")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 8
Potential serum and urine biomarkers in patients with lupus nephritis and the unsolved problems
Received 15 May 2016
Accepted for publication 7 July 2016
Published 19 September 2016 Volume 2016:8 Pages 81—91
DOI https://doi.org/10.2147/OARRR.S112829
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Song-Chou Hsieh,1 Chang-Youh Tsai,2 Chia-Li Yu3
1Department of Internal Medicine, National Taiwan University Hospital, National Taiwan University College of Medicine, 2Section of Allergy, Immunology & Rheumatology, Taipei Veterans General Hospital, 3Department of Internal Medicine, Institute of Molecular Medicine, National Taiwan University College of Medicine, Taipei, Taiwan
Abstract: Lupus nephritis (LN) is one of the most frequent and serious complications in the patients with systemic lupus erythematosus. Autoimmune-mediated inflammation in both renal glomerular and tubulointerstitial tissues is the major pathological finding of LN. In clinical practice, the elevated anti-dsDNA antibody titer concomitant with reduced complement C3 and C4 levels has become the predictive and disease-activity surrogate biomarkers in LN. However, more and more evidences suggest that autoantibodies other than anti-dsDNA antibodies, such as anti-nucleosome, anti-C1q, anti-C3b, anti-cardiolipin, anti-endothelial cell, anti-ribonuclear proteins, and anti-glomerular matrix (anti-actinin) antibodies, may also involve in LN. Researchers have demonstrated that the circulating preformed and in situ-formed immune complexes as well as the direct cytotoxic effects by those cross-reactive autoantibodies mediated kidney damage. On the other hand, many efforts had been made to find useful urine biomarkers for LN activity via measurement of immune-related mediators, surface-enhanced laser desorption/ionization time-of-flight mass spectrometry proteomic signature, and assessment of mRNA and exosomal-derived microRNA from urine sediment cell. Our group had also devoted to this field with some novel findings. In this review, we briefly discuss the possible mechanisms of LN and try to figure out the potential serum and urine biomarkers in LN. Finally, some of the unsolved problems in this field are discussed.
Keywords: anti-dsDNA antibodies, serum biomarkers, urine biomarkers, THP
Introduction
Systemic lupus erythematosus (SLE) is an archetype of systemic autoimmune disease characterized by the presence of diverse autoantibodies and self-reactive T lymphocytes that cause multiple tissue and organ damage. Lupus nephritis (LN) is one of the most important and devastating complications in patients with SLE. Despite remarkable progression in treatment, up to 25% of SLE patients progress to end-stage renal failure 10 years after the onset of renal damage.1 Nowadays, renal biopsy remains the gold standard for establishing the tissue diagnosis, prognosis, and guidance of the therapeutic decision in LN. However, renal biopsy cannot be routinely conducted serially, and the obtained small-size specimens are unable to reflect the global renal pathological status of the LN.2 In contrast, the clinically available routine tests such as measurement of 24-hour urine protein, the cell composition of urine sediments, and the fluctuation of serum anti-dsDNA antibodies concomitant with reduced complement C3 and C4 levels have long been applied in monitoring LN activity in daily practice.3,4 However, these clinical parameters lack enough sensitivity and specificity to reflect the real-time renal immunopathological activity and the extent of tissue damage. Particularly, these situations would be further confounded by the preexisting chronic inflammation. It is believed that urine is an ideal specimen for finding potential biomarkers of LN due to easy accessibility and can directly reflect the real-time status of the kidney inflammation and tissue damage. In addition, LN is considered an immune-mediated inflammation in both glomerular and tubulointerstitial tissues due to aberrant systemic and intrarenal immunity.5–9 Accordingly, a bunch of immune products including protein molecules, mRNAs, and microRNAs related to cytokines/chemokines/growth factors and their soluble receptors, adhesion molecules, enzymes, and activated endothelial/epithelial products have been successively discovered as surrogate urine biomarkers in LN.10–20 Unfortunately, none of these urine immune-related molecules has been validated hitherto in clinical practice.
Possible immunological mechanisms for lupus pathogenesis
It is conceivable that “breakdown of self-tolerance” is the hallmark of autoimmune diseases.21 The genetic and epigenetic predispositions would be the upstream causes for aberrant T and B cell signaling.22–28 As illustrated in Figure 1, the genetic predisposing loci for SLE include MHC-class II (HLA-DR2, HLA-DR3, HLA-DQ6, etc), MHC-class III (C4A null gene), and other extra-MHC loci that involve in immune complex (IC) process, signal transduction, cell apoptosis and its clearance, and the signaling pathways of Toll-like receptors, NOD-like receptors, and type I interferon expression.29–34 Of equal importance is the abnormal epigenetic regulations of cytokines/chemokines/growth factors including DNA methylation (DNA methyltransferase)/demethylation (activation-induced cytidine deaminase), and histone modifications (histone acetyl- and deacetyltransferase).35–39 Recently, deranged posttranscriptional regulation of mRNAs by microRNAs was found involved in LN.39–46 In addition, certain cell membrane defects (low phosphatidyl-serine content),47,48 low enzyme activity (low serum DNase 1 activity),49 aberrant T cell signaling,50,51 poor bioenergetics,52–54 excessive oxidative stress due to mitochondrial dysfunction,54–60 and exacerbated polymorphonuclear neutrophil (PMN) NETosis61–64 may also involve in lupus pathogenesis. These multiple abnormalities would increase cell apoptosis in patients with SLE. The low complements and C-reactive protein production may further impair necrotic cell debris clearance. It is conceivable that complement system is deeply involved in the pathogenesis of SLE in multiple ways. Complements are implicated in phagocytosis and clearance of apoptotic cells.65 Hereditary homogenous deficiency of the early components of the complement classical pathway, especially C1q, is strongly associated with susceptibility to SLE.66–68 Furthermore, antibodies against some complement components, C3b and C1q, are found part of autoantibody responses association with development of LN.69,70 As a result, increased nucleic acids released from excessive cell necrosis by delayed apoptotic cell clearance provide neoepitopes or act as pathogen-associated molecular pattern-like or danger-associated molecular pattern-like molecules to stimulate intracellular TLRs and NLRP-3 inflammasomes. These activations subsequently enhance production of proinflammatory cytokines IL-1β, IL-6, IL-8, IL-17, TNF-α, and type I interferon from innate immune system.71–74 Besides these endogenous defects in SLE, some environmental factors such as infections, chemicals, heavy metals, or drugs can boost, initiate and sustain the overt autoimmune reactions.75,76 Finally, autoantibodies from B cells and proinflammatory cytokines from T and innate immune cells elicit diverse tissue/organ damage.
