastatic disease at diagnosis portend a dismal prognosis with a 5-year overall survival of <20%.23,24
Molecularly targeted therapy
While there have been clear advancements in the use of genetic profiling for the diagnosis of ES, the translation of genomic knowledge into therapeutic advances has been more challenging. Unlike kinase fusions, the transcription factor fusions such as EWSR1–FLI1 have not been amenable to direct inhibition to date. Furthermore, some evidence suggests that EWS–FLI1-directed oncogenesis in ES involves the fusion oncoprotein downregulating rather than upregulating genetic pathways, making targeting downstream effects of the fusion a challenge as well.25 A number of targeted therapy efforts are ongoing in attempts to overcome these challenges (Table 2).
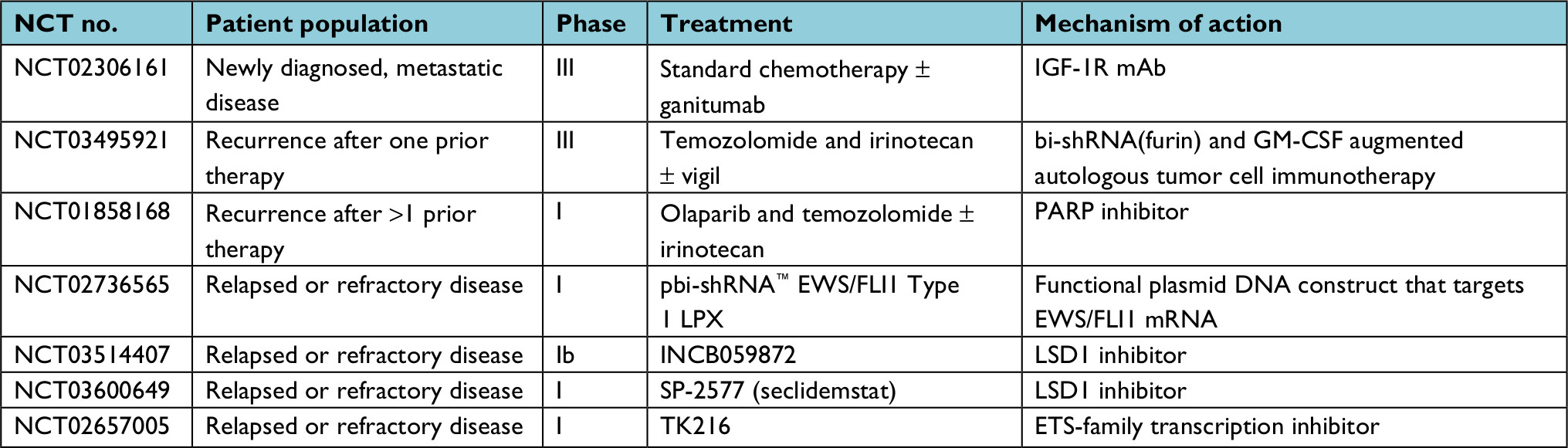 | Table 2 Current clinical trials for advanced Ewing sarcoma Abbreviations: GM-CSF, granulocyte-macrophage colony-stimulating factor; IGF-1R, insulin-like growth factor one receptor; mAb, monoclonal antibody. |
Insulin-like growth factor one receptor (IGF-1R) inhibitors
The insulin and IGF-1R pathway has a role in normal cellular development and has been associated with the development of malignancies such as prostate cancer,26 breast cancer,27,28 and colon cancer.29 Since the IGF-1R pathway is deregulated by the EWSR1–FLI1 translocation, it has come under the spotlight as a potential target for therapy. Based on promising preclinical evidence, several Phase I studies of this drug class have been completed with encouraging results and good tolerability.30–32 In a 2016 review of IGF-1R studies in ES, 311 ES patients were treated with IGF-1R inhibitors as monotherapy on Phase I or Phase II trial. Of these, 2 (0.6%) had a complete response, 31 (9.9%) had a partial response, and 66 (21%) had stable disease.33–35 Although preclinical data and an earlier Phase II trial suggested combination therapy may help overcome resistance,36 a Phase II trial utilizing an IGF-1R inhibitor with an mTOR inhibitor unfortunately did not show any responses in the 43 eligible patients, including 11 ES patients.34,37
Given the modest disease activity of IGF-1R inhibitors as monotherapy, current efforts focus on whether there is a role for the addition of this targeted therapy to standard cytotoxic chemotherapy. A Phase III trial assessing the IGF-1R monoclonal antibody ganitumab in combination with chemotherapy in first-line treatment for metastatic ESFT is ongoing (NCT02306161).
