")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Pre-Clinical Pharmacokinetics, Tissue Distribution and Physicochemical Studies of CLBQ14, a Novel Methionine Aminopeptidase Inhibitor for the Treatment of Infectious Diseases
Authors Ekpenyong O, Gao X, Ma J, Cooper C, Nguyen L, Olaleye OA, Liang D, Xie H
Received 11 November 2019
Accepted for publication 16 March 2020
Published 30 March 2020 Volume 2020:14 Pages 1263—1277
DOI https://doi.org/10.2147/DDDT.S238148
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Oscar Ekpenyong, Xiuqing Gao, Jing Ma, Candace Cooper, Linh Nguyen, Omonike A Olaleye, Dong Liang, Huan Xie
Department of Pharmaceutical and Environmental Health Sciences, College of Pharmacy and Health Sciences, Texas Southern University, Houston, TX, USA
Correspondence: Huan Xie
Department of Pharmaceutical and Environmental Health Sciences, College of Pharmacy and Health Sciences, Texas Southern University, 3100 Cleburne Street, Houston, TX 77004, USA
Email [email protected]
Introduction: CLBQ14, a derivative of 8-hydroxyquinoline, exerts its chemotherapeutic effect by inhibiting methionine aminopeptidase (MetAP), the enzyme responsible for the post-translational modification of several proteins and polypeptides. MetAP is a novel target for infectious diseases. CLBQ14 is selective and highly potent against replicating and latent Mycobacterium tuberculosis making it an appealing lead for further development.
Methods: The physicochemical properties (solubility, pH stability and lipophilicity), in vitro plasma stability and metabolism, pre-clinical pharmacokinetics, plasma protein binding and tissue distribution of CLBQ14 in adult male Sprague-Dawley rats were characterized.
Results: At room temperature, CLBQ14 is practically insoluble in water (< 0.07 mg/mL) but freely soluble in dimethyl acetamide (> 80 mg/mL); it has a log P value of 3.03 ± 0.04. CLBQ14 exhibits an inverse Z-shaped pH decomposition profile; it is stable at acidic pH but is degraded at a faster rate at basic pH. It is highly bound to plasma proteins (> 91%), does not partition to red blood cells (B/P ratio: 0.83 ± 0.03), and is stable in mouse, rat, monkey and human plasma. CLBQ14 exhibited a bi-exponential pharmacokinetics after intravenous administration in rats, bioavailability of 39.4 and 90.0%, respectively from oral and subcutaneous route. We observed a good correlation between predicted and observed rat clearance, 1.90 ± 0.17 L/kg/h and 1.67 ± 0.08 L/kg/h, respectively. Human hepatic clearance predicted from microsomal stability data and from the single species scaling were 0.80 L/hr/kg and 0.69 L/h/kg, respectively. CLBQ14 is extensively distributed in rats; following a 5 mg/kg intravenous administration, lowest and highest concentrations of 15.6 ± 4.20 ng/g of heart and 405.9 ± 77.11 ng/g of kidneys, respectively, were observed. In vitro CYP reaction phenotyping demonstrates that CLBQ14 is metabolized primarily by CYP 1A2.
Conclusion: CLBQ14 possess appealing qualities of a drug candidate. The studies reported herein are imperative to the development of CLBQ14 as a new chemical entity for infectious diseases.
Keywords: CLBQ14, 8-hydroxyquinoline, methionine aminopeptidase, clioquinol, pharmacokinetics, tissue distribution, physicochemical, drug development
Introduction
The global burden of infectious disease on public health is enormous. In 2016 alone, there were over 15.5 million visits to physician offices with infectious and parasitic diseases as primary diagnosis.1 In the last decade, there has been a dramatic increase in bacterial pathogens (amongst other micro-organisms) presenting with multi resistance to common antibacterial agents either by mutations or acquisition of genetic elements carrying resistance gene.2 Despite the increase in the infectious disease burden, research and development of new drugs for infectious disease has failed to meet the clinical need for novel antibiotics. The discovery and development of more effective and safe chemotherapeutic agents is expedient to combating this infectious disease burden.
The metalloprotease, methionine amino peptidase (MetAP), has been reported as a novel therapeutic target for infectious diseases.3–11 It has been validated as a target for various infectious diseases caused by methicillin-resistant Staphylococcus aureus and Escherichia coli,3 Mycobacterium tuberculosis (Mtb),4–7 Cryptosporidium parvum,8 Plasmodium falciparum,9 Leishmania donovani,10 Enterococcus faecalis,11,12 Rickettsia prowazekii,13 and Acinetobacter baumannii.14 It has also been investigated as a potential therapeutic target for several other diseases including cancer and rheumatoid arthritis.15–19
MetAP plays a crucial role in initiating protein and polypeptide synthesis by catalyzing the cleavage of the N-terminal methionine, a process known as N-terminal methionine excision (NME).20–22 NME is universal and is a pre-requisite for post-translational modification, stability and localization of newly synthesized proteins and polypeptides.21,22 Targeting MetAP for infectious diseases is very appealing as NME is an essential process in prokaryotes. Two classes of MetAP have been identified in prokaryotes and eukaryotes: MetAP1 and MetAP2 in eukaryotes, and homologs of either classes in prokaryotes.23–25
The molecule, 7-bromo-5-chloroquinolin-8-ol (CLBQ14, structure shown in Figure 1), has been identified as a potent and selective inhibitor of MetAPs in Mtb (MtMetAP1a and MtMetAP1c).2,17 Its efficacy profile has been previously reported;5 it has great potency against replicating Mtb; an increased potency against aged non-growing Mtb and exhibited great selectivity for both MtMetAPs over human MetAPs.4–7 Another derivative of 8-hydroxyquinoline, clioquinol, was previously marketed as an oral amebicide but was later withdrawn due to neurotoxicity. Its mechanism of action was unknown for a long time; however, recent studies have shown that exerts its chemotherapeutic effects by inhibiting MetAPs.5 Structurally, CLBQ14 differs from clioquinol only by the halogen substitution at position C7. While clioquinol has iodine at position C7, CLBQ14 has a bromine substitution. At 25°C, CLBQ14 is acidic in nature and has a calculated pKa of 2.23 ± 0.30 and a calculated log P value of 3.92 ± 0.39.26
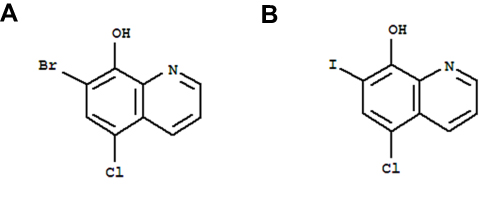 |
Figure 1 Chemical Structure of (A) CLBQ14 and (B) Clioquinol. CLBQ14 and CQ are congeners of 8-hydroxyquinoline that differ from each other only by the halogen at position C7. |
Identifying a new chemical entity (NCE) with desirable pharmacokinetic properties is one of the major hurdles during drug discovery and development. Early evaluation of the absorption, distribution, metabolism and excretion (ADME) of an NCE is essential to speeding up the discovery and development process. Also, detailed preclinical in-vitro and in-vivo metabolic stability and pharmacokinetic evaluation of new therapeutic candidate is one of the regulatory requirements prior to clinical studies. We previously reported the development of an LC-MS/MS assay for the quantification of CLBQ14 and successfully applied it to estimate the intravenous pharmacokinetics of CLBQ14 following a 2, 5 and 10 mg per kg single IV bolus doses to SD rats; its pharmacokinetic parameters were estimated using a two compartmental model analysis.29 In this study, we investigated the ADME properties of CLBQ14: intravenous (IV), oral (PO) and subcutaneous (SC) pharmacokinetic disposition, plasma and microsomal stability as well as the cytochrome P450 (CYP) enzymes involved in CLBQ14 metabolism. We evaluated the physicochemical properties of the molecule including solubility, lipophilicity and pH driven stability. We also assessed the plasma protein binding (PPB) of the CLBQ14, its blood-plasma partitioning as well as its tissue distribution following a single IV bolus dose in rats.
Highlights
- The physicochemical properties, in vitro/in vivo pharmacokinetics and tissue distribution of CLBQ14 was investigated.
- CLBQ14 is practically insoluble in water but is freely soluble in dimethyl acetamide; it has a log P value of 3.03 and is more stable at acidic pH than basic pH.
- It had a biphasic pharmacokinetic disposition following intravenous administration of CLBQ14, and oral and subcutaneous bioavailability was 39% and 90%, respectively.
- Tissue distribution studies revealed that CLBQ14 is distributed extensively to the body, with the lowest and highest accumulations in the heart and kidneys, respectively.
Materials and Methods
Materials
CLBQ14 (purity ≥ 98%) was purchased from TCI Chemicals (Tokyo, Japan). LC-MS grade water and acetonitrile, clioquinol, formic acid, trifluoroacetic acid (TFA), ethanol, Tween 20, Tween 80, soybean oil, paraffin oil, olive oil, dimethyl sulfoxide (DMSO), N’N dimethyl acetamide (DMA), polyethylene glycol 400 (PEG 400), glycerol, 1-octanol, 0.85% sodium chloride solution, phosphate buffered saline tablets, and CD-1 mouse, Sprague Dawley (SD) rat, cynomolgus monkey and human microsomes were purchased from Sigma Aldrich (St. Louis, MO). Transcutol High Purity (HP)®, Labrasol® and Capyrol 90® were gifts from Gattefosse (Lyon, France). Recombinant human cytochrome P450 enzymes (rhCYP) were purchased from BD Biosciences (San Jose, CA). Heparin (1000 units/mL) and pharmaceutical grade normal saline were purchased from Hospira (Lake Forest, IL). Human plasma was purchased from Gulf Coast Blood Center (Houston, TX) and fresh rat plasma was collected from male SD rats (Envigo RMS Inc., Indianapolis, IN) and stored at −80°C until use. CD-1 mouse and cynomolgus monkey plasma were purchased from BioIVT (Westbury, NY). All chemicals and reagents were used as received.