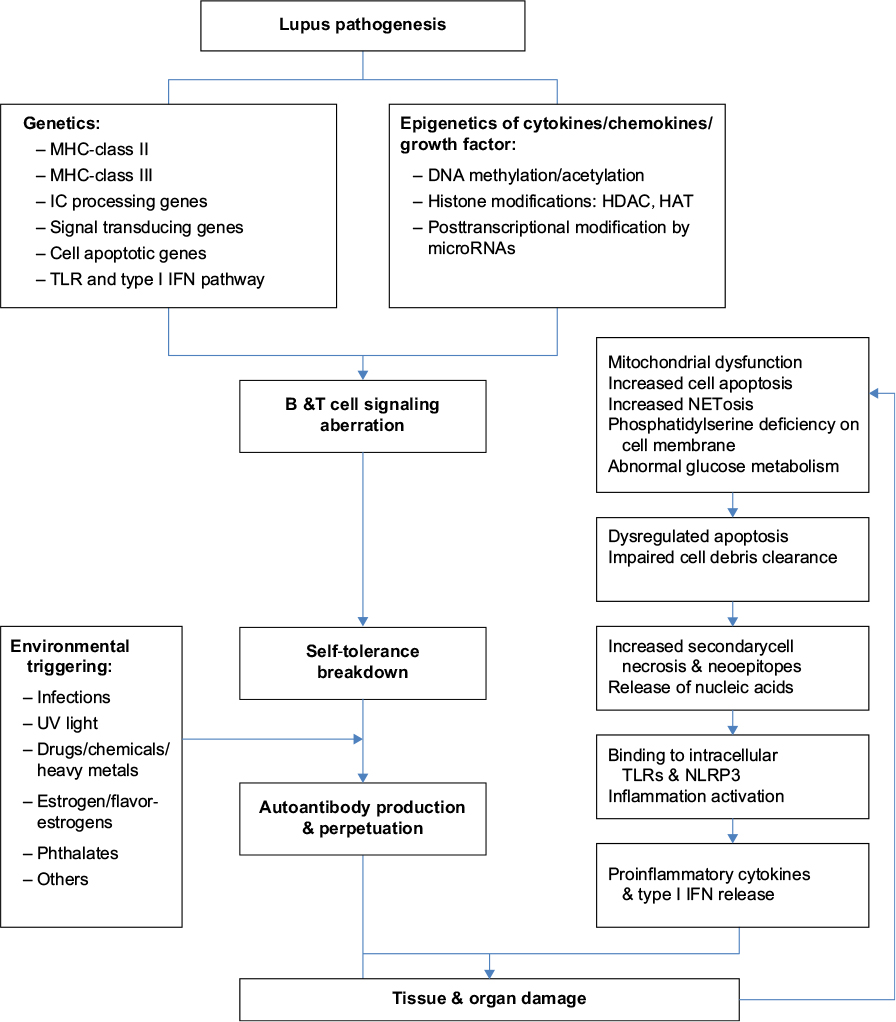 | Figure 1 The etiopathogenetic factors contributing to breakdown of self-tolerance and autoimmune reactions that cause tissue/organ damage in patients with systemic lupus erythematosus. Abbreviations: IC, immune complex; UV, ultraviolet. |
Potential serum biomarkers specific for tissue/organ damage in SLE
Classically, deposition of serum preformed antigen–antibody IC in glomerular basement membrane activates complements and inflammation that is traditionally regarded as an important extrarenal mechanism for LN. As listed in Table 1, recent studies reveal that intricate serum multiple autoantibodies and immune factors may involve in a particular tissue/organ damage. Alternatively, one specific autoantibody may cause multiorgan damage. It is worthy to notice that many pathological autoantibodies including anti-dsDNA, anti-cardiolipin, anti-ribosomal P, anti-SSA/Ro, anti-Sm, anti-endothelial cell, anti-epithelial cell, anti-glomerular matrix, and anti-glomerular basement membrane antibodies concomitant with reduced serum complement C3 and C4 levels have been found correlated with LN.77–79 It is believed that the cross-reactive property of these diverse pathological autoantibodies exhibits redundant and pleiotropic cytotoxic effects on different tissues/organs quite similar to those of cytokines/chemokines on cellular functions. Besides, elevated serum concentration of TNF-α/soluble TNF-RII, IL-6/IL-6R, and soluble VCAM-I has also been proved to be correlated with lupus nephropathy.20,80 By contrast, the presence of anti-SSB/La autoantibodies can ameliorate and prevent renal damage in patients with SLE by an unknown mechanism.
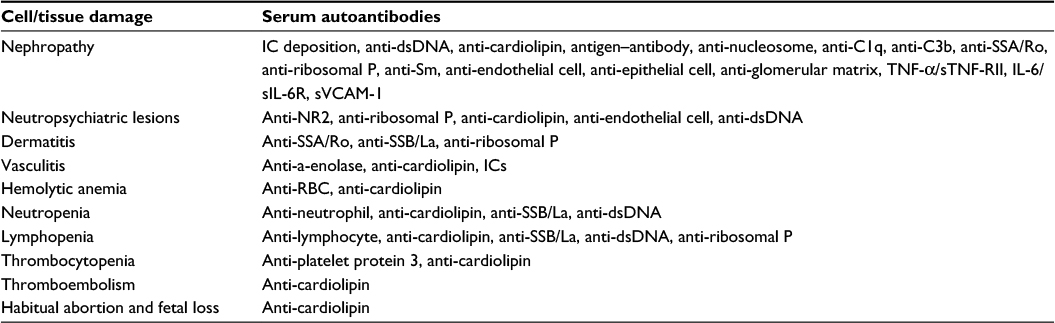 | Table 1 Potential serum autoantibody and protein biomarkers specific for tissue and cell damage in systemic lupus erythematosus Abbreviations: IC, immune complex; UV, ultraviolet; NR2, N-methyl-D-asparatate receptor subtype 2a/2b. |
Immunopathological mechanisms in LN
Renal pathology reveals that autoimmune reactions occur in both glomerular and tubulointerstitial tissues in LN.9 As shown in Table 2, immunofluorescence and electron microscopic studies have showed the deposition of immunoglobulins and complements on both sides of basement membrane of glomeruli. These intrarenal IC depositions may derive from either circulating preformed or in situ-formed ICs in glomeruli.9 Further investigations have explored that two mechanisms of cytotoxic autoantibodies can cause glomerular damage via charge–charge interaction81–83 and direct binding to cross-reactive glomerular autoantigens to form in situ IC.84–86 On the other hand, the infiltration of both innate immune cells (monocytes/macrophages/dendritic cells and PMNs) and adaptive immune cells (Th1, Th2, and Th17) into glomerular parenchymal tissues indicates cellular autoimmunity occurrence in lupus glomeruli (Table 2).
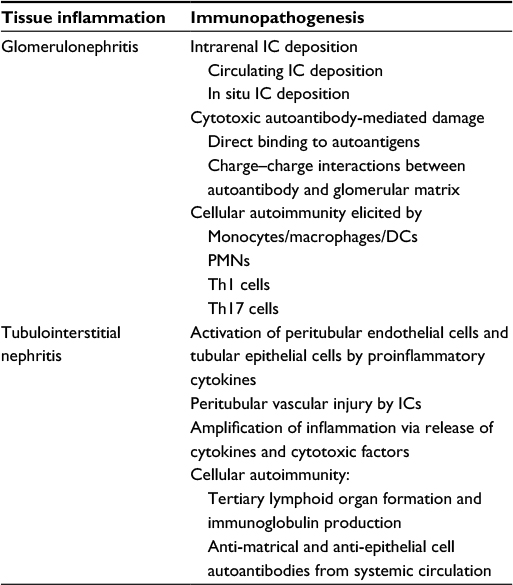 | Table 2 Immunopathology and possible immunopathogenesis of lupus nephritis Abbreviation: IC, immune complex. |
On the part of tubulointerstitial involvement in LN, several distinctive pathological findings are noted in Table 2: 1) activation of peritubular endothelial cells and tubular epithelial cells by proinflammatory cytokine stimulation, 2) peritubular vascular injury by IC deposition or cytotoxic autoantibodies, 3) amplification of inflammation via release of cytokines and cytotoxic factors by activated immune cells, 4) cellular autoimmunity manifested by tertiary lymphoid organ formation leading to immunoglobulin production, and 5) effects by anti-matrical and anti-epithelial cell autoantibodies from systemic circulation. In conclusion, lupus tubulointerstitial nephritis is caused mainly by autoantibodies against matrix, endothelial and epithelial cells, and proinflammatory cytokines from systemic circulation as well as local production.9 However, more investigations are required to confirm it.
