")
Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 12
Potential Roles of Glucagon-Like Peptide-1 and Its Analogues in Dementia Targeting Impaired Insulin Secretion and Neurodegeneration
Authors Mehan S , Bhalla S, Siddiqui EM, Sharma N, Shandilya A, Khan A
Received 8 November 2021
Accepted for publication 16 February 2022
Published 7 March 2022 Volume 2022:12 Pages 31—59
DOI https://doi.org/10.2147/DNND.S247153
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Thomas Müller
Sidharth Mehan,1 Sonalika Bhalla,1 Ehraz Mehmood Siddiqui,1 Nidhi Sharma,1 Ambika Shandilya,1 Andleeb Khan2
1Neuropharmacology Division, Department of Pharmacology, ISF College of Pharmacy, Moga, Punjab, India; 2Department of Pharmacology & Toxicology, College of Pharmacy, Jazan University, Jazan, Kingdom of Saudi Arabia
Correspondence: Sidharth Mehan, Neuropharmacology Division, Department of Pharmacology, ISF College of Pharmacy, Moga, 142001, Punjab, India, Tel +91 8059889909 ; +91 9461322911, Email [email protected]; [email protected]
Abstract: Dementia is a chronic, irreversible condition marked by memory loss, cognitive decline, and mental instability. It is clinically related to various progressive neurological diseases, including Parkinson’s disease, Alzheimer’s disease, and Huntington’s. The primary cause of neurological disorders is insulin desensitization, demyelination, oxidative stress, and neuroinflammation accompanied by various aberrant proteins such as amyloid-β deposits, Lewy bodies accumulation, tau formation leading to neurofibrillary tangles. Impaired insulin signaling is directly associated with amyloid-β and α-synuclein deposition, as well as specific signaling cascades involved in neurodegenerative diseases. Insulin dysfunction may initiate various intracellular signaling cascades, including phosphoinositide 3-kinase (PI3K), c-Jun N-terminal kinases (JNK), and mitogen-activated protein kinase (MAPK). Neuronal death, inflammation, neuronal excitation, mitochondrial malfunction, and protein deposition are all influenced by insulin. Recent research has focused on GLP-1 receptor agonists as a potential therapeutic target. They increase glucose-dependent insulin secretion and are beneficial in neurodegenerative diseases by reducing oxidative stress and cytokine production. They reduce the deposition of abnormal proteins by crossing the blood-brain barrier. The purpose of this article is to discuss the role of insulin dysfunction in the pathogenesis of neurological diseases, specifically dementia. Additionally, we reviewed the therapeutic target (GLP-1) and its receptor activators as a possible treatment of dementia.
Keywords: dementia, insulin signaling, neurodegeneration, GLP-1 activators
Introduction
Dementia is an irreversible, slowly progressive syndrome characterized by cognitive decline and memory impairment due to anatomical changes in the brain.1 It is primarily associated with Alzheimer’s disease, vascular dementia, frontotemporal dementia, Creutzfeldt-Jakob disease, mixed dementia, Lewy body dementia, Parkinson-related dementia, and Huntington’s disease.2 More than 55 million people worldwide have dementia, with 10 million new cases diagnosed each year. Alzheimer’s disease causes 60–70% of all dementia cases.3 Most surveys conducted globally found annual incidence rates of 10–15 per 100,000 people, and its prevalence could reach 2% in people aged 65 and up.4 Dementia is now the seventh leading cause of death worldwide and has become one of the most common causes of old age disability and dependency. Dementia is classified into three stages based on severity and progression: early, middle, and late stages. It typically begins with forgetfulness, but patients may develop difficulties with recognition and aggression as the disease progresses.1 Dementia has significant physical, psychological, social, and economic consequences that affect dementia patients, caregivers, families, and society.5 It affects memory, thinking, orientation, comprehension, calculation, learning capacity, language, and judgment does not affect consciousness. Changes in mood, emotional control, behaviour, or motivation are frequently associated with and occasionally precede cognitive function impairment.1