Physicochemical Properties
Solubility
The thermodynamic solubility of CLBQ14 in water, ethanol, PEG 400, propylene glycol monocapryrate type I (PGMC 1), Capyrol 90® (propylene glycol monocapryrate type II), DMSO, DMA, Tween 80®, Tween 20®, paraffin oil, soybean oil, and olive oil was assessed by the shake-flask method. Briefly, excess amount of CLBQ14 was added to each of the selected solvents in a scintillation vial. The vials were allowed to shake on a mechanical reciprocating shaker at room temperature for 72 h after solvent saturation was attained. The samples were centrifuged at 18,879 rcf for 10 min and subsequently filtered through a 0.22 μm filtration unit. The resulting filtrate was analyzed by HPLC-UV to determine the amount of CLBQ14 dissolved in the solvents. The experiment was conducted in triplicate.
Solution State pH Stability
The pH dependent stability of CLBQ14 was assessed in aqueous media of pH ranging from 1 to 10. Standard USP buffers were prepared and used for this experiment: 0.2 M hydrochloride buffer was used for pH 1–2, 0.2 M acetate buffer for pH 3–5, 0.2 M phosphate buffer for pH 6–8 and carbonate buffer for 9–10. Tween 80 (0.2% v/v) was added to each buffer solution to enhance the aqueous solubility of CLBQ14 and prevent its precipitation. CLBQ14 dissolved in DMSO was added to each buffer system placed in a water bath at 37°C. Aliquots were withdrawn at predetermined time points and analyzed by HPLC-UV for the amount of CLBQ14 remaining.
Lipophilicity
The lipophilicity of CLBQ14 indicated by the log P value (octanol – water partition coefficient) was determined by the shake-flask method. Briefly, equal volumes of 1-octanol and water (representing a two-phase lipid and aqueous system) were placed in a glass bottle and allowed to shake on a reciprocating shaker for 24 h to allow equilibration. CLBQ14 (1 mg) dissolved in DMSO was added to the two-phase system and allowed to shake continuously at room temperature, allowing CLBQ14 to partition between the two layers for 72 h. The two phases were separated using a separatory funnel, centrifuged at 18,879 rcf for 10 mins. The concentration of CLBQ14 in the 1-octanol and aqueous phase was determined by LC-MS/MS. The experiment was conducted in triplicate.
The log P was calculated by the equation:
(1)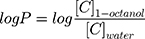
where 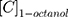 and
and 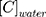 are the concentrations of CLBQ14 in the 1-octanol and water phase respectively.
are the concentrations of CLBQ14 in the 1-octanol and water phase respectively.
In vitro Studies
Stability in Plasma
CLBQ14 (1 μM) was incubated in CD-1 mouse, SD rat, monkey and human plasma respectively at 37°C for up to 4 hrs. At predetermined time points, aliquots (50 µL) were withdrawn and quenched with acetonitrile containing clioquinol as internal standard (IS). The quenched samples were centrifuged at 18,879 rcf for 5 mins and the supernatant analyzed by LC-MS/MS for the amount of CLBQ14 remaining.
Blood-Plasma Partition
CLBQ14 added to fresh whole SD rat blood to obtain a final concentration of 1 μM was incubated at 37°C for 1 hr. An aliquot was withdrawn and centrifuged for 15 mins at 4°C and 1750 rcf to separate plasma. The plasma and whole blood were analyzed by LC-MS/MS for the amount of CLBQ14. The blood-plasma (B/P) ratio was determined as a ratio of the mean peak area ratio of CLBQ14 in blood to that in plasma separated from the spiked blood samples.
Plasma Protein Binding
The plasma protein binding (PPB) of CLBQ14 was evaluated in SD rat plasma. The fraction bound was characterized using the centrifugal ultrafiltration technique. Briefly, 270 μL of blank rat plasma was spiked with 30 μL of standard solutions of CLBQ14 to yield final concentrations of 5, 25 and 50 μg/mL. The plasma samples were incubated at 37°C for 1 hr, and then transferred to Amicon Ultra® – 0.5 mL Centrifugal Filters (30 kDa, EMD Millipore, Billerica, MA) for ultrafiltration. The concentration of CLBQ14 in the filtrate and concentrate was determined by HPLC-UV. Prior to the determination of CLBQ14 in the concentrate, CLBQ14 was extracted by protein precipitation using acetonitrile containing IS. This experiment was conducted in triplicate.
PPB was calculated by the equation:
(2)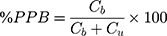
where 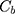 and
and 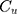 are CLBQ14 concentrations in the concentrate (bound) and filtrate (unbound), respectively.
are CLBQ14 concentrations in the concentrate (bound) and filtrate (unbound), respectively.
Microsome Metabolic Stability
CLBQ14 (3 µM) was incubated in liver microsomes from CD-1 mouse, SD rat, cynomolgus monkey and human. The incubation (total volume: 0.5 mL) consisted of liver microsomes (0.5 mg/mL), NADPH (5 mM) in phosphate buffer, and 50 mM phosphate buffer (pH 7.4). Following three minutes of preincubation of CLBQ14 and liver microsomes in phosphate buffer at 37°C, the reactions were initiated by the addition of NADPH. Aliquots (50 µL) were withdrawn at 0, 5, 15, 30, 45 and 60 mins and immediately quenched with 200 µL of acetonitrile containing IS. Concomitant NADPH-free control incubations were prepared similarly; aliquots were withdrawn at 0 and 60 mins. The quenched samples were centrifuged for 10 mins at 4000 rcf to pellet the proteins, and the supernatant analyzed by LC-MS/MS for the amount of CLBQ14 remaining.
CYP Reaction Phenotyping
CLBQ14 was incubated with recombinant human cytochrome P450 enzymes (rhCYP). The incubation (total volume: 250 µL) consisting of 200 pmol of the rhCYP and 1 µM of CLBQ14 in a phosphate cocktail (100 mM of K2HPO4 and 10 mM MgCl2 at pH 7.4) was pre-incubated before the reaction. The reaction was initiated by the addition of 1 mM of NADPH in phosphate cocktail. The final organic solvent content in the incubation was limited to ≤0.1% v/v for DMSO and ≤1% v/v for acetonitrile. Aliquots (40 µL) were withdrawn at 0, 5, 15, 30, 60 mins and the reactions were terminated by the adding 160 µL of acetonitrile containing IS. Negative controls were prepared either without NADPH, with insect control cells or both. The quenched samples were vortexed and centrifuged for 10 min at 4000 rcf to pellet the protein; the supernatant was transferred for LC-MS/MS analysis for the amount of CLBQ14 remaining. The pseudo transition 273.9 → 167.0 corresponding to parent Q1/Q3 shifts of [M+16]+/[M+16]+ was monitored for any hydroxy metabolite of CLBQ14 (OH-CLBQ14).
Dosing Formulation
Co-solvent systems of varying compositions and ratios of solvents containing up to 10 mg/mL of CLBQ14 were evaluated. Based on the solubility data, DMA, PEG 400, Tween 80 were selected as potential co-solvents for the formulation. The robustness of the various formulations and their ability to maintain the aqueous solubility of CLBQ14 was evaluated by challenging the formulations with varying proportions of normal saline. Each system was diluted with normal saline at a ratio of 1:2, 1:5, 1:10, 1:20 (v/v) and observed for signs of CLBQ14 precipitation within 4 hrs. The optimal formulation was selected based on the solubility of CLBQ14, precipitation upon dilution with aqueous media, toxicity of the solvent, and stability of CLBQ14 in the formulation.
Animal Studies
Adult male SD rats (body weight 300–350 g) purchased from Envigo RMS (Indianapolis, IN) were kept in an environmentally controlled room (average temperature: 25°C, average humidity 50% and 12 h dark-light cycle) for at least one week before experiments. The rats were fed ad libitum, except when fasted at least 12 hrs prior to oral dosing. The study protocol was approved by the Texas Southern University Animal Care and Use Committee. All experimental procedures involving animals were performed in accordance with the National Institute of Health’s “Guide for the Care and Use of Laboratory Animals, 8th Edition27”.
Pharmacokinetics and Bioavailability Studies
The study rats were cannulated under anesthesia one day prior to dosing. In a parallel group design, the rats were divided into 3 groups (n = 3) and administered either a single 2 mg/kg IV bolus dose, 20 mg/kg oral dose (PO) or a 10 mg/kg subcutaneous dose (SC) of CLBQ14. Prior to dosing, the co-solvent formulations were diluted with 5 times of normal saline for IV and SC or pure water for PO. Serial blood samples of approximately 250 μL were withdrawn at 0.08, 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24 and 48 hrs after dosing and placed in heparinized micro-centrifuge tubes. Urine samples were also collected for 24 hrs. The blood samples were centrifuged, and the supernatant plasma was collected and stored at –80°C until analysis. The concentration of CLBQ14 in the plasma and urine samples were determined by LC-MS/MS.