Anti-dsDNA antibodies cross-react with different autoantigens other than dsDNA expressed on renal tissues to mediate cytostatic effects
Although a number of autoantibodies exist in SLE serum, anti-dsDNA antibody is considered the most important and unique one in mediating LN. Sun et al84,85 and Tsai et al87 demonstrated that mouse monocloncal antibody against dsDNA cross-reacted with acidic ribosomal P proteins P0, P1, and P2, expressed on the cell surface of glomerular mesangial cells, liver, spleen, brain tissues, and different blood cells. After binding with glomerular mesangial cells, the cytostatic effect was found correlated with the titer of anti-dsDNA. This finding is consistent with the observation that serum titer of anti-dsDNA can reflect the global lupus disease activity and particularly the LN. Further investigations suggest that both cross-reactivity and charge–charge interactions render anti-dsDNA antibodies binding with cross-reactive antigens and negatively charged molecules other than dsDNAs.82–86 As shown in Table 3, widely distributed molecules on glomerular mesangial cells, epithelial cells, endothelial cells, basement membranes, glomerular matrix, or extracellular proteins are the targets of anti-dsDNA. Despite these, the serum titer of anti-dsDNA is neither specific nor sensitive enough to predict, monitor, or prognosticate the severity of LN in the case of acute renal inflammation superimposing on chronic renal damage. Accordingly, searching for more reliable biomarkers is mandatory for clinical practice. Since cellular autoimmune reactions occur in LN (Table 2), the immune-related molecules excreted from inflamed renal tissues to urine may probably become the potential urine surrogate biomarkers in LN other than the serum autoantibodies or renal immunopathological findings.7–9
 | Table 3 Anti-dsDNA antibodies cross-react with different surface-expressed molecules or directly bind to negatively charged extracellular matrical proteins via electrostatic force Abbreviations: HSP70, human heat shock protien 70; β2GPI, β2-glycoprotien I. |
The excreted protein molecules in the urine of normal and LN patients
Normal human urine contains scant amount of albumin, certain proteolytic enzymes, corticosteroid metabolites, hormones and metabolites, modulators of urinary stone formation, and THP (which acts as a binding molecule for urinary tract microbial pathogens and different cytokines/chemokines) as shown in Table 4. Proteinuria, increased urine sediments with different cell components, and the presence of cellular/hyaline casts are considered the abnormal urine findings in LN. However, these abnormal findings are totally nonspecific and cannot be used for differentiating acute inflammation from acute inflammation superimposing on chronic renal damage. Among the excreted protein molecules in normal urine, it is noteworthy to realize that THP is synthesized only by the tubular epithelial cells in the thick ascending limb of Henle’s loop and proximal convoluted tubules. The daily excreted amount of THP in normal urine is ~50–150 mg/day.88,89 The major physiological function of THP is preventing the attachment of pathological microbes and then penetrating into urinary tract interstitium.90–93 Another important physiological function of THP relies on its capacity to nonspecifically bind with a variety of circulating protein molecules, cytokines, and chemokines in the kidneys.94,95 Decreased excretion or deranged functions of THP molecule in urine may reflect renal tubular cell damage by any cause.
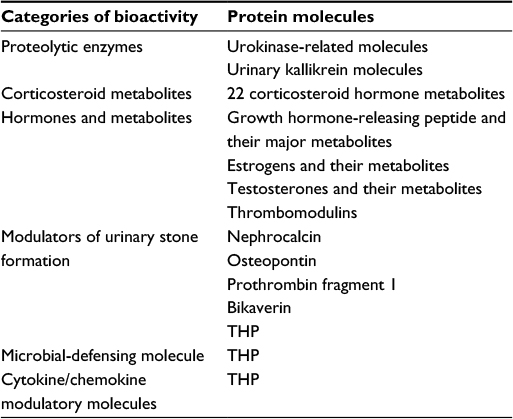 | Table 4 The excreted protein molecules in normal human urine |
Potential urine biomarkers in LN
From the clinical point of view, urine is considered the ideal source for finding the potential biomarkers in LN due to its easy accessibility and can directly reflect the real-time pathological status of kidneys. Taking into consideration the autoimmune nature of LN, many immune-related molecules are excreted into urine from the inflamed and damaged kidneys of LN. Recent investigations demonstrated that glomerular mesangial cells work as the intrarenal fixed tissue macrophage-like contractile cells and are the first affected cells in LN.96,97 Many authors have showed that not only innate and adaptive immune cells but also renal parenchymal cells are activated to proliferate by immunological reactions in both glomerular and tubulointerstitial tissues in LN.98,99 In chronic LN, tissue damage, tissue atrophy, and eventually fibrosis lead to endstage kidney disease.
Tsai et al10 reported increased excretion of soluble IL-2 receptors and free light-chain immunoglobulins in the urine of patients with active LN. These findings suggest that both T and B cells in LN are engaged in cell proliferation, differentiation, and maturation by autoimmune mechanism. Then, Tsai et al13 and Boenisch et al100 further demonstrated increased excretion of β2-microglobulin, IL-6, and IL-8 and decreased excretion of THP in the urine of patients with active LN. These results further suggest that the proinflammatory cytokines produced from activated monocytes/macrophages, neutrophils, and even renal parenchymal cells can reflect the degree of inflammation in LN. The increased urine b2-microglobulin excretion also reflects both immune cell activation and tissue destruction in LN. However, the most unique finding of this study indicates that decreased THP excretion can be considered a potential biomarker for tubulointerstitial inflammation/damage in LN. Later, Yang et al101 further reported increased 24-hour urine NGAL in active LN. NGAL, a 25 kDa protein molecule capable of binding and transporting small hydrophobic molecules (such as iron), is secreted by many cell populations including PMNs, macrophages, and epithelial and endothelial cells.102,103 Increased urine NGAL excretion was reported in both acute and chronic LN.104,105 As listed in Table 5, not only a number of serum protein molecules are excreted in the LN urine but also a vast amount of in situ-produced immune-related molecules in renal tissues such as proinflammatory/anti-inflammatory cytokines, chemokines, soluble cytokine/chemokine receptors, soluble adhesion molecules, and tissue growth factors have been successively discovered in LN urine. Besides, many authors have found that specific mRNAs and exosomal microRNAs are obtained from urinary sediment cells of patients with LN.14,15,17,46,106 The urine proteomic signature analysis by using surface-enhanced laser desorption/ionization time-of-flight mass spectrometry has also been successfully applied for urinary biomarker identification in LN.107 The enzyme-linked immunosorbent assay measurement of NAG was has been found as another biomarker of tubulointerstitial nephritis as well as THP.108–110 Unfortunately, none of these florid urine biomarkers has been validated to date in clinical application.