PARP inhibition
The PARP pathway is essential for genomic stability via detection of single-strand breaks and signaling of enzymatic DNA repair. A large systematic drug screen utilizing hundreds of cell lines identified ES as being exquisitely sensitive to the PARP inhibitor olaparib.38 Mouse xenograft studies additionally support PARP inhibition, particularly in combination with irinotecan and temozolomide as a viable therapeutic strategy for this disease.39 In a Phase II trial, 12 patients with ES who previously progressed on standard chemotherapy were treated with olaparib monotherapy at a dose of 400 mg by mouth twice daily. Although the drug was well tolerated, no complete or partial responses were seen in any of the patients. Stable disease was seen in 4 of the 12 patients (range 10.9–18.4 weeks), and median progression-free survival was 5.7 weeks.40 An ongoing trial is assessing combination therapy with olaparib with temozolomide and irinotecan in recurrent/refractory ES (NCT01858168).
LSD1 inhibition
LSD1, as a member of the NuRD co-repressor complex, is recruited by EWS–FLI1 to exert transcriptional regulation. In both in vitro and in vivo disease models, inhibition of LSD1 leads to a reversal of the EWS–FLI1 transcriptional profile and shows significant single-agent activity.41,42 Reversible, as opposed to irreversible LSD1 inhibition, in particular, seems promising based on xenograft models.42 As a result of these preclinical efforts, several LSD1 inhibitors have recently begun testing in ES in early phase monotherapy trials (Table 2).
Immunotherapy
Immunotherapy breakthroughs in a number of adult cancer types have sparked the interest in investigating this approach in ES as well. Unfortunately, early experience with checkpoint inhibitor therapy in ES has not shown significant efficacy to date. Most notably, 0 of 13 ES patients treated with pembrolizumab on the SARC028 trial experienced an objective response.43 The limited success with the use of immune checkpoint inhibitors in ES has been attributed to low mutational burden and absence of PD-L1 expression in the tumor.44
Utilization of cellular and vaccine immunotherapy strategies has become a topic of interest in ES.45 Recently, a pilot study utilizing an autologous vaccination strategy has shown promising response data46 and has led to an ongoing randomized trial (NCT03495921) with other approaches continuing to be developed. Anecdotal clinical efficacy has been reported with transgenic T-cell infusion.47 Chimeric antigen receptor T cells targeting IGF1R or ROR1 are also being investigated for their use in ES. Thus far, there has been success in ES mouse models leading to a prolonged overall survival, but results have been limited to preclinical studies.48
Prognosis and disease monitoring
Genetic prognostic markers
Prognostic biomarkers are useful for identifying patients who may not respond well to conventional therapy. As discussed above, the genomic landscape of ESFT is typified by a low somatic mutational burden and few recurrently mutated genes. Of the three most highly recurrent mutations in STAG2, CDKN2A, and TP53, there are conflicting data at present on the prognostic utility of these findings. Initial landscape genomics studies suggested that STAG2 mutation, particularly the combination of STAG2 and TP53 mutations, is a poor prognostic indicator;4,12 however, this finding was not confirmed in a follow-up study using an independent cohort.49 Similarly, there are conflicting studies regarding whether CDKN2A deletion is associated with poor prognosis.50,51 Given the conflicting data regarding prognostic implications of the most common genetic alterations, further research is necessary prior to incorporation in standard clinical practice.
Liquid biopsy
Of growing interest in precision oncology is the use of “liquid biopsy” techniques to monitor disease response and for early detection of relapse. Given that presence of TET–ETS fusion is a requisite for ESFT, detection and monitoring of the fusion transcript or from circulating tumor DNA (ctDNA) in the peripheral blood is a logical approach.
Several efforts utilizing RT-PCR methodology have shown the ability to detect fusion transcript in the peripheral blood or bone marrow of patients with both localized and metastatic disease.52–56 The prognostic value and utility of this finding, however, was not conclusive between studies, and this technique has not been widely adopted.