The absolute bioavailability (F%) of CLBQ14 was estimated from the following equation:
(3)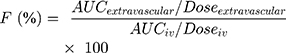
where AUCextravascular is the area under the plasma concentration vs time curve following extravascular administration (PO or SC), Doseextravascular is the corresponding PO or SC dose, AUCiv is the area under the plasma concentration vs time curve following IV administration and Doseiv is the intravenous dose.
Tissue Distribution Studies
Adult male SD rats cannulated under anesthesia prior to dosing were administered a single 5 mg/kg IV bolus dose of CLBQ14. Plasma and major organs such as liver, lungs, spleen, thymus, kidney and heart were collected 2 hrs after dosing. The organs were homogenized in three times volume of phosphate buffered saline and analyzed by LC-MS/MS for the concentration of CLBQ14. Tissue concentration of the CLBQ14 (ng/g) was calculated by following equation:28
(4)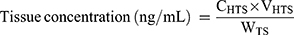
where, CHTS is concentration (ng/mL) of CLBQ14 in homogenized tissue sample; VHTS is volume of homogenized tissue sample (mL) and WTS is weight of tissue sample (g).
The tissue to plasma ratio (Kp) was calculated by the equation below:
(5)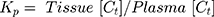
where Tissue [Ct] and Plasma [Ct]are tissue and plasma concentration at time t (2 hrs).
Drug Concentration Analysis
Samples from physicochemical studies, in vitro metabolism, pharmacokinetics and tissue distribution studies were analyzed either by a HPLC-UV method or by LC-MS/MS. HPLC-UV was deployed to analyze samples from physicochemical studies while LC-MS/MS was deployed for samples from in vitro metabolism, pharmacokinetics and tissue distribution studies. Analysis of CLBQ14 was performed using appropriate standard curve (matrix matched) or comparison of peak area ratios.
HPLC-UV Assay Development and Qualification
A simple, sensitive and reliable HPLC-UV method was developed and qualified for the quantification of CLBQ14 in solution. Chromatographic analysis was performed using a Waters 2487 Dual λ Absorbance Detector (Waters, Milford, MA) at a detection wavelength of 254 nm. Chromatographic separation was achieved with a Hypersil BDS C18 column (3.0 μm, 4.6×100 mm, Thermo Fisher Scientific, Waltham, MA) using a Waters HPLC system comprising of a 600 Pump and a 717 Plus Auto-sampler. The mobile phase, an isocratic solvent system comprising of 60% acetonitrile in water with 0.1% TFA, was employed at a flow rate of 1 mL/min. Clioquinol was used as IS at 20 µg/mL.
Stock solutions of CLBQ14 and IS were prepared in HPLC grade acetonitrile (final concentration of 1 mg/mL) and stored at –20°C until use. Standard samples of CLBQ14 were prepared in the mobile phase with concentration ranging from 1–100 μg/mL. Quality control (QC) samples of low (5 μg/mL), medium (20 μg/mL) and high (80 μg/mL) concentration of CLBQ14 were prepared in mobile phase. An injection volume of 20 μL was employed for chromatographic analysis, and the assay run time was 6 mins. Data acquisition and analysis was performed with Empower software v1.0 (Waters Corporation, Milford, MA).
Linear calibration curves were generated by plotting the peak area ratio of CLBQ14 to IS against known standard concentrations of CLBQ14. The slope, intercept, and coefficient of determination were estimated using least squares linear regression method with a weighting of 1/x2. The lower limit of quantification (LLOQ) was evaluated based on the signal-to-noise ratio of at least 5:1. The assay intra-day accuracy and precision was determined by analyzing six replicates of QC samples using a calibration curve constructed on the same day. The inter-day accuracy and precision were determined by analyzing six replicates of QC samples using calibration curves constructed on three different days. The accuracy of the assay was established by calculating the relative error (% RE) from the theoretical CLBQ14 concentrations, while the precision was reflected by the coefficient of variation (%CV).
LC-MS/MS Assay
A previously reported LC-MS/MS method was adopted for the determination of CLBQ14 in studies requiring the sensitivity and selectivity of mass spectrometry, Clioquinol was used as IS at 150 ng/mL.29 Briefly, chromatographic separation was performed with a Shimadzu Nexera X2 UHPLC System (Columbia, MD) equipped with a Waters XTerra® MS C18 column (3.5 μm, 125Å, 2.1×50 mm, Milford, MA) at room temperature. A binary solvent system: Solvent A – 0.2% formic acid in LC-MS grade water and Solvent B – 0.2% formic acid in LC-MS grade acetonitrile was employed at a flow rate of 0.5 mL/min. Ten microliters of sample was injected, and all samples were analyzed using gradient elution: initial 20% B, 70% B at 0.80 min, 95% from 2.80–3.80 min, and 40% B from 4.00–5.50 min. Detection was performed using a 4000 QTRAP® hybrid triple quadrupole LIT mass spectrometer (AB Sciex, Redwood City, CA) equipped with a Turbo V™ ion source. Data acquisition for CLBQ14 and IS was performed by electrospray ionization (ESI) using multiple reaction monitoring (MRM) operated in positive mode. The transitions [M+H]+ m/z 257.919 → m/z 151.005 for CLBQ14 and [M+H]+ m/z 305.783 → m/z 178.900 for IS (clioquinol) were selected for the final MRM method based on intensity.
Plasma Sample Preparation for LC-MS/MS Analysis
Standards and test samples were prepared by simple protein precipitation. Blank plasma (45 µL) was spiked with 5 µL of standard solution and vortexed. Precipitant (200 µL of acetonitrile containing IS) was added, vortexed and centrifuged for 10 mins at 18,879 rcf to pellet the proteins. The supernatant was injected for LC-MS/MS analysis. Test samples were prepared in similar manner with 50 µL of plasma samples.
Pharmacokinetic Data Analysis
Non-compartmental pharmacokinetic analysis (NCA) was performed to estimate the area under the plasma concentration – time curve (AUC), mean residence time (MRT), volume of distribution of the central compartment (VD), the plasma clearance (Cl) and elimination half-life (T1/2). These pharmacokinetic parameters were estimated using Phoenix WinNonlin v7.0 software (Pharsight Corporation, Mountain View, CA, USA). The maximum plasma concentration (Cmax) and the corresponding time (Tmax) were recorded from experimental observations. The AUC0-t and AUC0-∞ were estimated by linear up-log-down trapezoidal rule and extrapolation to time infinity. The weighting schemes (uniform, 1/Y, 1/Y2, and 1/Yhat*Yhat) were evaluated based on correlation coefficients and observed/predicted fits of the plasma concentration vs time plot for the most suitable weighting for the data analysis. A two-compartment model analysis was performed to estimate the absorption half-life for extravascular route of administration; the weighting schemes were also assessed for the most suitable for the data analysis.
The metabolic half-life (t1/2, m) for CLBQ14 was estimated from microsomal stability data using the equation for first order kinetics (Equation 6). The slope of the log-linear curve of the concentration of CLBQ14 remaining versus time was calculated by linear regression and used as the rate constant for metabolism (km). The microsomal intrinsic clearance (Cl’int, m) was calculated using Equation 7.
(6) (7)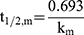
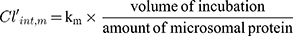
The hepatic intrinsic clearance (Clint, h) of CLBQ14 in various species was estimated from the microsomal data using Equation 8. Forty-five (45) mg of microsomal protein per gram of liver tissue was applied for all species and 87 g, 40 g, 32 g and 26 g of liver tissue per kilogram of body weight was used for mouse, rat, monkey and human respectively.30
(8) (9)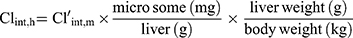
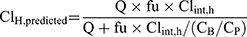
The in vivo hepatic clearance of CLBQ14 in various species was predicted by the “well-stirred” model shown in Equation 9, where Q is the hepatic blood flow, Clint, h is the intrinsic clearance determined using Equation 8, fu is the fraction unbound in plasma, and CB/CP is the blood-to-plasma ratio.31,32 Hepatic blood flow of 90, 55, 44, and 21 mL/min/kg was used for mouse, rat, monkey and human.
In addition, human hepatic clearance (ClH, human) for CLBQ14 was also predicted by single species scaling using liver blood flow. ClH, human was estimated using the liver blood flow (LBF) equations proposed by Ward and Smith:33
(10)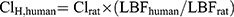
Clrat was derived from the in vivo pharmacokinetic study in SD rats and the LBF values mentioned above was applied for SD rat and human respectively.
Results
HPLC-UV Chromatographic Conditions and Assay Qualification
Several mobile phase compositions and columns were evaluated for the most appropriate peak shape and signal intensity. Optimal symmetrical peak shape was obtained with 60% acetonitrile in water with 0.1% TFA compared to formic acid, phosphate buffer, and ammonium acetate. Peak tailing observed with Xterra MS C18 (150x4.6 mm, 3.5 μm, Waters, Milford, MA) was circumvented by using a Hypersil BDS C18 column (4.6×100 mm, 3.0 μm, Thermo Fisher Scientific, Waltham, MA). The retention times for CLBQ14 and IS were 3.43 and 4.69 mins respectively (Figure 2); the total assay run time was 6 mins.