 | Table 5 Potential urinary biomarkers in lupus nephritis Abbreviation: SELDI-TOF-MS, surface-enhanced laser desorption/ionization time-of-flight mass spectrometry. |
Comparison of sensitivity and specificity of part of the potential serum and urine biomarkers in LN
To become an ideal and useful disease biomarker in clinical practice, both sensitivity and specificity should be high enough (>80%). Table 6 lists the comparison of sensitivity and specificity of different published serum80,111–113 and urine19,114–117 biomarkers related to LN. However, only serum anti-dsDNA antibodies fulfill the aforementioned criteria as a useful disease biomarker in patients with LN. Urine NGAL may probably fulfill the criteria of an LN disease activity biomarker, but it is not the routine test. Regarding miR-29c, only one report demonstrated in the literatures despite high sensitivity and specificity.115 Further investigation is necessary to confirm this possibility.
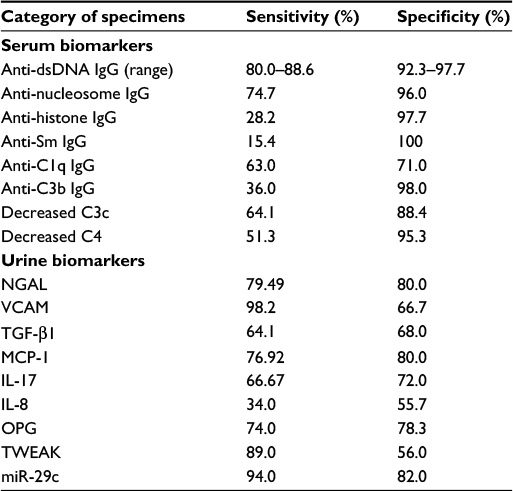 | Table 6 The reported sensitivity and specificity of some potential serum and urine biomarkers in lupus nephritis |
THP, a unique urine glycoprotein, acts as a nonspecific binder for modulating immune reaction in urinary system
THP was discovered by Tamm and Horsfall in 1950.92 This unique glycoprotein containing 25%–30% carbohydrate moieties is synthesized only in the thick ascending limb of Henle’s loop and the early distal convoluted tubule of the kidney.90,91 The major physiological function of THP is initially identified as a potent inhibition of viral hemagglutination in urine against New Castle, mumps, and influenza viruses for preventing pathogen attachment and then invasion into urinary interstitial tissues.92,93 Recent studies revealed that THP is a natural nonspecific binder of cytokines and chemokines with different binding affinities for modulating the fate of these immune factors in circulation or their in situ production in the kidneys.94,95 Table 7 summarizes the capacity of THP to bind with different protein molecules including serum proteins, neutrophil granular enzymes, and various cytokines/chemokines. In physiological and certain pathological conditions, the sugar moieties in THP side chains are altered, or the amount in urine excretion is reduced.118–123 Evidence suggests that mutations in THP gene may lead to congenital cystic kidney diseases or familial juvenile hyperuricemic nephropathy.124–127 Table 8 lists the alterations in THP glycosylation or defective production in different pathological conditions. Although we found reduced urine excretion of THP in active lupus tubulointerstitial nephritis,100 it is still not elucidated whether the molecular structure or the functions of THP are altered or not in LN. In addition, THP had been reported as a ligand for TLR4128 and scavenger receptors on macrophages/dendritic cells,129,130 and SREC1 on endothelial cells.130,131 The THP receptor binding activates these phagocytes to increase uptake and clearance of the engulfed bacteria and CpG DNAs. Accordingly, it is possible that reduced THP production in patients with LN would delay CpG DNAs clearance in the damaged kidney. This delayed clearance of CpG DNAs facilitates anti-nucleic acid antibody production that may further exacerbate kidney damage in SLE patients. However, the cause–effect relationships among less THP production, increased CpG DNA levels, facilitation of anti-dsDNA antibodies production, and occurrence of tubulointerstitial LN are not clear at the present time.
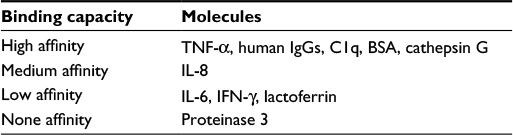 | Table 7 Capacity of THP to bind with different molecules |
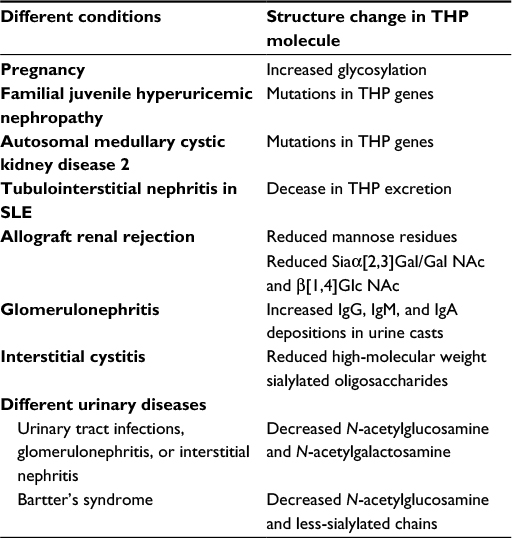 | Table 8 Changes of THP structure in physiology and different pathological conditions Abbreviation: SLE, systemic lupus erythematosus. |
Unsolved problems in identifying useful urine biomarkers in LN
To become the useful biomarkers for prediction, monitoring, and prognosis of a particular disease in clinical practice, one should consider the accessibility, specificity and sensitivity of the test, and the popularity of the specimen should be considered. The reported urine biomarkers in LN mostly are immune-related molecules produced by innate and adaptive immune reactions.131 The dilemma in selecting the ideal urine biomarkers of LN in clinical practice remains unless the following problems are solved:
- Which urine specimen is better for detecting the specific biomarker? Spot urine vs 24-hour urine.
- Which biomarker can accurately reflect the real-time kidney inflammation and tissue damage in acute vs chronic inflammation vs acute inflammation superimposing on chronic kidney damage.
- What is the specific urine biomarker for acute fulminate renal inflammation such as rapidly progressive glomerulonephritis?
- Does THP per se play a role in modulating balance of proinflammatory/anti-inflammatory cytokines in LN? Is the glycosylation of THP in LN altered or not?
- Is it necessary to reevaluate the roles of different nephritogenic autoantibodies in LN?
- How to design a laboratory screen strip for rapid evaluation of the renal inflammation by a drop of urine?
- What is the crucial role of glomerular mesangial cells in the pathogenesis of LN?
- What are the roles of Th17 cells and PMNs in initiating and sustaining the progression of LN?
- What is the role and acting mechanism of renal parenchymal cells in LN?