Dementia, associated with several neurodegenerative diseases such as Huntington’s disease (HD), causes genetic defects that decline a patient’s memory, thinking, and emotional state.6 Tau protein accumulation around brain cells causes the formation of neurofibrillary tangles via amyloid-β protein. Plaques may cause Alzheimer’s disease, or they may be a byproduct of the disease process and related to dementia.2 Additionally, when an abnormal infectious protein called a prion is expressed and accumulated in the brain at high levels, it causes irreversible damage to neuronal cells. This condition is known as Creutzfeldt-Jakob dementia, characterized by unusual anatomical changes in the brain, particularly in the frontal and temporal lobes.7 Frontotemporal dementia is marked by atrophy in these brain areas and is associated with personality, behaviour, and language.8 In Parkinson’s disease, α-syn abnormal accumulation is found in substantia nigra neurons, leading to cognitive problems like memory loss and dementia. These are the Lewy bodies’ deposits that lead to Lewy body dementia.7 Neurocognitive complications caused by gradual vascular changes are estimated to have a global impact on mental abilities.9
According to the literature, the pancreas is primarily responsible for insulin production that reaches all body areas. Previously, the brain was thought to be an organ unaffected by insulin, but recent research has revealed the presence of insulin and insulin receptors in certain brain regions.10 Insulin activity in neurons is dependent on its ability to cross the blood-brain barrier (BBB) via receptor-mediated transport and diffusion across the BBB.10 According to post-mortem studies, the brain’s hypothalamus region has the highest insulin concentration. The presence of insulin receptors in the brain suggests that insulin affects the CNS independent of glucose consumption.11 Insulin regulates the brain-liver axis centrally by acting on specific brain stem nuclei containing pre-autonomic nuclei responsible for sympathetic and parasympathetic regulation of liver function. Furthermore, the neuronal insulin level is determined by the rate at which it crosses the blood-brain barrier (BBB) via a saturable (receptor-mediated) transport and diffusion mechanism located outside of the BBB.10
Insulin primarily affects peripheral tissues like muscle, adipose tissue, and liver. Furthermore, it has been discovered that peripheral insulin can reach the brain via a receptor-mediated transport pathway found in the blood-brain barrier.12 When its receptor is activated, it causes phosphorylation and activation of the AKT and ERK pathways, resulting in the mobilization of the glucose transporter 4 (GLUT4) to the cell membrane and increased glucose uptake by these cells. However, it predominantly stimulates the insulin-insensitive glucose transporters GLUT1 (in astrocytes and blood-brain barrier endothelial cells) and GLUT3 (in glial cells and neurons).13 Moreover, insulin and its receptors have also been implicated in neurite outgrowth and axon guidance via the P13K/AKT signaling pathway activation. It plays a critical role in learning and memory, reproduction, neuron development and maturation, and energy balance via controlling the brain’s neuromodulator, neurotrophic, and neuroprotective activities.14 It strongly influences the hypothalamus, inhibiting glucose release from the liver. Insulin binds to its receptors in many brain regions throughout the central nervous system, including the cortex, olfactory bulb, hypothalamus, cerebellum, and hippocampus (CNS).15 Previously, it was considered that irregularities in the molecular signaling pathway of insulin played a crucial role in Alzheimer’s disease etiology. As a result, intracellular pathways like PI3K, JNK, and MAPK are activated. Along with it, insulin interacts with its receptor, causing autophosphorylation of key tyrosine residues in the beta subunit, some of which are detected by the Src homology two domain of the PI3K regulatory subunit p85 (SH2 domain)16 (Figure 1).