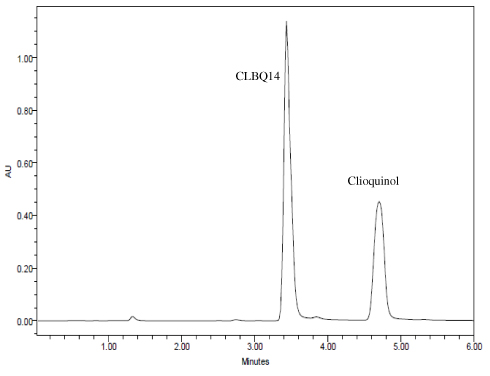 |
Figure 2 Representative HPLC-UV Chromatograms for CLBQ14 and clioquinol (IS) in Solution. |
The calibration curves of CLBQ14 in solution was linear in the concentration range of 1–100 μg/mL with correlation coefficient greater than 0.999. The accuracy and precision data, summarized in Table 1, shows that they were well within the 15% acceptance range. Our HPLC-UV analytical method was validated as accurate and precise for the quantification of CLBQ14 in solution of concentration ranging from 1–100 μg/mL.
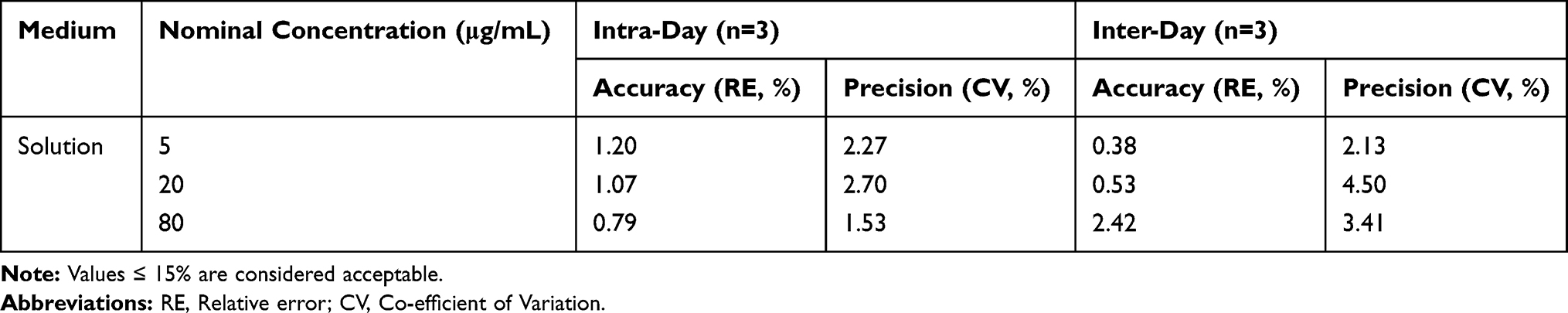 |
Table 1 Intra- and Inter-Day Accuracy and Precision of HPLC-UV Method |
Physicochemical Properties
Physicochemical characteristics of CLBQ14 were evaluated in this study. These properties are imperative to the selection of a suitable formulation for the in vivo delivery of CLBQ14 and elucidation of the pharmacokinetic properties and biodistribution of the compound. The solubility, pH driven degradation and lipophilicity of the compound were assessed.
Solubility
The solubility of CLBQ14 in water (pH 7), ethanol, PEG 400, Capyrol 90®, PGMC 1, DMSO, DMA, 1-octanol, glycerol, Tween 80®, Tween 20®, paraffin oil, soybean oil, and olive oil, determined by the shake-flask method, is depicted in Figure 3. The compound is practically insoluble in water (0.07 mg/mL) but is soluble in DMA (>80 mg/mL).
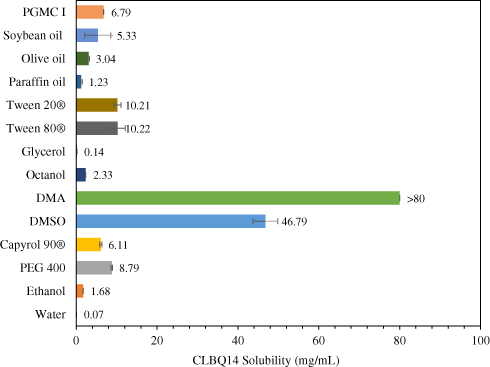 |
Figure 3 Solubility of CLBQ14 in various solvents at room temperature. Mean of n = 3. |
pH Stability
As shown in Figure 4 and Table 2, the pH stability data suggests that CLBQ14 is relatively stable in aqueous media at 37oC for up to 48 hrs. However, beyond 48 hrs, there is a slow and late onset degradation of CLBQ14 in aqueous media of pH greater than 5. Its degradation appears to be biphasic and follows a first order kinetics across various pH ranges. The pH degradation rate profile, depicted in Figure 5, is a plot of the rate constant for CLBQ14 degradation in various buffers versus the corresponding pH. The shape of the rate profile suggests that CLBQ14 undergoes a more rapid degradation at higher pH compared to pH 1–5.
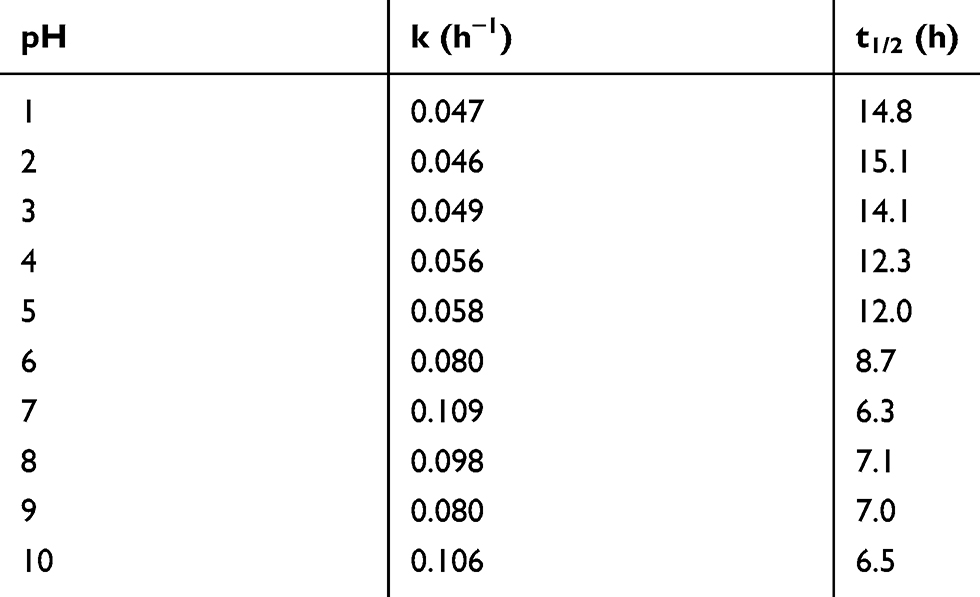 |
Table 2 Degradation Rate Constants and Half-Life of CLBQ14 at Various pH |
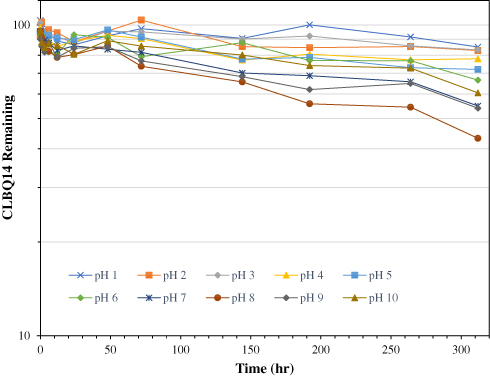 |
Figure 4 Stability of CLBQ14 in various pH buffers at 37oC. Data is expressed as mean of n = 3. |
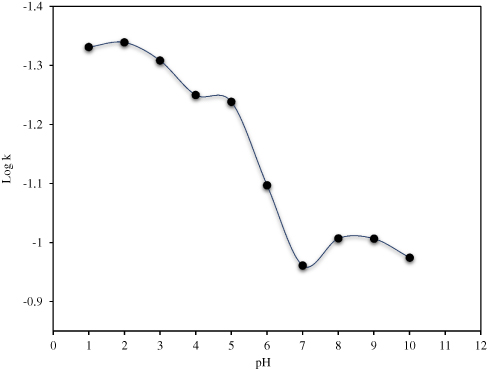 |
Figure 5 pH degradation rate profile for CLBQ14 in various USP buffers at 37 °C. |
Lipophilicity
The octanol – water partition coefficient, denoted by log P, is a measure of the lipophilicity of a molecule. Lipophilicity significantly impacts the absorption of a compound, its biodistribution, penetration of essential biological barriers and membranes, liver metabolism and elimination (ADME properties). Although it is not the only determining factor, log P also helps in predicting the likely transport of a compound around the body, it affects the formulation of the drug, dosing, clearance mechanism, and the toxicity.
In this study, the log P of CLBQ14 was estimated using the shaker method. CLBQ14 has an experimental log P value of 3.03 ± 0.04 which is analogous with predicted value reported in literature (3.92 ± 0.39 at room temperature).26 Although Lipinski’s rule of 5 considers a log P value less than 5 as desirable for good oral activity, for good oral absorption, a log P value of 1.35–1.8 is considered ideal.34,35 The experimental log P value of CLBQ14 suggests that there might be solubility and consequently absorption concerns when administered orally.
In vitro Studies
Plasma Stability
When incubated in mouse, rat, rhesus monkey and human plasma, mean + SD CLBQ14 recoveries of 97.6 ± 2.27%, 101.1 ± 13.04%, 105.3 ± 4.25%, and 103.7 ± 0.99%, respectively, was observed after 4 hrs. The data suggest that CLBQ14 is stable in plasma from all the species, at least for up to 4 hrs, ruling out any concerns for ex vivo biotransformation or degradation.