Conclusion
In clinical practice, biomarkers can be classified into predictive, diagnostic, and prognostic categories. SLE, a complex polygenic autoimmune disease, can be triggered by environmental factors leading to chronic immune dysregulation. LN is a serious complication with high mortality and morbidity in SLE patients. Currently reported serum or urine biomarkers for LN are no less than immune-related molecules that fail to specifically reflect the real-time disease activity of kidneys. Since the upstream etiological mechanisms for LN are the deranged genetic and epigenetic regulations, the genome-wise singlenucleotide polymorphism/mutant analysis in well-defined LN subpopulations with different disease severity will be the best strategy for searching the predictive and prognostic biomarkers in LN.
Disclosure
The authors declare no conflicts of interest in this work.
References
Mok CC. Therapeutic options for resistant lupus nephritis. Semin Arthritis Rheum. 2006;36(2):71–81. | ||
Schwartz N, Michaelson JS, Putterman C. Lipocalin-2, TWEAK, and other cytokines as urinary biomarkers for lupus nephritis. Ann N Y Acad Sci. 2007;1109(8):265–274. | ||
Balow JE. Clinical presentation and monitoring of lupus nephritis. Lupus. 2005;14(1):25–30. | ||
Enghard P, Riemekasten G. Immunology and the diagnosis of lupus nephritis. Lupus. 2009;18(4):287–290. | ||
Lech M, Anders HJ. The pathogenesis of lupus nephritis. J Am Soc Nephrol. 2013;24(9):1357–1366. | ||
Sarhsyan SA, Serkova NJ, Renner B, et al. Detection of glomerular complement C3 fragments by magnetic resonance imaging in murine lupus nephritis. Kidney Int. 2012;81(2):152–159. | ||
Hooke DH, Hancock WW, Gee DC, Kraft N, Atkins RC. Monoclonal antibody analysis of glomerular hypercellularity in human glomerulonephritis. Clin Nephrol. 1984;22(4):163–168. | ||
Nolasco FE, Cameron JS, Hartley B, Coelho A, Hildreth G, Beuben R. Intraglomerular T cells and monocytes in nephritis: study with monoclonal antibodies. Kidney Int. 1987;31(5):1160–1166. | ||
Lorenz G, Desai J, Anders HJ. Lupus nephritis: update on mechanisms of systemic autoimmunity and kidney immunopathology. Curr Opin Nephrol Hypertens. 2014;23(3):211–217. | ||
Tsai CY, Wu TH, Sun KH, Lin WM, Yu CL. Increased excretion of soluble interleukin 2 receptors and free light chain immunoglobulins in the urine of patients with active lupus nephritis. Ann Rheum Dis. 1992;51(2):168–172. | ||
Tesar V, Masek Z, Rychlik I, et al. Cytokine and adhesion molecules in renal vasculitis and lupus nephritis. Nephrol Dial Transplant. 1998;13(7):1662–1667. | ||
Tesar V, Jirsa M Jr, Zima T, et al. Soluble cytokine receptor in renal vaseculitis and lupus nephritis. Med Sci Monit. 2002;8(1):BR24–BR29. | ||
Tsai CY, Wu TH, Yu CL, Lu JY, Tsai YY. Increased excretion of beta 2-microglobulin, IL-6, and IL-8 and decreased excretion of Tamm-Horsfall glycoprotein in urine of patients with active lupus nephritis. Nephron. 2000;85(3):207–214. | ||
Chan RW, Tam LS, Li EK, et al. Inflammatory cytokine gene expression in the urinary sediment of patients with lupus nephritis. Arthritis Rheum. 2003;48(5):1326–1331. | ||
Chan RW, Lai FM, Li EK, et al. Expression of chemokine and fibrosing factor messenger RNA in the urinary sediment of patients with lupus nephritis. Arthritis Rheum. 2004;50(9):2882–2890. | ||
Rovin BH, Song H, Birmingham DJ, Herbert LA, Yu CY, Nagaraja HN. Urine chemokines: biomarkers of human systemic lupus erythematosus activity. Am Soc Nephrol. 2005;16(2):467–473. | ||
Arihingsanon Y, Phumesin P, Benjachat T, et al. Measurement of urinary chemokine and growth factor messenger RNAs: a noninvasive monitoring in lupus nephritis. Kidney Int. 2006;69(4):747–753. | ||
Li Y, Tucci M, Narain S, et al. Urinary biomarkers in lupus nephritis. Autoimmun Rev. 2006;5(6):383–388. | ||
Wu T, Xie C, Wang HW, et al. Elevated urinary VCAM-1, P-selection, soluble TNF receptor-1, and CXC chemokine ligand 16 in multiple murine lupus strains and human lupus nephritis. J Immunol. 2007;79(10):7166–7175. | ||
Mok CC. Biomarkers for lupus nephritis: a critical appraisal. J Biomed Biotechnol. 2010;2010:638413. | ||
Liao X, Reihl AM, Luo XM. Breakdown of immune tolerance in systemic lupus erythematosus by dendritic cells. J Immunol Res. 2016;2016:6269157. | ||
Costa-Reis P, Sullivan KE. Genetics and epigenetics of systemic lupus erythematosus. Curr Rheumatol Rep. 2013;15(9):369–377. | ||
Deng Y, Tsao BP. Advances in lupus genetics and epigenetics. Curr Opin Rheumatol. 2014;26(5):482–492. | ||
Niewold TB. Advances in lupus genetics. Curr Opin Rheumatol. 2015;27(5):440–447. | ||
Ceccarelli F, Perricone C, Borgianic P, et al. Genetic factors in systemic lupus erythematosus: contribution to disease phenotype. J Immunol Res. 2015;2015:745647. | ||
Ghodke-Puranik Y, Niewold TB. Immunogenetics of systemic lupus erythematosus: a comprehensive review. J Autoimmun. 2015;64(11):125–136. | ||
Zhang Z, Zhang R. Epigenetics in autoimmune diseases: pathogenesis and prospects for therapy. Autoimmun Rev. 2015;14(10):854–863. | ||
Relle M, Weinmann-Henke J, Scorletti E, Cavagna L, Schwarting A. Genetics and novel aspects of therapies in systemic lupus erythematosus. Autoimmun Rev. 2015;14(11):1005–1018. | ||
Blomberg S, Eloranta ML, Magnusson M, Alm GV, Ronnblom L. Expression of the markers BDCA-2 and BDCA-4 and production of interferon-alpha by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Rheum. 2003;48(9):2524–2532. | ||
Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005;52(5):1491–1503. | ||
Kwok SK, Lee JY, Park SH, et al. Dysfunctional interferon-alpha production by peripheral plasmacytoid dendritic cells upon Toll-like receptor-9 stimulation in patients with systemic lupus erythematosus. Arthritis Res Ther. 2008;10(2):R29. | ||
Tang Y, Luo X, Cui H, et al. MicroRNA-146A contributes to abnormal activation of the type 1 interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60(4):1065–1075. | ||