More recently, technological advances in detection of plasma-derived cell-free tumor DNA by capture-based NGS or digital droplet PCR (ddPCR) have reignited the interest in plasma-based detection of the ES fusion as a disease monitoring tool with intriguing, although early, results. In a 94-patient ES cohort using an NGS hybrid capture assay, ctDNA was identified in 53% of newly diagnosed patients with localized disease.57 The presence of detectable ctDNA by this assay was associated with worse 3-year event-free survival (48.6% vs 82.1%, P=0.006) and overall survival (79.8% vs 92.6%, P=0.01).
Several groups have now shown ddPCR to be a highly sensitive and dynamic plasma-based detection tool for the EWS fusion.58–60 Though modest in patient numbers, these initial efforts have consistently shown nearly 100% sensitivity for detection in patients with measurable disease. Furthermore, the quantity of ctDNA was noted to be dynamic, correlating well with disease burden. Interestingly, ctDNA detection by this method can precede radiographic disease progression, suggesting a role for minimal residual disease monitoring.
While these initial efforts are intriguing, further investigation is needed to clarify how these technologies might be incorporated into clinical practice. At present, ctDNA detection in ES patients is limited to the research setting, but it is increasingly being included as a translational component in collaborative group trials.
Conclusion
Advancements in molecular and genetic techniques have significantly furthered our biological understanding of ES. ES is typified by a driving TET–ETS fusion with an otherwise relatively quiet genome. Detection of one of several characteristic fusions, most commonly EWSR1–FLI1, is the gold standard for diagnosis. Despite a lack of driving kinase pathway mutations, functional genomics studies have helped to identify recurrently aberrant pathways as well as genetic and epigenetic vulnerabilities that have led to ongoing investigations with precision therapy approaches. Detection of ctDNA in the plasma of ES patients represents a potential novel disease monitoring tool. Germline genomics studies have recently identified a higher than anticipated burden of pathogenic syndromic mutations in ES patients, often with clinical implications. Although the driving mutation and the gene expression profile have been more clearly defined, there has been a limited role of precision medicine in ES when compared to other malignancies. Continued efforts toward precision medicine approaches are actively being pursued in ES to better refine prognosis, identify germline markers of disease susceptibility, influence therapeutic selection, effectively monitor disease activity in real time, and to identify genetic and immunotherapeutic targets for therapeutic development.
Disclosure
ASB – a family member has provided expert testimony for GlaxoSmithKline. The other authors report no conflicts of interest in this work.
References
Fletcher CDM, Bridge JA, Hogendoorn P, Mertens F. WHO Classification of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2013. | ||
Siegel RL, Miller KD, Jemal A, Statistics C. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. | ||
Subbiah V. Prospects and pitfalls of personalizing therapies for sarcomas: from children, adolescents, and young adults to the elderly. Curr Oncol Rep. 2014;16(9):401. | ||
Brohl AS, Solomon DA, Chang W, et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014;10(7):e1004475. | ||
Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326–1341. | ||
Tirode F, Surdez D, Ma X, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014;4(11):1342–1353. | ||
Delattre O, Zucman J, Melot T, et al. The Ewing family of tumors – a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med. 1994;331(5):294–299. | ||
Shern JF, Chen L, Chmielecki J, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4(2):216–231. | ||
Sand LG, Szuhai K, Hogendoorn PC. Sequencing overview of Ewing sarcoma: a journey across genomic, epigenomic and transcriptomic landscapes. Int J Mol Sci. 2015;16(7):16176–16215. | ||
Kovar H. Blocking the road, stopping the engine or killing the driver? Advances in targeting EWS/FLI-1 fusion in Ewing sarcoma as novel therapy. Expert Opin Ther Targets. 2014;18(11):1315–1328. | ||