Plasma Protein Binding and Blood-to-Plasma Ratio
The binding of CLBQ14 to plasma protein was evaluated at different concentrations using ultrafiltration. At 5, 25 and 50 μg/mL, the fraction of CLBQ14 bound to plasma protein (fu) were 91.0 ± 0.6%, 96.3 ± 1.4, and 98.3 ± 0.3 respectively. Data is expressed as mean ± standard deviation. CLBQ14 appear to be highly bound to plasma protein, with fraction unbound in plasma (fu) less than 0.09. This atypical plasma protein data may be due to the limited solubility of the unbound drug in the aqueous component of plasma. The protein component in plasma increasingly becomes a reservoir for CLBQ14 as its concentration increase.
The blood-plasma partitioning of CLBQ14 was assessed in vitro at a concentration of 1 μM. CLBQ14 has a B/P ratio of 0.83 ± 0.03 (mean ± SEM). Since B/P ratio is derived from the ratio of two independent variables, the standard error was calculated as a square root of the sum of square of the ratio of SEM to mean in blood and plasma multiplied by the B/P ratio. The data suggest that CLBQ14 does not partition into red blood cells during circulation.
Microsomal Metabolic Stability
The mean ± SD percentage of CLBQ14 remaining after a 60 min incubation in mouse, rat, rhesus monkey and human liver microsomes were 3.7 ± 1.36%, 26.8 ± 2.37%, 17.5 ± 0.86% and 35.5 ± 0.33% respectively (Table 3). As shown in Figure 6, the turnover of CLBQ14 is fastest in mouse microsome but slower in human microsomes. In general, small animals tend to metabolize drugs more rapidly than human as the relative amount of hepatic metabolizing enzymes, CYP/gram of body weight, is higher in small animals than in humans.42 Overall, the microsomal stability data suggest that the compound is primarily eliminated by liver metabolism, corroborating the observation in in vivo pharmacokinetic studies where 0.01% of the administered dose was excreted unchanged in urine.29
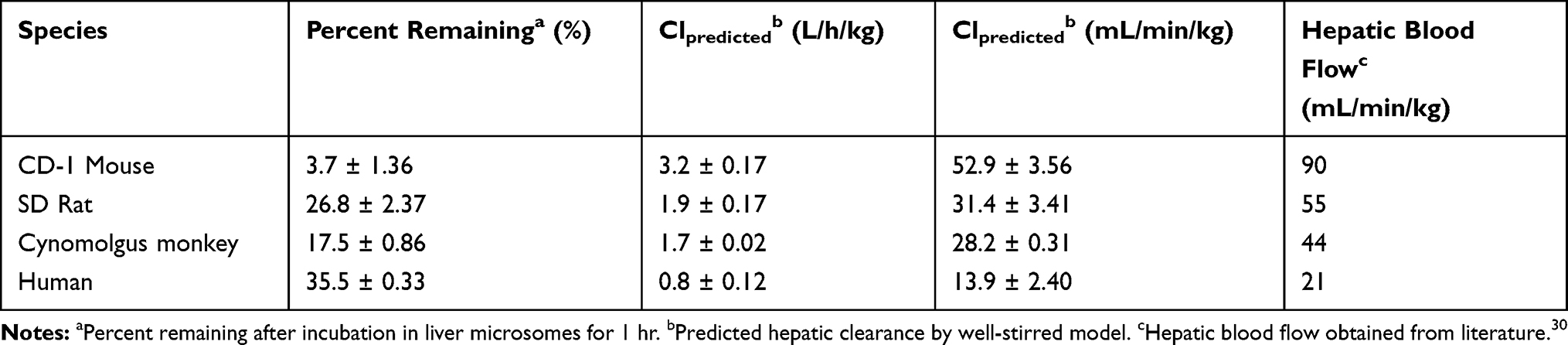 |
Table 3 Microsomal Stability and Predicted Plasma Clearance of CLBQ14 |
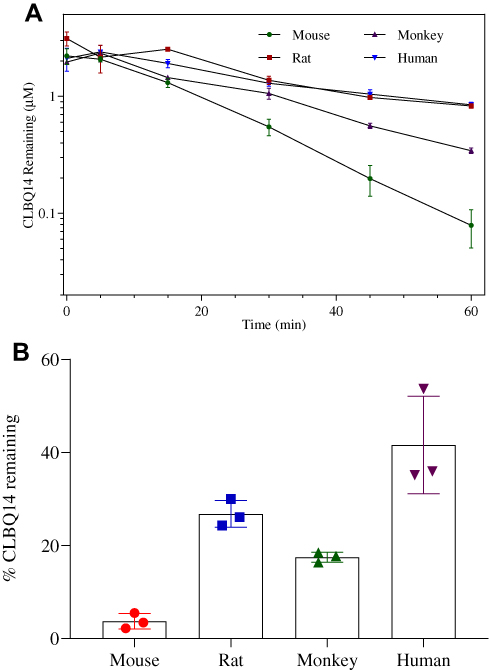 |
Figure 6 Microsomal stability of CLBQ14 following incubation in liver microsomes from CD-1 mouse, SD rat, cynomolgus monkey and human at 37 °C for 60 mins. (A) Rate of CLBQ14 disappearance. (B) Percent CLBQ14 remaining after incubation. (Error bar = standard deviation). |
CYP Reaction Phenotyping
To determine the major CYP enzymes involved in the metabolism of CLBQ14, the compound was incubated with rhCYPs enzymes; the disappearance of the parent compound was monitored, and the percentage of the parent remaining was calculated. Any isoform resulting in >50% turnover of the compound was considered as a potential contributor to the metabolism of the CLBQ14. The disappearance of CLBQ14 is depicted in Figure 7A, and the percent remaining after incubation for 1 hr is shown in Figure 7B. CYP1A2 and CYP2D6 appear to be the primary contributors to the metabolism of CLBQ14. Detection of OH-CLBQ14 peaks in CYP1A2 samples suggests that hydroxylation is the primary CLBQ14 metabolic reaction catalyzed by CYP1A2.
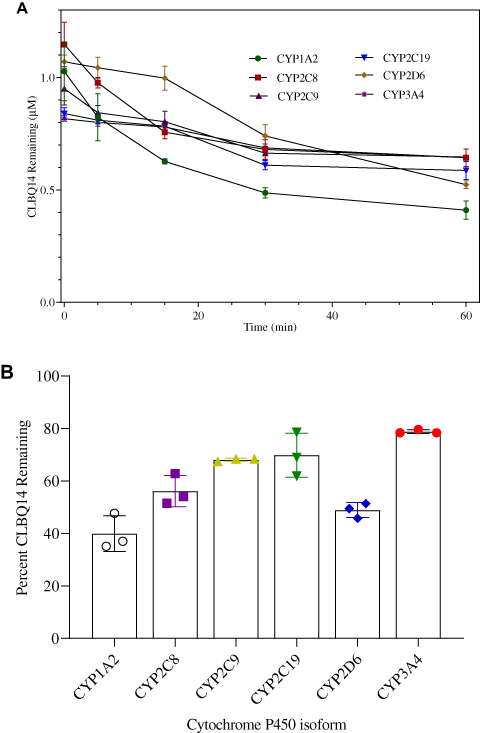 |
Figure 7 CYP Phenotyping of CLBQ14 following incubation in rhCYPs at 37°C for 60 mins. (A) CLBQ14 disappearance. (B) Percent CLBQ14 remaining after incubation. (Error bar = standard deviation). |
Dosing Solution
The optimal formulations for the in vivo delivery of CLBQ14 were selected based on the solubility of CLBQ14, precipitation of the API from aqueous media when diluted with normal saline or sterile water, and minimal toxicity of excipients. A co-solvent systems (APT1) comprising of 5% v/v DMA, 35% v/v PEG 400, 60% v/v Tween 80® and 5 mg/mL of CLBQ14 was selected as the optimal formulation for IV administration of CLBQ14. This formulation was diluted 5 times with normal saline prior to IV administration. On the other hand, a co-solvent formulation (PTT6) comprising of 10% v/v Transcutol HP®, 20% v/v PEG 400, 70% Tween 80®, and 7.5 mg/mL of CLBQ14 was selected as the optimal formulation for PO and SC administration. Also, this formulation is to be diluted with sterile water or normal saline prior to PO and SC administration, respectively.
In vitro plasma precipitation screening was performed to evaluate the tonicity of the selected formulations with respect to plasma. Different dilutions of the optimal cosolvent formulation was spiked at varying ratios to rat plasma placed in a water bath at 37ºC for 4 hrs. Normal saline was used as a negative control. No precipitation was observed in all but one of the samples diluted 1:2 with normal saline.