Karonitsch T, Feierl E, Steiner CW, et al. Activation of the interferon-gamma signaling pathway in systemic lupus erythematosus peripheral blood mononuclear cells. Arthritis Rheum. 2009;60(5):1463–1471. | ||
Wu L, Qin Y, Xia S, et al. Identification of cyclin-dependent kinase 1 as a novel regulator of type 1 interferon signaling in systemic lupus erythematosus. Arthritis Rheumatol. 2016;68(5):1222–1232. | ||
Sekigawa I, Okawa M, Ogasawara H, Kaneko H, Hishikawa T, Hashimoto H. DNA methylation in systemic lupus erythematosus. Lupus. 2003;12(2):78–85. | ||
Dai Y, Zhang L, Hu C, Zhang Y. Genome-wide analysis of histone H3 lysine 4 trimethylation by CHIP-chip in peripheral blood mononuclear cell of systemic lupus erythematosus patient. Clin Exp Rheumatol. 2010;28(2):158–168. | ||
Absher DM, Li X, Waite LL. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+T-cell populations. PLoS Genet. 2013;9(8):e1003678. | ||
Zhang Y, Zhao M, Sawalha AH, Richardson B, Lu Q. Impaired DNA methylation and its mechanisms in CD4(+)T cells of systemic lupus erythematosus. J Autoimmun. 2013;41(3):92–98. | ||
Zhao M, Liu S, Luo S, et al. DNA methylation and mRNA and microRNA expression of SLE CD4+T cells correlate with disease phenotype. J Autoimmun. 2014;54(11):127–136. | ||
Pan W, Zhu S, Yuan M, et al. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. 2010;184(12):6773–6781. | ||
Stagabis E, Bertsias G, Verginis P, et al. Identification of novel microRNA signatures links to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann Rheum Dis. 2011;70(8):1496–1506. | ||
Perez-Hernandez J, Forner MJ, Pinto C, Chaves FJ, Cortes R, Redon J. Increased urinary exosomal microRNAs in patients with systemic lupus erythematosus. PLoS One. 2015;10(9):e0138618. | ||
Wang W, Mou S, Wang L, et al. Up-regulation of serum miR-130b-3p level is associated with renal damage in early lupus nephritis. Sci Rep. 2015;5(8):12644. | ||
Duroux-Richard I, Cuenca J, Ponsolles C, et al. MicroRNA profiling of B cell subsets from systemic lupus erythematosus patients reveals promising novel biomarkers. Int J Mol Sci. 2015;16(8):16953–16965. | ||
Rai G, Rai R, Saeidian AH, Rai M. Microarray to deep sequencing: transcriptome and miRNA profiling to elucidate molecular pathway in systemic lupus erythematosus. Immunol Res. 2016;64(1):14–24. | ||
Abulaban KM, Fall N, Nurna R, et al. Relationship of cell-free urine microRNA with lupus nephritis children. Pediatr Rheumatol Online J. 2016;14(1):4–10. | ||
Niwa Y, Sakane T, Ozaki Y, Kanoh T, Taniguchi S. Phospholipid base exchange activity in the leukocyte membranes of patients with inflammatory disorders. Am J Pathol. 1987;127(2):317–326. | ||
Niwa Y, Miyachi Y, Sakane T, Kanoh T, Taniguchi S. Methyltransferase and phospholipase A2 activity in the cell membrane of neutrophils and lymphocytes from patients with Behcet’s disease, systemic lupus erythematosus and rheumatoid arthritis. Clin Chim Acta. 1998;174(1):1–14. | ||
Yeh TM, Chang HC, Liang CC, Wu JJ, Liu MF. Deoxyribonuclease inhibitory antibodies in systemic lupus erythematosus. J Biomed Sci. 2003;10(5):544–551. | ||
Fujii Y, Fujii K, Tanaka Y. Attempt to correct abnormal signal transduction in T lymphocyte from systemic lupus erythematosus patients. Autoimmun Rev. 2006;5(2):143–144. | ||
Moulton VR, Lo MS, Tsokos GC. Methods and protocols to study T cell signaling abnormalities in human systemic lupus erythematosus. Methods Mol Biol. 2012;900:25–60. | ||
Wu T, Xie C, Han J, et al. Metabolic disturbances associated with systemic lupus erythematosus. PLoS One. 2012;7(6):e37210. | ||
Li KJ, Wu CH, Hsieh SC, Lu MC, Tsai CY, Yu CL. Deranged bioenergetics and defective redox capacity in T lymphocytes and neutrophils are related to cellular dysfunction and increased oxidative stress in patients with active systemic lupus erythematosus. Clin Dev Immunol. 2012;2012:548516. | ||
Lee HT, Lin CS, Lee CS, Tsai CY, Wei YH. Increased 8-hydroxy-2-deoxyguanosine in plasma and decreased mRNA expression of human 8-oxoguanine DNA glycosylase 1, anti-oxidant enzymes, mitochondrial biogenesis-related proteins and glycolytic enzymes in leukocytes in patients with systemic lupus erythematosus. Clin Exp Immunol. 2014;176(1):66–77. | ||
Vyshkina T, Sylvester A, Sadiq S, et al. Association of common mitochondrial DNA variants with multiple sclerosis and systemic lupus erythematosus. Clin Immunol. 2008;129(1):31–35. | ||
Lee HT, Lin CS, Chen WS, Liao HT, Tsai CY, Wei YH. Leukocyte mitochondrial DNA alteration in systemic lupus erythematosus and its relevance to the susceptibility to nephritis. Int J Mol Sci. 2012;13(7):8853–8868. | ||
Peri A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol. 2013;9(11):674–686. | ||
Shah D, Nahajan N, Sah S, Nath SK, Paudyal B. Oxidative stress and its biomarkers in systemic lupus erythematosus. J Biomed Sci. 2014;17(3):21–23. | ||
Gheita TA, Kenawy SA. Measurement of malondialdehyde, glutathione and glutathione peroxidase in SLE patients. Methods Mol Biol. 2014;1134:193–199. | ||
Jafari SM, Salimi S, Nakhaee A, et al. Pro-oxidant-antioxidant balance in patients with systemic lupus erythematosus and its relationship with clinical and laboratory findings. Autoimmun Dis. 2016;2016:4343514. | ||
Knight JS, Kaplan MJ. Lupus neutrophils: “NET” gain in understanding lupus pathogenesis. Curr Opin Rheumatol. 2012;24(5):441–450. | ||
Yu Y, Su K. Neutrophil extracellular traps and systemic lupus erythematosus. J Clin Cell Immunol. 2013;4(4). doi: 10.4172/2155-9899.1000139. | ||
Smith CK, Vivekanandan-Giri A, Tang C, et al. Neutrophil extracellular trap-derived enzymes oxidize high density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. 2014;66(9):2532–2544. | ||
Lood C, Blanco LP, Purmalek MM, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–153. | ||
Kang YH, Urban BC, Sim RB, Kishore U. Human complement factor H molecules C1q-mediated phagocytosis of apoptotic cells. Immunobiology. 2012;217(4):455–464. | ||
Walport MJ, Davies KA, Botto M. C1q and systemic lupus erythematosus. Immunobiology. 1998;199(2):265–285. | ||