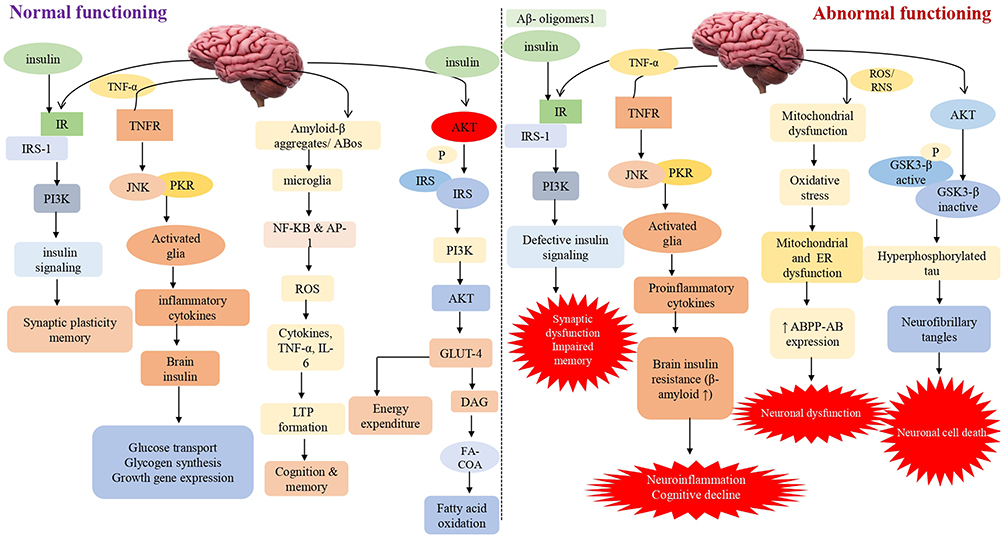 |
Figure 1 Impaired insulin signaling/ secretion involved in the progression of dementia This schematic diagram depicts both normal and abnormal insulin signaling in the brain. Insulin activates signaling pathways such as AKT, PI3K, JNK, NF-KB, GLUT4 and GSK3. Synaptic plasticity and memory are both linked to the PI3K pathway. The PI3K pathway is linked to synaptic plasticity and memory. As a result, aberrant insulin binding can cause neuroinflammation by activating proinflammatory cytokines and lowering cognitive capacities via JNK and NF-KB. Long-term memory loss is caused by oxidative stress, mitochondrial dysfunction, and inflammation caused by GSK3 and its hyperphosphorylation. It also elevates the expression of caspase-3 and 9, which ultimately leads to neuronal cell death. |
GLP-1 (glucagon-like peptide-1) is a gastrointestinal hormone secreted by intestinal L cells in response to food intake. GLP-1 regulates blood glucose levels by increasing glucose-dependent insulin secretion, reducing glucagon secretion, delaying gastric emptying, and decreasing food intake.17 The GLP-1 receptor (GLP-1R) in type 2 diabetes is responsible for activating adenylyl cyclase (AC) to produce cAMP and protein kinase A (PKA) to phosphorylate and activate CREB.18 It expresses a nuclear transcription factor (CREB) in a constitutively active state that regulates the expression of genes involved in neuronal survival and function. These drugs were designed, and FDA approved for diabetes treatment in 2005 and discoveries of their neurotrophic properties were made in the early 2000s.19 In a previous study, GLP-1 agonists are beneficial in CNS diseases, including Parkinson’s disease. Astrocyte cultured AGEs produce oxidative stress and cytokines. Astrocytes were considered because they produce GLP-1, GLP-1R and pro-inflammatory cytokines like TNF, which contribute to neuronal damage and the formation of Alzheimer’s disease lesions.17
According to our findings, a GLP-1 receptor analogue (Liraglutide) inhibited AGE-mediated ROS generation, inflammatory cytokine secretion, caspase activation, and cell death by involving GLP-1R signaling via cAMP/PKA.18 The GLP-1 analogue cross the blood-brain barrier and exerts its anti-inflammatory and neuroprotective action. Research studies suggest these agents reduces amyloid plaque deposition in a mouse model of Alzheimer’s disease and are currently being tested in clinical trials in Alzheimer’s patients. Earlier, we investigated the effect of GLP-1 activators in diabetes, but recent studies suggest its therapeutic effect in treating neurodegenerative diseases.20
Throughout this article, we will explore the physiological and pathological effects of dementia and other neurodegenerative diseases on insulin signaling. Research into GLP-1 activators and their potential role in various neurodegenerative disorders has revealed some remarkable outcomes.