Pharmacokinetics and Bioavailability
The plasma concentration versus time profiles for CLBQ14 in SD rats following IV, PO and SC administration are shown in Figure 8. The pharmacokinetic parameters for CLBQ14 in rats, estimated by NCA using Phoenix WinNonlin v7.0, are summarized in Table 4. A uniform weighting factor was used for the NCA of the data. CLBQ14 displayed a highly variable pharmacokinetic disposition following oral administration. A Cmax of 1.7 ± 0.65 mg/L (attained at a Tmax of 30 mins) was observed after a single 20 mg/kg PO dose of CLBQ14. The rate and extent of the oral absorption of CLBQ14 are depicted by the absorption half-life and the absolute bioavailability, respectively. CLBQ14 has an oral absorption half-life of 8.3 ± 1.44 mins indicating rapid GI absorption. The mean ± SD absolute oral bioavailability of CLBQ14, calculated using Equation 3, was 39.4 ± 12.64%. On the other hand, following a single 10 mg/kg SC dose of CLBQ14, a Cmax ± SD of 0.9 ± 0.12 mg/mL was reached after about 55 mins (Tmax). The rate and extent of the subcutaneous absorption of CLBQ14 are depicted by the absorption half-life and the absolute bioavailability respectively. CLBQ14 has a subcutaneous absorption half-life of 23.3 ± 0.6 mins. The absolute subcutaneous bioavailability of CLBQ14 was 90.0 ± 7.20%. Expectedly, the elimination of CLBQ14 follows a first order kinetics, and the plasma concentrations declined exponentially after Cmax was attained in both routes. The human hepatic clearance predicted from single species (SD rat) scaling of in vivo clearance and liver blood flow was 0.69 L/h/kg while the ClH, human from liver microsomal stability data was 0.8 L/hr/kg.
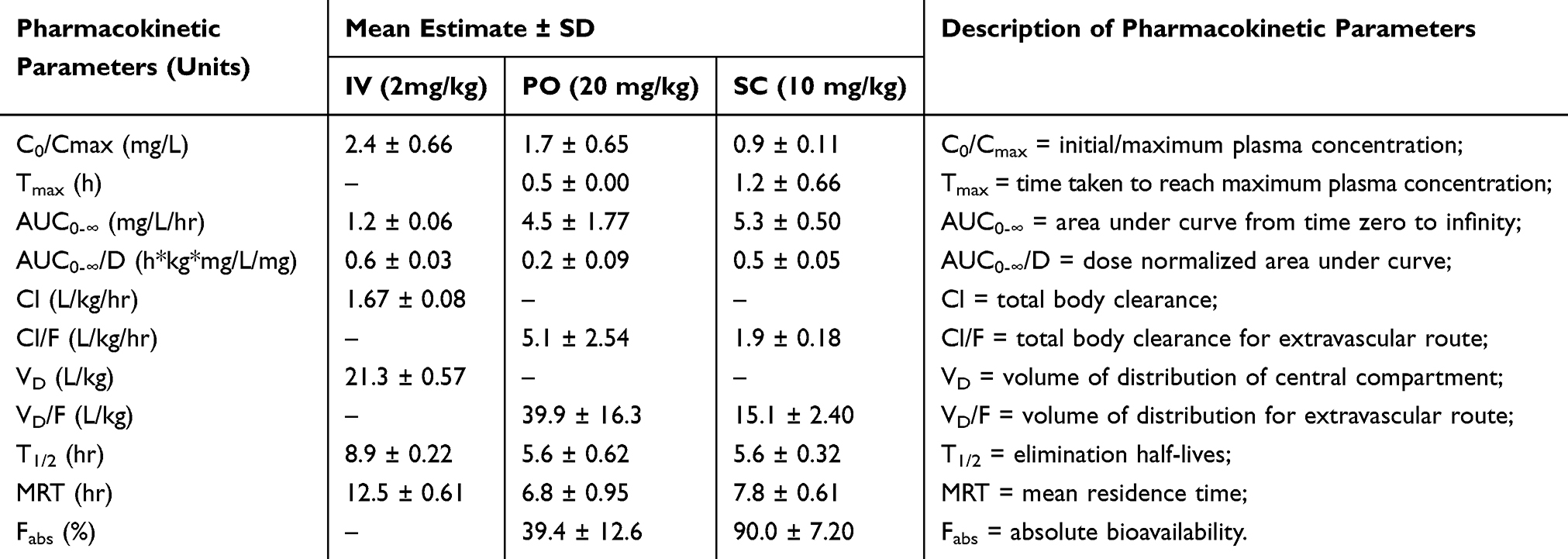 |
Table 4 CLBQ14 Pharmacokinetic Parameters Generated by Non-Compartmental Analysis |
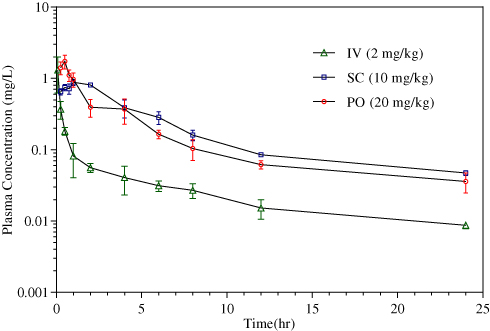 |
Figure 8 Concentration versus time profile of CLBQ14 following the administration of 2 mg/kg IV bolus, 10 mg/kg PO and 20 mg/kg SC doses to SD rats (n=3 each, error bar = standard deviation). |
Tissue Distribution Study
The tissue concentration and Kp value for each tissue are depicted in Figure 9 and Table 5 respectively. CLBQ14 was detected in all the tissues sampled suggesting that it is well distributed to all the major organs sampled in this study. Tissue exposure was observed in the descending order kidneys > liver > lungs > thymus > pancreas > spleen > heart. The tissue distribution data suggest that CLBQ14 will be suitable for infectious disease of major organs provided the microbe is susceptible to CLBQ14.
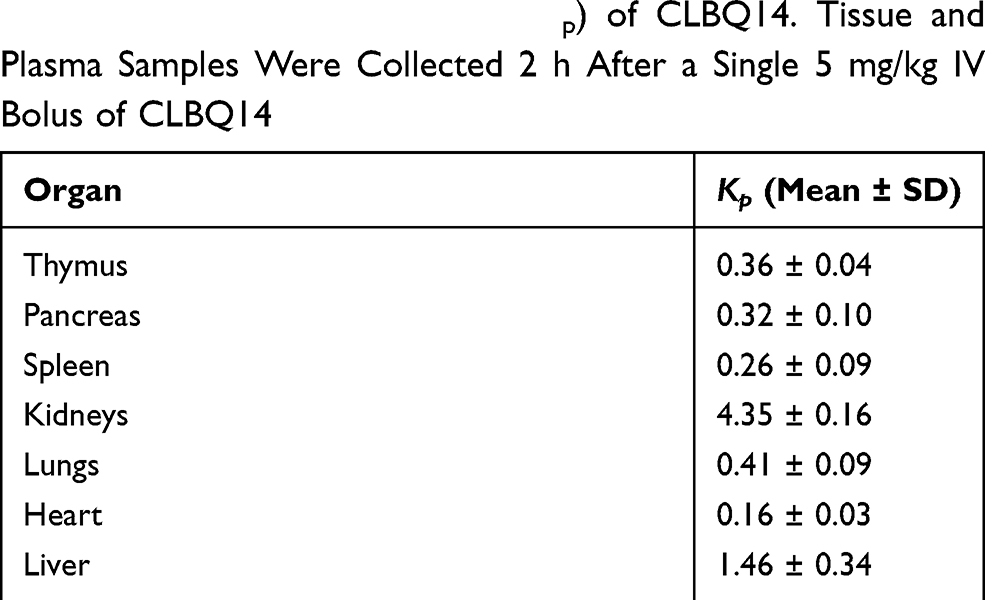 |
Table 5 Tissue-to-Plasma Ratio (Kp) of CLBQ14. Tissue and Plasma Samples Were Collected 2 h After a Single 5 mg/kg IV Bolus of CLBQ14 |
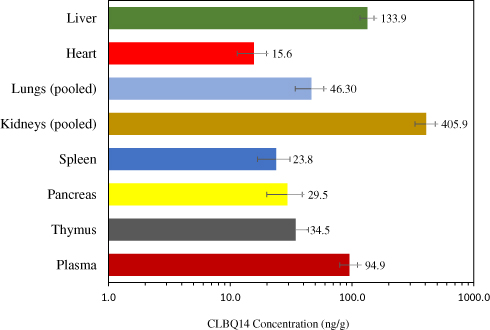 |
Figure 9 Tissue distribution of CLBQ14 at 2 hrs following an IV bolus dose of 5 mg/kg to SD rats (error bar = standard deviation). |
Discussion
In general, there is an increasing global awareness on the need for new infectious disease drugs; it is expedient to discover and develop new chemotherapeutic agents to combat the global infectious disease burden. Pivoting to tuberculosis, no new drug was approved for its treatment for almost 50 years until the recent approval of bedaquiline. In search for new targets, MetAP, which catalyzes NME – a process essential for protein and polypeptide synthesis, has been identified as a potential target for tuberculosis and several other infectious diseases. Through high throughput screening, various analogues of 8-hydroxyquinoline have been identified as effective inhibitors of MetAP.5 Notably, CLBQ14, a congener of 8-hydroxyquinoline has been shown to have in vitro activity against MetAPs in Mtb and is being developed for the treatment of tuberculosis and potentially other infectious diseases. Preclinical efficacy studies in Mtb shows that CLBQ14 has great potency against replicating Mtb; an increased potency against aged non-growing Mtb and exhibits great selectivity for both MtMetAPs over human MetAPs.5 Early evaluation of the ADME/physicochemical properties, and tissue distribution of CLBQ14 is expedient in its discovery and development process. This report highlights these important studies; the data generated will be invaluable to the development and potential advancement of CLBQ14 to the clinic.