Robson MG, Cook HT, Botto M, et al. Accelerated nephrotoxic nephritis is exacerbated in C1q-deficient mice. J Immunol. 2001;166(11):6820–6828. | ||
Lewis MJ, Botto M. Complement deficiencies in human and animals: links to autoimmunity. Autoimmunity. 2006;39(5):367–378. | ||
Vasilev V, Noe R, Dragon-Durey MA, et al. Functional characterization of autoantibodies against complement component C3 in patients with lupus nephritis. J Biol Chem. 2015;290(42):25343–25355. | ||
Birmingham DJ, Bitter JE, Ndnkwe EG, et al. Relationship of circulating anti-C3b and C1q IgG to lupus nephritis and its flare. Clin J Am Soc Nephrol. 2016;11(1):47–53. | ||
Kahlenberg JM, Kaplan MJ. The inflammasome and lupus: another innate immune mechanism contributing to disease pathogenesis? Curr Opin Rheumatol. 2014;26(5):475–481. | ||
Yang Q, Yu C, Yang Z, et al. Deregulated NLRP3 and NLRP1 inflammasomes and their correlations with disease activity in systemic lupus erythematous. J Rheumatol. 2014;41(3):444–452. | ||
Yang CA, Huang ST, Chiang BL. Sex-dependent differential activation of NLRP3 and AIM2 inflammasomes in SLE macrophages. Rheumatology. 2015;54(2):324–331. | ||
Yang CA, Chiang BL. Inflammasomes and human autoimmunity: a comprehensive review. J Autoimmun. 2015;61(7):1–8. | ||
Samarkos M, Vaiopoulos G. The role of infections in the pathogenesis of autoimmune diseases. Curr Drug Targets Inflamm Allergy. 2005;4(1):99–103. | ||
Mu Q, Zhang H, Luo XH. SLE: another autoimmune disorder influenced by microbes and diet? Front Immunol. 2015;6(11):608–707. | ||
Deocharan B, Qing X, Lichauco J, Puttman C. Alpha-actinin is a cross-reactive renal target for pathogenic anti-DNA antibodies. J Immunol. 2002;168(6):3072–3078. | ||
Pesickova SS, Rysava R, Lenicek M, et al. Prognostic value of anti-CRP antibodies in lupus nephritis in long-term follow up. Arthritis Res Ther. 2015;17(1):371–377. | ||
Monori G, Quaglini S, Rachice A, et al. The value of a panel of autoantibodies for predicting the activity of lupus nephritis at time of renal biopsy. J Immunol Res. 2015;2015:106904. | ||
Yung S, Chan TM. Mechanism of kidney injury in lupus nephritis: the role of anti-dsDNA antibodies. Front Immunol. 2015;6:475. | ||
Li Z, Schettino EW, Padlan EA, Ikematsu H, Casali P. Structure-function analysis of a lupus anti-dsDNA autoantibody: central role of the heavy chain complementary-determining region 3 Arg in binding of double- and single-stranded DNA. Eur J Immunol. 2000;30(7):2015–2026. | ||
Guth AH, Zhang X, Smith D, Detanico T, Wysocki LJ. Chromatin specificity of anti-double-stranded DNA antibodies and a role for Arg residues in the third complementarity determining region of the heavy chain. J Immunol. 2003;171(11):6260–6266. | ||
Song YC, Sun GH, Lee TP, et al. Arginines in the CDR of anti-dsDNA autoantibodies facilitate cell internalization via electrostatic interactions. Eur J Immunol. 2008;38(11):3178–3190. | ||
Sun KH, Liu WT, Tsai CY, Tang SJ, Han SH, Yu CL. Anti-dsDNA antibodies cross-react with the ribosomal P proteins expressed on the surface of glomerular mesangial cells to exert cytostatic effect. Immunology. 1995;85(2):262–269. | ||
Sun KH, Liu WT, Tang SL, et al. The expression of acidic ribosomal phosphoproteins on the surface of different tissues in autoimmune and normal mice which are the target molecules for anti-dsDNA antibodies. Immunology. 1996;87(3):362–371. | ||
Li Y, Fang X, Li QZ. Biomarker profiling for lupus nephritis. Genomics Proteomics Bioinformatics. 2013;11:158–165. | ||
Tsai CY, Wu TH, Sun KH, Liao TS, Lin WM, Yu CL. Polyclonal IgG anti-dsDNA antibodies exert cytotoxic effect on cultured rat mesangial cells by binding to cell membrane and augmenting apoptosis. Scand J Rheumatol. 1993;22(4):162–171. | ||
Sikri KL, Foster CL, Mac Hugh N, Marshall RD. Localization of Tamm-Horsfall glycoprotein in the human kidney using immuno-fluorescence and immuno-electron microscopical techniques. J Anal. 1981;132(Pt 4):597–605. | ||
Hoyer JR, Seiler HW. Pathophysiology of Tamm-Horsfall protein. Kidney Int. 1979;16(3):279–289. | ||
Fletcher AP, Neuberger A, Ratcliffee WA. Tamm-Horsfall urinary glycoprotein: the chemical composition. Biochem J. 1970;120(2):417–424. | ||
Stevenson FK, Kent PW. Subunits of Tamm-Horsfall glycoprotein. Biochem J. 1970;116(5):791–796. | ||
Tamm I, Horsfall FL Jr. Characterization and separation of an inhibitor of viral hemagglutination present in urine. Proc Soc Exp Biol Med. 1950;74(1):106–108. | ||
Pak J, Pu Y, Zhong ZT, Hasty DL, Wu XR. Tamm-Horsfall protein binds to type I fimbriated Escherichia coli and prevents E. coli binding to uroplakin 1b receptors. J Biol Chem. 2001;276(13):9924–9930. | ||
Wu TH, Hsieh SC, Yu CY, Lee YF, Tsai CY, Yu CL. Intact protein core-structure is essential for protein binding, mononuclear cell proliferation and neutrophil phagocytosis-enhancing activities of normal human Tamm-Horsfall glycoprotein. Int Immunopharmacol. 2008;8(1):90–99. | ||
Wu TH, Li KJ, Siao SC, et al. The binding affinity and molecular basis of the structure-functional relationship between urinary Tamm-Horsfall glycoprotein and tumor neurosis factor-α. Molecules. 2012;17(10):11978–11989. | ||
Kurogi Y. Mesangial cell proliferation inhibitors for the treatment of proliferative glomerular disease. Med Res Rev. 2003;23(1):15–31. | ||
Scindia YM, Deshmukh US, Bagavant H. Mesangial pathology in glomerular disease: targets for therapeutic intervention. Adv Drug Deliv Rev. 2010;62(14):1337–1343. | ||
Striker LJ, Doi T, Elliot S, Striker GE. The contribution of glomerular mesangial cells to progressive glomerulosclerosis. Semin Nephrol. 1989;9(4):318–328. | ||
Sterzel RB, Schulze-Lohoff E, Weber M, Goodman SL. Interactions between glomerular mesangial cells, cytokines, and extracellular matrix. J Am Soc Nephrol. 1992;2(10 Suppl):S126–S131. | ||
Boenisch M, Hurst R, Huber S, Koehn J, Krapfenbauer K. Improved prognostic diagnosis of systemic lupus erythematosus in an early stage of disease by a combination of different predictive biomarkers identified by proteome analysis. EBMA J. 2014;5(1):5. | ||