Impaired Insulin Signaling in the Progression of Dementia and Related Neurocomplications
Role of Amyloid-Beta Deposition
Dementia was first described in the second decade of the twentieth century but was not associated with high incidence or mortality rates. Most professionals are familiar with dementia symptoms and the functional decline that coincides with the restrictions placed on these patients’ daily activities. According to various studies, Alzheimer’s disease is the most prevalent type of dementia.21 The most frequently discovered neuropathological entities are beta-amyloid plaques, extracellular complexes of the beta-amyloid (Aβ) peptide, and intracellular neurofibrillary tangles, composed of hyperphosphorylated tau protein.22 Considering the symptoms, most healthcare professionals are unaware of the connection between dementia, Alzheimer’s disease, insulin receptor function, and signaling pathway dysfunction. Dementia is classified as sporadic or familial. The most common type appears to be sporadic dementia, caused by environmental and genetic factors. The APO E gene is one of the most similar intrinsic risk factors. On the other hand, the rare and sporadic form of AD is caused by a mutation in the amyloid precursor protein (APP), presenilin 1 (PSEN1), or presenilin 2 (PSEN2) (PSEN2).23 Alternate APP cleavage, which results in insoluble amyloid-beta (Aβ) fibrils, is one of the pathogenic processes underlying dementia. Synaptic signaling gets disrupted when Aβ fibrils infiltrate into synaptic clefts. When oligo polymerizes and clumps, plaques are formed.24,25 A40 and A42 are two types of Aβ polymers that play a significant role in the pathogenesis of dementia. Accumulation of A40/A42 disrupts ion channel function, alters calcium homeostasis, increases mitochondrial oxidative stress, impairs energy metabolism, and disrupts glucose homeostasis, all of which contribute to neuronal integrity degradation and cell death26 (Figure 2).
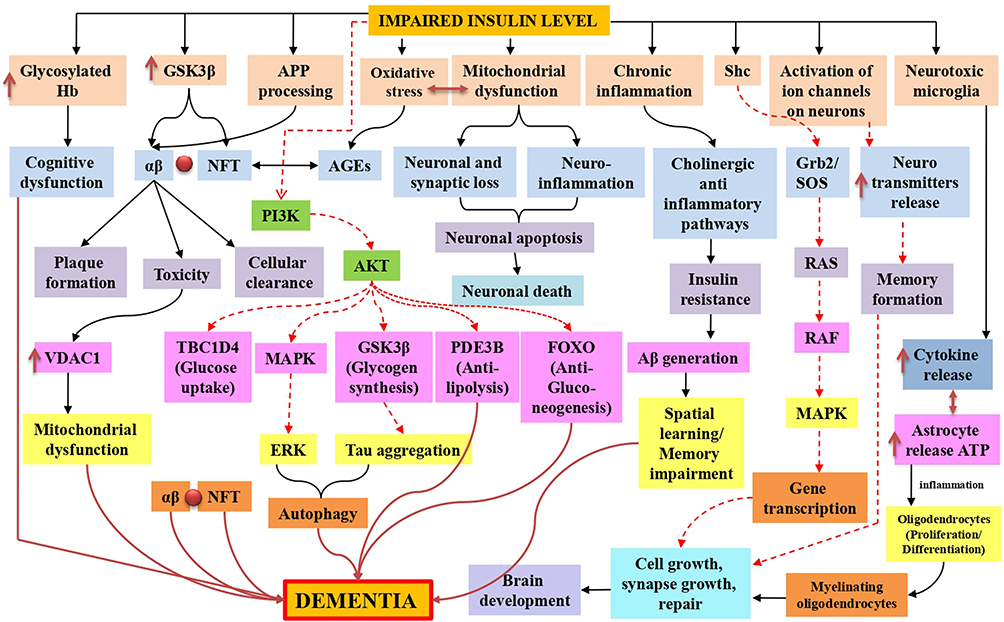 |
Figure 2 Insulin resistance in dementia is represented schematically by mitochondrial dysfunction, which leads to synaptic damage and neurodegeneration, glycosylated haemoglobin in intoxicated cognitive function due to failure in glucose transport for neurons, oxidative stress-induced amyloid beta and phosphorylated tau lineups via advanced glycation end products, inflammation caused by mitochondrial dysfunction and toxicities of amyloid-beta and glycation end products. Additionally, in both normal and impaired insulin signaling, Tau hyperphosphorylation was also observed, stimulates AKT, and inhibits tau hyperphosphorylation by inactivating GSK-3. Furthermore, the TOR pathway was also active for cellular growth and kept autophagy at a low level for cellular function. Black normal flow lines represent normal working mechanism; dotted red lines show inhibition of the pathway, and normal dark red flow lines show the overall cause of dementia. |
Another pathogenic mechanism underlying dementia progression is hyperphosphorylation of Tau proteins. Tau protein aids in the organisation of microtubules, making them more stable. Hyperphosphorylated tau proteins form paired helical filaments, a critical component of neurofibrillary tangles.27–29
Memory loss is the primary symptom for the majority of people. Memory loss can occur either before or concurrently with the onset of cognitive impairment. Later in the disease’s course, patients may develop executive dysfunction and visuospatial impairment. These flaws create covertly.30 Dementia is a type of memory loss distinct from other types of memory loss. In most patients, semantic and episodic memory is impaired, particularly recollection of recent events, dependent on the hippocampus and other medial temporal lobe components. The subcortical system is involved in procedural memory and motor coordination and is not affected until late in the disease.31–34
Insulin receptors are abundant in astrocytes and neuronal synapses of the cerebral cortex, hippocampus, olfactory bulb, hypothalamus, septum, and amygdala. In this case, insulin signaling is involved in synaptogenesis and synaptic remodelling. In addition to its immediate effects on the specific receptor, insulin has additional neuromodulatory effects that affect intellectual ability.35 Insulin, for instance, insulin may enhance the generation of long-term excitability, which is considered a necessary biological substrate for memory consolidation, by modulating the expression of the N-methyl-D-aspartate receptor on cell membranes. Insulin also regulates acetylcholine and norepinephrine, which is associated with memory.36 Insulin may also enhance cortical cerebral glucose metabolism in areas of the brain associated with cognition and memory. Given insulin’s multiple effects on specific brain regions, including those in the medial temporal lobe associated with memory and learning, it’s unsurprising that evidence linking insulin to cognitive performance is progressing.37 Preclinical and clinical studies state that insulin treatment improves cognition.37,38 By contrast, Aβ regulates insulin transmission in the brain. In preparations of mouse hippocampus slices, soluble Aβ adheres to the insulin receptor, impairing its signaling ability and inducing long-term potentiation.38 These adverse effects were avoided by insulin pretreatment to the tissues before exposure to Aβ.