As expected of halogenated organic compounds, CLBQ14 has poor solubility in water and other common solvents. However, we developed a suitable co-solvent formulation significantly improving its aqueous solubility to enable the in vivo characterization of the molecule. We employed Transcutol HP and Tween 80 as excipients for oral and subcutaneous delivery. Transcutol HP has been used as excipient in approved medication for oral administration, for example in Lysanxia, Pilosuryl and Urosiphon oral solutions. In animal studies, 5% solution of Transcutol HP administered orally to rats for 90 days was found to be safe with a NOAEL of 1%; acute administration of 5 g/kg is also considered safe with LD50 >5g/kg.37 Also, the amount of Tween 80 in our dosing solutions is within the safe limit of 2.5 mg/kg. Our formulation is to be diluted 5X with water before dosing, the final concentration of Tween 80 is 12% v/v; at the dosing volume of 2 mL/kg of diluted formulation, Tween 80 is significantly low (0.24 mL/kg).
CLBQ14 has an experimental log P value of 3.03 ± 0.04 which suggests that the compound is lipophilic in nature. Its lipophilicity may significantly impact its absorption, distribution, penetration of biological barriers and membranes, liver metabolism, renal excretion and potentially its toxicity. Its log P value also raised solubility and/or absorption concerns from extravascular routes of administration. However, our rat PO and SC bioavailability data suggest otherwise. On the other hand, we assessed the pH driven decomposition of CLBQ14 at pH ranging from 1–10. CLBQ14 appears to exhibit as inverse Z-shaped pH decomposition profile; it is mostly stable at acidic pH but is degraded at a faster rate at basic pH. Although the mechanism of degradation is unclear, we hypothesize that higher pH deprotonates the phenol, inducing oxidation of the oxygen and this leads to the kicking out of the chlorine atom. Nonetheless, the decomposition profile suggests that the molecule will most likely be stable in stomach pH, and no special formulation considerations will be warranted for oral delivery.
Our studies revealed that CLBQ14is highly bound to plasma proteins and this binding appears to be slightly dependent on the plasma concentration of CLBQ14. As described by O’Brien et al (2013), PPB values ≥ 99% can be classified as extremely high, 90 – 99% high, 70 – 90% moderate and ≤ 70% low. CLBQ14 has PPB values up to 99% which suggests that PPB is likely to have a significant impact on the disposition of the molecule.36 It has been well reported that PPB plays a crucial role in the pharmacokinetic and pharmacodynamic properties of a drug. Only unbound drug is available at the site of action for pharmacological activity. PPB influences the distribution of a drug from blood to tissues and from tissues to site of action. Also, drugs bound in plasma can serve as a reservoir for free drugs. In addition, a high PPB points to a greater propensity for drug-drug interaction when co-administered with other molecules with significant protein binding. In the same vein, the BPP for CLBQ14 was measured to assess the degree of its affinity for the red blood cells (RBCs); BPP represents the extent of accumulation of a drug in RBC. For compounds with BPP >1, blood clearance determined by plasma data can be misleading, as the compound has a higher affinity for the RBC fraction of whole blood than plasma. It can be inferred from our PPB and BPP data that CLBQ14 has a higher affinity for plasma proteins than RBCs, hence its blood partitioning of 0.83.
In vitro studies revealed that the metabolism of CLBQ14 is predominantly driven by CYP1A2 and CYP2D6, but CYP2C8 may also be a contributor. We observed CLBQ14 turnover greater than 50% when incubated with CYP1A2 and CYP2D6 isoforms. In silico predictions suggest that positions 3 and 6 are soft spots and are favored sites for CYP metabolism.38–40 The biotransformation of CLBQ14 appears to be NADPH dependent as there was no significant change (from baseline) in the concentration of CLBQ14 in control samples from microsomal stability and CYP reaction phenotyping studies. Hydroxylation seems to be the primary CLBQ14 metabolic reaction catalyzed by CYP1A2; OH-CLBQ14 was detected in samples incubated with CYP1A2. CYP1A2 is not expressed in the intestine, implying that inter-subject variability in bioavailability driven by CYP1A2 metabolism in the gut may not be an issue for CLBQ14. However, CYP1A2 is induced by smoking, rifampin, carbamazepine and barbiturates pointing; there is a propensity for drug interactions in such patients. On the other hand, although CYP2D6 is thought to be non-inducible, and only accounts for 4% of the CYPs, it is still responsible for the metabolism of 30% of the drugs on the market.41 Its inhibition by very low levels of quinidine, ritonavir the HIV protease inhibitor as well as serotonin re-uptake inhibitors has been well reported.42,43 These point to the propensity for CLBQ14 interactions with co-administered drugs. Furthermore, variability in the functional activity of CYP2D6 due to genetic polymorphism may also impact the metabolism of CLBQ14; this will guide any future clinical studies of the molecule.
One of the aims of this study was to determine the bioavailability of CLBQ14 following extravascular administration. Dose selection was purely exploratory and non-empirical; selected doses were conventional for PO and SC pharmacokinetic studies. In a parallel group design, the IV, PO and SC pharmacokinetic properties of CLBQ14 was assessed in adult male SD rats. Although CLBQ14 has a high affinity for plasma proteins (PPB >90%), it has a Vss in rats that is greater than total body water. This result is consistent with molecules that have a high hepatic uptake (high rodent Vss and liver to plasma ratio). PO and SC bioavailability of CLBQ14 were 39.4% and 90% respectively. Since CLBQ14 has a poor aqueous solubility, we suspect that its extravascular systemic exposure is driven by a good permeability; however, this is yet to be confirmed in a PAMPA and/or Caco-2 model. Furthermore, we suspect that its SC bioavailability was enhanced by Transcutol HP which is a well-known permeability enhancer.44 Following oral administration to SD rats, CLBQ14 is rapidly absorbed (T1/2absorption = 8.3 mins). Its tissue concentration in rats at 2 hrs post dose is less than plasma concentration except in kidneys and liver, both are highly perfused organs. Interestingly, although CLBQ14 has a high affinity for the kidneys, it is largely not excreted unchanged in urine – 0.01% of the administered dose was excreted unchanged in urine in previous pharmacokinetic studies.29 The higher concentration in the kidney may be driven by its higher perfusion; whether this will pose any safety risk is to be seen in safety lead optimization studies. In spite of this, the extensive distribution of CLBQ14 will be beneficial in chemotherapy targeting these tissues if this observation translates to humans.
Lastly, the in vivo clearance of CLBQ14 was predicted by physiologically based scaling from the in vitro metabolism of CLBQ14 by liver microsomes. We observed a good correlation between predicted and observed clearance in rats, 1.90 ± 0.17 L/kg/h and 1.67 ± 0.08 L/kg/h respectively. The predicted hepatic clearance in the different species can be ranked from highest to lowest in the order CD-1 mouse, SD rat, cynomolgus monkey and human. A higher clearance in smaller animals is quite in line with the relative amount of hepatic enzymes in each species; smaller animals have higher CYPs/gram body weight and tend to have higher clearance than larger mammals. On the other hand, when compared to hepatic blood flow, predicted hepatic clearance of CLBQ14 can be characterized as intermediate to moderate in mice, rats, monkeys and humans. Simultaneously, the human hepatic clearance predicted from microsomal stability data and from the single species scaling using liver blood flow and in vivo clearance in SD rats were 0.80 L/hr/kg and 0.69 L/h/kg, respectively, giving us great confidence in our predictions. Although first-in-human studies is a long way out and will require elaborate interspecies pre-clinical studies, we believe that the studies detailed in this work will nonetheless provide essential preliminary information on the physicochemical and ADME properties of the molecule.
Conclusion
CLBQ14, a derivative of 8-hydroxyquinoline has been identified and characterized as an inhibitor of MetAP, a promising new target for the treatment of infectious diseases. In this study, we investigated the physicochemical properties of CLBQ14, its pharmacokinetic disposition and bioavailability in rat following IV, PO and SC administration and its tissue distribution. CLBQ14 is practically insoluble in water but is freely soluble in DMA; it is more stable in acidic pH than basic pH. It is lipophilic in nature, highly bound to plasma proteins but does not partition into red blood cells. CLBQ14 has a biphasic pharmacokinetic disposition with oral and subcutaneous bioavailability of 39 and 90%, respectively, in rats. It is rapidly and extensively distributed to most essential organs following IV administration. This study provides additional information about the pre-clinical profile of CLBQ14; this will be expedient in future interspecies pre-clinical studies and subsequent clinical studies.
Acknowledgments
Oscar Ekpenyong is currently affiliated with the Department of Pharmacokinetics, Pharmacodynamics and Drug Metabolism, Merck & Co., Inc., South San Francisco, CA, USA. Candace Cooper is currently affiliated with Technical Services, Pfizer & Co., Inc., McPherson, KS, USA.
Funding
This study was funded partially by the National Institute of Health SC3 (grant number 1SC3GM102018), by the National Institute of Health’s Research Centers in Minority Institutes Program (RCMI, grant number G12MD007605) and by the Cancer Prevention & Research Institute of Texas (CPRIT, grant number RP180748).
Disclosure
Dr Oscar Ekpenyong reports a patent for the formulation of CLBQ14 pending to Omonike Olaleye, Huan Xie, and Oscar Ekpenyong. The authors report no other conflicts of interest in this work.