Ballinger ML, Goode DL, Ray-Coquard I, et al. Monogenic and polygenic determinants of sarcoma risk: an international genetic study. Lancet Oncol. 2016;17(9):1261–1271. | ||
Brohl AS, Patidar R, Turner CE, et al. Frequent inactivating germline mutations in DNA repair genes in patients with Ewing sarcoma. Genet Med. 2017;19(8):955–958. | ||
Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373(24):2336–2346. | ||
Lee JM, Ledermann JA, Kohn EC. PARP inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014;25(1):32–40. | ||
Delattre O, Zucman J, Plougastel B, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359(6391):162–165. | ||
Sankar S, Lessnick SL. Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet. 2011;204(7):351–365. | ||
Kawamura-Saito M, Yamazaki Y, Kaneko K, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet. 2006;15(13):2125–2137. | ||
Solomon DA, Brohl AS, Khan J, Miettinen M. Clinicopathologic features of a second patient with Ewing-like sarcoma harboring CIC–FOXO4 gene fusion. Am J Surg Pathol. 2014;38(12):1724–1725. | ||
Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR–CCNB3 gene fusion. Nat Genet. 2012;44(4):461–466. | ||
Kousar Jahan SK, Mayanna M, Kavitha BL, Patil A, Kumari P, Jahan SK. A rare case of translocation (12;22) (p13;Q) in Ewing’s sarcoma. Indian J Med Paediatr Oncol. 2014;35(1):89–92. | ||
Shukla N. Ewing sarcoma: a tough road to clinically relevant biomarkers. Pediatr Blood Cancer. 2015;62(5):741–742. | ||
Arnaldez FI, Helman LJ. New strategies in Ewing sarcoma: lost in translation? Clin Cancer Res. 2014;20(12):3050–3056. | ||
Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol. 2000;18(17):3108–3114. | ||
Oberlin O, Deley MC, Bui BN, et al. Prognostic factors in localized Ewing’s tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer. 2001;85(11):1646–1654. | ||
Owen LA, Kowalewski AA, Lessnick SL. EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing’s sarcoma. PLoS One. 2008;3(4):e1965. | ||
Nickerson T, Chang F, Lorimer D, Smeekens SP, Sawyers CL, Pollak M. In vivo progression of LAPC-9 and LNCaP prostate cancer models to androgen independence is associated with increased expression of insulin-like growth factor I (IGF-I) and IGF-I receptor (IGF-IR). Cancer Res. 2001;61(16):6276–6280. | ||
Cullen KJ, Yee D, Sly WS, et al. Insulin-like growth factor receptor expression and function in human breast cancer. Cancer Res. 1990;50(1):48–53. | ||
Gooch JL, van den Berg CL, Yee D. Insulin-like growth factor (IGF)-I rescues breast cancer cells from chemotherapy-induced cell death – proliferative and anti-apoptotic effects. Breast Cancer Res Treat. 1999;56(1):1–10. | ||
Hassan AB, Macaulay VM. The insulin-like growth factor system as a therapeutic target in colorectal cancer. Ann Oncol. 2002;13(3):349–356. | ||
Olmos D, Postel-Vinay S, Molife LR, et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a Phase 1 expansion cohort study. Lancet Oncol. 2010;11(2):129–135. | ||
Tolcher AW, Rothenberg ML, Rodon J, et al. A Phase I pharmacokinetic and pharmacodynamic study of AMG 479, a fully human monoclonal antibody against insulin-like growth factor type 1 receptor (IGF-1R), in advanced solid tumors. J Clin Oncol. 2007;25(18 Suppl):3002. | ||
Kurzrock R, Patnaik A, Aisner J, et al. A Phase I study of weekly R1507, a human monoclonal antibody insulin-like growth factor-I receptor antagonist, in patients with advanced solid tumors. Clin Cancer Res. 2010;16(8):2458–2465. | ||
van Maldegem AM, Bovée JV, Peterse EF, Hogendoorn PC, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171–180. | ||
Malempati S, Weigel B, Ingle AM, et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(3):256–262. | ||
Tap WD, Demetri G, Barnette P, et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol. 2012;30(15):1849–1856. | ||
Schwartz GK, Tap WD, Qin LX, et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, Phase 2 trial. Lancet Oncol. 2013;14(4):371–382. | ||
Wagner LM, Fouladi M, Ahmed A, et al. Phase II study of cixutumumab in combination with temsirolimus in pediatric patients and young adults with recurrent or refractory sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2015;62(3):440–444. | ||
Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–575. | ||
Stewart E, Goshorn R, Bradley C, et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep. 2014;9(3):829–840. | ||
Choy E, Butrynski JE, Harmon DC, et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. BMC Cancer. 2014;14(1):813. | ||
Sankar S, Bell R, Stephens B, et al. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene. 2013;32(42):5089–5100. | ||
Sankar S, Theisen ER, Bearss J, et al. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin Cancer Res. 2014;20(17):4584–4597. | ||
Tawbi HA, Burgess M, Bolejack V, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, Phase 2 trial. Lancet Oncol. 2017;18(11):1493–1501. | ||
Spurny C, Kailayangiri S, Jamitzky S, et al. Programmed cell death ligand 1 (PD-L1) expression is not a predominant feature in Ewing sarcomas. Pediatr Blood Cancer. 2018;65(1):e26719. | ||
Rossig C. Cellular immunotherapy strategies for Ewing sarcoma. Immunotherapy. 2014;6(5):611–621. | ||
Ghisoli M, Barve M, Mennel R, et al. Three-year follow up of GMCSF/bi-shRNA (furin) DNA-transfected autologous tumor immunotherapy (Vigil) in metastatic advanced Ewing’s sarcoma. Mol Ther. 2016;24(8):1478–1483. | ||
Thiel U, Schober SJ, Einspieler I, et al. Ewing sarcoma partial regression without GvHD by chondromodulin-I/HLA-A*02:01-specific allorestricted T cell receptor transgenic T cells. Oncoimmunology. 2017;6(5):e1312239. | ||
Huang X, Park H, Greene J, et al. IGF1R- and ROR1-specific CAR T cells as a potential therapy for high risk sarcomas. PLoS One. 2015;10(7):e0133152. | ||
Bahrami A, Wu J, Dyer MA, Delattre O, Pappo AS. Loss of STAG2 expression and prognosis in Ewing sarcoma family of tumors. J Clin Oncol. 2015;33(15 Suppl):10024. | ||
Honoki K, Stojanovski E, McEvoy M, et al. Prognostic significance ofp16INK4a alteration for Ewing sarcoma. Cancer. 2007;110(6):1351–1360. | ||
Lerman DM, Monument MJ, McIlvaine E, et al. Tumoral TP53 and/or CDKN2A alterations are not reliable prognostic biomarkers in patients with localized Ewing sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2015;62(5):759–765. | ||
Fagnou C, Michon J, Peter M, et al. Presence of tumor cells in bone marrow but not in blood is associated with adverse prognosis in patients with Ewing’s tumor. Société Française d’Oncologie Pédiatrique. J Clin Oncol. 1998;16(5):1707–1711. | ||
de Alava E, Lozano MD, Patiño A, Sierrasesúmaga L, Pardo-Mindán FJ. Ewing family tumors: potential prognostic value of reverse-transcriptase polymerase chain reaction detection of minimal residual disease in peripheral blood samples. Diagn Mol Pathol. 1998;7(3):152–157. | ||
West DC, Grier HE, Swallow MM, Demetri GD, Granowetter L, Sklar J. Detection of circulating tumor cells in patients with Ewing’s sarcoma and peripheral primitive neuroectodermal tumor. J Clin Oncol. 1997;15(2):583–588. | ||
Athale UH, Shurtleff SA, Jenkins JJ, et al. Use of reverse transcriptase polymerase chain reaction for diagnosis and staging of alveolar rhabdomyosarcoma, Ewing sarcoma family of tumors, and desmoplastic small round cell tumor. J Pediatr Hematol Oncol. 2001;23(2):99–104. | ||
Zoubek A, Ladenstein R, Windhager R, et al. Predictive potential of testing for bone marrow involvement in Ewing tumor patients by RT-PCR: a preliminary evaluation. Int J Cancer. 1998;79(1):56–60. | ||
Shulman DS, Klega K, Imamovic-Tuco A, et al. Detection of circulating tumour DNA is associated with inferior outcomes in Ewing sarcoma and osteosarcoma: a report from the Children’s Oncology Group. Br J Cancer. 2018;119(5):615–621. | ||
Hayashi M, Chu D, Meyer CF, et al. Highly personalized detection of minimal Ewing sarcoma disease burden from plasma tumor DNA. Cancer. 2016;122(19):3015–3023. | ||
Shukla NN, Patel JA, Magnan H, et al. Plasma DNA-based molecular diagnosis, prognostication, and monitoring of patients with EWSR1 fusion-positive sarcomas. JCO Precis Oncol. 2017 (1):1–11. | ||
Krumbholz M, Hellberg J, Steif B, et al. Genomic EWSR1 fusion sequence as highly sensitive and dynamic plasma tumor marker in Ewing sarcoma. Clin Cancer Res. 2016;22(17):4356–4365. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.