Synthetic soluble Aβ oligomers cause synaptic spine atrophy in primary hippocampal cultured neurons by inhibiting plasma membrane insulin receptors. Additionally, pretreatment with insulin was effective in reversing this.39 Synaptotoxic effects are another mechanism by which insulin and Aβ may work together to modify disease. The absence of synapses is the first structural flaw that becomes apparent. Insulin inhibits the attachment of Aβ to synapses, thereby preserving synaptic stability. Soluble oligomeric forms of Aβ are synaptotoxic. Insulin also inhibits oligomer formation, which appears to have significant protective effects; one operational consequence seems to be protection against Aβ-induced loss of long term potentiation stability.40 A process of synaptic remodelling suspected to be involved in memory consolidation.40,41
Tau Hyperphosphorylation in Dementia
Intraneuronal neurofibrillary tangles (NFTs) are composed of helical strands of hyperphosphorylated tau protein-coupled together. Hyperphosphorylation of tau results in aggregation, which results in the production of NFTs and neurodegeneration.42 The primary risk factor for dementia is increasing age.43 Other common disorders associated with ageing include diabetes mellitus, impaired glucose tolerance, insulin resistance, and a decline in pancreatic endocrine activity and insulin secretion. Insulin signaling may also play a role in regulating longevity and brain ageing. Compared to age-matched controls, patients with memory impairment had lower CSF insulin, impaired insulin-like signal transduction, insufficient brain insulin and insulin-like growth factor I expression and function,44 and impaired glucose metabolism. Insulin may also act as a neurotrophic and regulatory peptide in the human brain by regulating tau phosphorylation in neuronal cells.45 (Figure 2). Returning the mice to normothermia partially alleviated this massive hyperphosphorylation, indicating that it is primarily caused by hypothermia and not just associated with it. On the other hand, insulin treatment of diabetic rats completely restored tau phosphorylation to normal levels, demonstrating that a lack of insulin resulted in moderate hyperphosphorylation.46 It has been postulated that tau hyperphosphorylation occurred via kinase and/or phosphatase dysregulation, which causes aggregation and hinders attachment to microtubules, disrupting the microtubule network in dementia.47 In vitro, hyperphosphorylation of human tau causes microtubule disruption and tau aggregation leading to dementia-like symptoms.48,49
Genetic Alteration in Dementia
Insulin resistance and insulin disintegrating enzymes are increasingly implicated in the pathogenesis of dementia, associated neuronal cytoskeletal abnormalities and Aβ deposits in the brain. Reports demonstrating reduced brain growth and increased tau phosphorylation in animals lacking either the insulin receptor substrate-2 or the neuronal insulin receptor gene have driven this fresh wave of curiosity.50,54 Insulin, IGF-I, and IGF-II peptides and receptors are all highly expressed in the brain. Dementia is associated with significant decreases in insulin and IGF-I mRNA expression and downregulation of related receptors.51, 52 Furthermore, it was discovered that genes encoding oxidative phosphorylation system subunits were differentially expressed in dementia patients, implying that mtDNA abnormalities may be responsible for the phenotypic variability.53 The correlation established between mtDNA mutations and the degree of nucleic acid injury in the cytoplasm,54 implying a possible link between neuronal oxidative damage and chromosomal defects, supports the idea that aberrant insulin contributes to genetic abnormalities.55 Dementia has also been linked to several hereditary factors. The APO E e4 allele expression has been shown to increase the risk of developing the disease. APO E is a polymorphic protein encoded by three alleles on chromosome 19. It exists in three isoforms (apo e2, e3, and e4) that appears to increase the risk of sporadic or familial late-onset diabetes.