References
1. Center for Disease Control and Prevention, Infectious Disease. FastStats: infectious/immune 2017. U.S. Department of Health & Human Services [
2. Roca I, Akova M, Baquero F, et al. The global threat of antimicrobial resistance: science for intervention. New Microbes New Infect. 2015;6:22–29. doi:10.1016/j.nmni.2015.02.007
3. Chai SC, Wang W-L, Ding D-R, et al. Growth inhibition of Escherichia coli and methicillin-resistant Staphylococcus aureus by targeting cellular methionine aminopeptidase. Eur J Med Chem. 2011;46(8):3537–3540. doi:10.1016/j.ejmech.2011.04.056
4. Olaleye O, Raghunand TR, Bhat S, et al. Methionine aminopeptidases from Mycobacterium tuberculosis as novel antimycobacterial targets. Chem Biol. 2010;17(1):86–97. doi:10.1016/j.chembiol.2009.12.014
5. Olaleye O, Raghunand TR, Bhat S, et al. Characterization of clioquinol and analogues as novel inhibitors of methionine aminopeptidases from Mycobacterium tuberculosis. Tuberculosis (Edinb). 2011;91(Suppl 1):S61–5. doi:10.1016/j.tube.2011.10.012
6. John SF, Aniemeke E, Ha NP, et al. Characterization of 2-hydroxy-1-naphthaldehyde isonicotinoyl hydrazone as a novel inhibitor of methionine aminopeptidases from Mycobacterium tuberculosis. Tuberculosis. 2016;101(Supplement):S73–S77. doi:10.1016/j.tube.2016.09.025
7. Zhang X, Chen S, Hu Z, et al. Expression and characterization of two functional methionine aminopeptidases from Mycobacterium tuberculosis H37Rv. Curr Microbiol. 2009;59(5):520–525. doi:10.1007/s00284-009-9470-3
8. Kang J-M, Ju H-L, Sohn W-M, et al. Characterization of the biochemical properties of two methionine aminopeptidases of Cryptosporidium parvum. Parasitol Int. 2012;61(4):707–710. doi:10.1016/j.parint.2012.05.008
9. Chen X, Chong CR, Shi L, et al. Inhibitors of Plasmodium falciparum methionine aminopeptidase 1b possess antimalarial activity. Proc Natl Acad Sci U S A. 2006;103(39):14548–14553. doi:10.1073/pnas.0604101103
10. Kumar R, Tiwari K, Dubey VK. Methionine aminopeptidase 2 is a key regulator of apoptotic like cell death in Leishmania donovani. Sci Rep. 2017;7(1):95. doi:10.1038/s41598-017-00186-9
11. Isichei A, Olaleye O. Characterization of CLBQ14 as a Methionine Aminopeptidase Inhibitor in Enterococcus Faecalis. Texas Southern University; 2017. Unpublished manuscript.
12. Kishor C, Gumpena R, Reddi R, et al. Structural studies of Enterococcus faecalis methionine aminopeptidase and design of microbe specific 2, 2ʹ-bipyridine based inhibitors. Med Chem Comm. 2012;3:1406–1412. doi:10.1039/c2md20096a
13. Helgren TR, Chen C, Wangtrakuldee P, et al. Rickettsia prowazekii methionine aminopeptidase as a promising target for the development of antibacterial agents. Bioorg Med Chem. 2017;25(3):813–824. doi:10.1016/j.bmc.2016.11.013
14. Yuan H, Chai SC, Lam CK, et al. Two methionine aminopeptidases from Acinetobacter baumannii are functional enzymes. Bioorg Med Chem Lett. 2011;21(11):3395–3398. doi:10.1016/j.bmcl.2011.03.116
15. Griffith EC, Su Z, Turk BE, et al. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem Biol. 1997;4(6):461–471. doi:10.1016/S1074-5521(97)90198-8
16. Ehlers T, Furness S, Philip Robinson T, et al. Methionine aminopeptidase Type-2 inhibitors targeting angiogenesis. Curr Top Med Chem. 2016;16(13):1478–1488. doi:10.2174/1568026615666150915121204
17. Shimizu H, Yamagishi S, Chiba H, et al. Methionine aminopeptidase 2 as a potential therapeutic target for human non-small-cell lung cancers. Adv Clin Exp Med. 2016;25(1):117–128. doi:10.17219/acem/60715
18. Chun E, Han CK, Yoon JH, et al. Novel inhibitors targeted to methionine aminopeptidase 2 (MetAP2) strongly inhibit the growth of cancers in xenografted nude model. Int J Cancer. 2005;114(1):124–130. doi:10.1002/(ISSN)1097-0215
19. Bernier SG, Lazarus DD, Clark E, et al. A methionine aminopeptidase-2 inhibitor, PPI-2458, for the treatment of rheumatoid arthritis. Proc Natl Acad Sci U S A. 2004;101(29):10768–10773. doi:10.1073/pnas.0404105101
20. Bradshaw RA, Brickey WW, Walker KW. N-Terminal processing: the methionine aminopeptidase and Nα-acetyl transferase families. Trends Biochem Sci. 1998;23(7):263–267. doi:10.1016/S0968-0004(98)01227-4
21. Giglione C, Vallon O, Meinnel T. Control of protein life-span by N-terminal methionine excision. EMBO J. 2003;22(1):13–23. doi:10.1093/emboj/cdg007
22. Giglione C, Boularot A, Meinnel T. Protein N-terminal methionine excision. Cell Mol Life Sci. 2004;61(12):1455–1474. doi:10.1007/s00018-004-3466-8
23. Arfin SM, Kendall RL, Hall L, et al. Eukaryotic methionyl aminopeptidases: two classes of cobalt-dependent enzymes. Proc Natl Acad Sci U S A. 1995;92(17):7714–7718. doi:10.1073/pnas.92.17.7714
24. Keeling PJ, Doolittle WF. Methionine aminopeptidase-1: the MAP of the mitochondrion? Trends Biochem Sci. 1996;21(8):285–286.
25. Lowther WT, Matthews BW. Structure and function of the methionine aminopeptidases. Biochim Biophys Acta. 2000;1477(1–2):157–167. doi:10.1016/S0167-4838(99)00271-X
26. National Center for Biotechnology Information. PubChem Compound Database; CID=82095. [cited March 10, 2017]. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/82095.
27. National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals.
28. Chhonker YS, Chandasana H, Kumar A, et al. Pharmacokinetics, tissue distribution and plasma protein binding studies of rohitukine: a potent anti-hyperlipidemic agent. Drug Res (Stuttg). 2015;65(7):380–387. doi:10.1055/s-0034-1387774
29. Ekpenyong O, Cooper C, Ma J, et al. A simple, sensitive and reliable LC-MS/MS method for the determination of 7-bromo-5-chloroquinolin-8-ol (CLBQ14), a potent and selective inhibitor of methionine aminopeptidases: application to pharmacokinetic studies. J Chromatogr B Analyt Technol Biomed Life Sci. 2018;1097–1098:35–43. doi:10.1016/j.jchromb.2018.08.027
30. Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10(7):1093–1095. doi:10.1023/A:1018943613122
31. Tamaki S, Komura H, Kogayu M, et al. Comparative assessment of empirical and physiological approaches on predicting human clearances. J Pharm Sci. 2011;100(3):1147–1155. doi:10.1002/jps.22321
32. Yang J, Jamei M, Yeo KR, et al. Misuse of the well-stirred model of hepatic drug clearance. Drug Metab Dispos. 2007;35(3):501–502. doi:10.1124/dmd.106.013359
33. Ward KW, Smith BR. A comprehensive quantitative and qualitative evaluation of extrapolation of intravenous pharmacokinetic parameters from rat, dog, and monkey to humans. I. Clearance. Drug Metab Dispos. 2004;32(6):603–611. doi:10.1124/dmd.32.6.603
34. Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1):3–25. doi:10.1016/S0169-409X(96)00423-1
35. Bhal SK. LogP—making sense of the value. Advanced Chemistry Development Inc. (ACD/Labs); 2011. Available from: https://www.acdlabs.com/download/app/physchem/making_sense.pdf.
36. O’Brien Z, Fallah Moghaddam M. Small molecule kinase inhibitors approved by the FDA from 2000 to 2011: a systematic review of preclinical ADME data. Expert Opin Drug Metab Toxicol. 2013;12(9):1597–1612. doi:10.1517/17425255.2013.834046
37. Gad SC, Cassidy CD, Aubert N, Spainhour B, Robbe H. Nonclinical vehicle use in studies by multiple routes in multiple species. Int J Toxicol. 2006;25(6):499–521. doi:10.1080/10915810600961531
38. Zaretzki J, Matlock M, Swamidass SJ. XenoSite: accurately predicting CYP-mediated sites of metabolism with neural networks. J Chem Inf. 2013;53(12):3373–3383.
39. Stewart JJP. MOPAC 2012, Stewart Computational Chemistry. Colorado Springs, CO, USA; 2012.
40. O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Chem Inf. 2011;3:33. doi:10.1186/1758-2946-3-33
41. Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2006;2(6):875–894. doi:10.1517/17425255.2.6.875
42. Smith DA, Jones BC. Speculations on the substrate structure–activity relationship (SSAR) of cytochrome P450 enzymes. Biochem Pharmacol. 1992;44:2089–2098. doi:10.1016/0006-2952(92)90333-E
43. Crewe HK, Lennard MS, Tucker GT, et al. The effect of selective serotonin re-uptake inhibitors on cytochrome P450 2D6 (CYP2D6) activity in human liver microsomes. 1992. Br J Clin Pharmacol. 2004;58:S744–S750. doi:10.1111/bcp.2004.58.issue-7
44. Osborne DW, Musakhanian J. Skin penetration and permeation properties of Transcutol®—neat or diluted mixtures. AAPS PharmSciTech. 2018;19(8):3512. doi:10.1208/s12249-018-1196-8
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.