Yang CC, Hsieh SC, Li KJ, et al. Urinary neutrophil gelatinase-associated lipocalin is a potent biomarker for renal damage in patients with systemic lupus erythematosus. J Biomed Biotechnol. 2012;2012:759313. | ||
Cowland JB, Borregaard N. Molecular characterization and pattern of tissue expression of the gene for neutrophil gelatinase-associated lipocalin from human. Genomics. 1997;45(1):17–23. | ||
Tong Z, Wu X, Ovcharenko D, Zhu J, Chen CS, Kehrer JP. Neutrophil gelatinase associated lipocalin as survival factor. Biochem J. 2005;391(Pt 2):441–448. | ||
Bolignano D, Donato V, Coppolino G, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a marker for kidney damage. Am J Kidney Dis. 2008;52(3):595–605. | ||
Rubinstein T, Pitashny M, Putterman C. The novel role of neutrophil gelatinase-B associated lipocalin (NGAL)/lipocalin-2 as a biomarker for lupus nephritis. Autoimmun Rev. 2008;7(3):229–234. | ||
Stypinska B, Paradowska-Gorycka A. Cytokines and microRNAs as candidate biomarkers for systemic lupus erythematosus. Int J Mol Sci. 2015;16(10):24194–24218. | ||
Suzuki M, Ross GF, Wiers K, et al. Identification of a urinary proteomic signature for lupus nephritis in children. Pediatr Nephrol. 2007;22(12):2047–2057. | ||
Erdener D, Aksu K, Bicer I, Doganavsargil E, Kutay FZ. Urinary N-acetyl-β-D-glucosaminidase (NAG) in lupus nephritis and rheumatoid arthritis. J Clin Lab Anal. 2005;19(4):172–176. | ||
Skalova S. The diagnostic role of urinary N-acetyl-β-D-glucosaminidase (NAG) activity in the detection of renal tubular impairment. Acta Medica (Hradec Kralove). 2005;48(2):75–80. | ||
Masuda T, Akimoto T, Ando Y, et al. Changes in the urinary excretion of β2-microglbulin (β2-MG) and N-acetyl-β-D-glucosaminidase (NAG) during treatment for lupus nephritis. Intern Med. 2008;47(4):287–290. | ||
Heidenreich U, Mayer G, Herold M, Klotz W, Al-Jazrawi KS, Lhotta K. Sensitivity and specificity of autoantibody tests in the differential diagnosis of lupus nephritis. Lupus. 2009;18(14):1276–1280. | ||
Torres-Salido MT, Cortes-Hernandez J, Vidal X, Pedrosa A, Vilardell-Tarres M, Ordi-Ros J. Neutrophil gelatinase-associated lipocalin as a biomarker for lupus nephritis. Nephrol Dial Transplant. 2014;29(9):1740–1749. | ||
Abdel Gawas ER, Mansour AI, Abdel Aziz YA, Soliman AF, Fawzy RM. Role of anti-nucleosome antibodies in the diagnosis of systemic lupus erythematosus and as a marker for lupus nephritis. Egypt J Immunol. 2014;21(1):57–65. | ||
El-Shehaby A, Darweesh H, EL-Khatib M, et al. Correlations of urinary biomarkers, TNF-like weak inducer of apoptosis (TWEAK), osteoprotegerin (OPG), monocyte chemoattractant protein-1 (MCP-1) and IL-8 with lupus nephritis. J Clin Immunol. 2011;31(5):848–856. | ||
Sole C, Cortes-Hernandez J, Felip ML, Vidal M, Ordi-Ros J. MiR-29c in urinary exosomes as a predictor of early renal fibrosis in lupus nephritis. Nephrol Dial Transplant. 2015;30(9):1488–1496. | ||
Wu C, Yang M, Zhou Z, et al. [Urinary soluble intercellular adhesion molecule-1 and vascular adhesion molecule-1: potential biomarkers of active lupus nephritis]. Nan Fang Yi Ke Da Xue Xue Bao. 2015;35(9):1272–1276. Chinese. | ||
Susianti H, Iriane VM, Dharmanata S, et al. Analysis of urinary TGFb1, MCP-1, NGAL, and IL-17 as biomarkers for lupus nephritis. Pathophysiology. 2015;22(1):65–71. | ||
Easton RL, Patankar MS, Clark GF, Morris HR, Dell A. Pregnancy associated changes in the glycosylation of Tamm-Horsfall glycoprotein. Expression of sialyl Lewis(x) sequences on core 2 type O-glycans derived from uromodulin. J Biol Chem. 2000;275(29):21928–21938. | ||
Fairley JK, Owen JE, Birch DF. Protein composition of urinary casts from healthy subjects and patients with glomerulonephritis. Br Med J (Clin Res Ed). 1983;287(6408):1838–1840. | ||
Olczak T, Olczak M, Kubicz A, Dulawa J, Kokot F. Composition of the sugar moiety of Tamm-Horsfall protein in patients with urinary diseases. Int J Clin Lab Res. 1999;29(2):68–74. | ||
Serafini-Cessi F, Malagolini N, Cavallone D. Tamm-Horsfall glycoprotein: biology and clinical relevance. Am J Kidney Dis. 2003;42(4):658–676. | ||
Wu TH, Hsieh SC, Li KJ, et al. Altered glycosylation of Tamm-Horsfall glycoprotein derived from renal allograft recipients lead to changes in the biological function. Transpl Immunol. 2008;18(3):237–245. | ||
Argade SP, Vanichsam C, Chenoweth M, Parsons CL. Abnormal glycosylation of Tamm-Horsfall protein in patients with interstitial cystitis. BJU Int. 2009;103(8):1085–1089. | ||
Iorember FN, Vehaskari VM. Uromodulin: old friend with new roles in health and disease. Pediatr Nephrol. 2014;29(7):1151–1158. | ||
Ekici AB, Hackenbeck T, Moriniere V, et al. Renal fibrosis is the common feature of autosomal dominant tubulointerstitial kidney diseases caused by mutations in mucin 1 or uromodulin. Kidney Int. 2014;86(3):589–599. | ||
Scolari F, Izzi C, Ghiggeri GM. Uromodulin: from monogenic to multifactorial diseases. Nephrol Dial Transplant. 2015;30(8):1250–1256. | ||
Venkat Raman G, Gast C, Marinaki A, Fairbanks L. From juvenile hyperuricemia to dysfunctional uromodulin: an ongoing metamorphosis. Pediatr Nephrol. Epub 2016 Feb 12. | ||
Saemann MD, Weichhart T, Zeyda M, et al. Tamm-Horsfall glycoprotein links innate immune cell activation with adaptive immunity via a Toll-like receptor-4-dependent mechanism. J Clin Invest. 2005;115(2):468–475. | ||
Peiser L, Mukhopadhyay S, Goeden S. Scavenger receptors in innate immunity. Curr Opin Immunol. 2002;14(1):123–128. | ||
Pfistershammer K, Klauser C, Leitner J, et al. Identification of the scavenger receptors SREC-1, Cla-1 (Sr-B1), and SR-A1 as cellular receptors for Tamm-Horsfall protein. J Leukoc Biol. 2008;83(1):131–138. | ||
Abulaban KM, Song H, Zhang X, et al. Predicting decline of kidney function in lupus nephritis using urine biomarkers. Lupus. 2016;25(9):1012–1018. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.