Moreover, mutations in the APP and presenilin (PS1 and PS2) genes have been linked with a small percentage of dementia cases.56 In the recent decade, extensive cell biology research has suggested that mutations in these genes cause dementia-like illness via a shared biological mechanism, culminating in aberrant Amyloid precursor protein (APP) metabolism.56 Recently, transgenic mice were used to assess the effects of the mutations listed above in vivo. Excessive expression of mutant dementia-related proteins in animals exhibits many neuropathological and behavioural characteristics as in humans. Transgenic animals with mutations in APP, PS1, and PS2 genes have been developed. These mutations are associated with the stimulation of astrocytes and microglial cells in the hippocampus and cerebral cortex.57 Additionally, animal models of Alzheimer’s disease-related dementia have been used to develop and test drugs that decrease Ab 42 peptide57 concentrations in the brain, thereby advancing the development of novel therapeutic techniques.58
Cholinergic Dysfunction in Dementia
Dementia is strongly intertwined with degenerative and functional diseases of the central nervous system, resulting in various neurochemical, neurophysiological, and anatomical abnormalities.59 Dementia patients were found to have lower test scores for memory, cognitive flexibility, fast data processing, and psychomotor efficacy.60 The link between poor insulin and the CNS greatly increases the risk of memory loss.59 Impaired glucose and energy metabolism61 hyperglycemia,62 reduced insulin sensitivity,44 insulin resistance and insulin/insulin receptor dysfunction,63 changes in hippocampal synaptic plasticity and transmission,64 increased advanced glycation end products (AGE) that cause memory dysfunction in dementia-like conditions,65 Poor insulin caused memory loss in rats, which was linked to increased cholinergic activity and oxidative stress, leading to dementia.66 Insulin resistance causes cognitive impairment in type 2 diabetes. A PPAR-c agonist has recently been linked to memory loss. Glucotoxicity is reported to be exerted by increased intracellular glucose oxidation in neurons.67 Insulin resistance causes cognitive issues in type 2 diabetes.
A PPAR-γ agonist has recently been linked to memory loss. It was also observed that glucotoxicity is caused by increased intracellular glucose oxidation in neurons.67 Oxidative damage to the rat hippocampus synapse has been linked to cognitive deficits.68 Cholinergic neurotransmission is the central process governing mental performance. In the nucleus basalis magnocellularis, cholinergic basal forebrain neurons innervate the cerebral cortex, amygdaloid complex, and hippocampus, required for cognitive and memory consolidation.70 The enzyme ChE is involved in one of the most critical pathways necessary for proper cholinergic function. Also, the hippocampus and cortex are innervated by cholinergic projections.68 Cholinergic abnormalities result in insufficient processing in the hippocampus area.
Furthermore, it has been discovered that cholinergic transmission is linked to recollection and cue perception.69 Due to the critical role of acetylcholinesterase in inhibiting acetylcholine-induced responses, using an acetylcholinesterase inhibitor may aid in memory.70 Numerous studies have linked increased ChE activity in the brain to cognitive deficits.69
Cognitive stimulation requires cholinergic stimulation from the nucleus basalis of Meynert (NBM).69,70 The deterioration of the NBM in dementia contributes to the cognitive impairment observed in dementia patients. The activity of choline acetyltransferase (ChAT) is significantly decreased in patients with the same severity of dementia, primarily in the neocortex. In other parts of the brain, there is less atrophy and neuronal dysfunction but more cholinergic loss.71,72 These findings are critical for elucidating the etiology of dementia and developing the most effective pharmacologic treatment strategy.
Neurotransmitters Dysfunction in Dementia
Alzheimer’s disease and senile dementia are associated with abnormal neurotransmitter levels in the brain.73 Because neurotransmitters have both excitatory and inhibitory effects in the brain, the proper balance of messenger types may be as critical as the total number of transmitters. Alzheimer’s disease and senile dementia are associated with abnormal neurotransmitter levels in the brain.73 Since neurotransmitters have both excitatory and inhibitory effects in the brain, the proper balance of messenger types may be as critical as the total number of transmitters. With increasing age, people naturally experience reduced transmitter generation and discharge, contributing to the mild forgetfulness associated with old age. Pathologies linked to inadequate insulin have been proposed as a probable cause of cognitive impairment due to neuron loss and long-term brain damage caused.74,75 Glucose synthesises neurotransmitters and serves as the brain’s primary energy source.76 Glucose is required for the amino-acid cycle, cortical neurons, and synaptosomes to function correctly.
In the brain, mitochondrial function is associated with synthesising and eliminating neurotransmitters. Because glutamate is the primary excitatory neurotransmitter in the brain77 and a direct precursor of the inhibitory neurotransmitter g-aminobutyric acid, mitochondria are connected to glutamate regulation (GABA).78 Glutamate is required for synaptic communication, but it must be cleared from the extracellular space as soon as active synapses release it to avoid unnecessary stimulation of its receptors, which could result in excitotoxicity.77 Recent evidence suggests that glutamate may contribute to the pathogenesis of various neurodegenerative diseases, including dementia and Alzheimer’s disease.79 Aspartate, like glutamate, is a major excitatory neurotransmitter in the brain that plays a critical role in converting brain energy.80 Insulin-induced hypoglycemia increases extracellular aspartate levels while decreasing glutamate and glutamine levels throughout the brain and in specific brain regions such as the hippocampus and striatum. Others discovered a significant increase in aspartate’s extracellular level in the hippocampus and striatum, as well as a minor increase in glutamate and GABA. Due to their excitotoxic properties at higher extracellular concentrations, glutamate and aspartate have been implicated in the neuronal injury that occurs in the brain following a hypoglycemic episode. Numerous studies on neurotransmitter abnormalities in dementia have been conducted over the last twenty years, resulting in a better understanding of the relationship between neurotransmitter irregularities and dementia.81–83 According to studies, decreased neurotransmitter activity does not always correlate with the severity of Lewy body pathology.84 Neurotransmitter synthesis modifications occur early in the disease process in several brain areas affected by dementia, implying that axonal transport fidelity is compromised before the apparent neuronal loss.
Numerous muscarinic receptors regulate the excitation and suppression of acetylcholine (ACh) discharge by the presynaptic neuron. Notably, the presence of multiple muscarinic receptor subtypes on the post-synaptic cell can result in a complicated reflex in the post-synaptic neuron in response to the presynaptic cell’s release of Ach. Nicotine has a long history of being associated with memory and learning, as well as having a beneficial effect on attentiveness and alertness.84–87 When nicotinic receptors on presynaptic cells are stimulated, ACh and other neurotransmitters involved in cognition and behaviour are released.88 The 4ß2 receptors are the most abundant high-affinity receptors in the central nervous system (CNS), whereas the homomeric seven receptors have a low affinity. Based on high-affinity nicotine binding in the brain, individuals with dementia were compared.84,89 Nicotine adherence is significantly reduced in the substantia nigra. Dementia patients demonstrated significant neuronal loss. This indicates that, as is the case in other brain areas, the loss of cholinergic activity in the substantia nigra precedes the degradation and loss of neurons in dementia. Interestingly, the parahippocampal gyrus region with the highest LB density did not correspond to the region with tremendous loss of nicotinic receptor in dementia. Individuals with significant receptor loss in the edentulous granular area may experience cognitive impairment.85,90
Neuronal Apoptosis in Dementia
Apoptosis occurs in the absence of insulin and as a long-term consequence of diabetes. It has been linked to type 1 diabetes,91 diabetic retinopathy,92 and nephropathy.93 On the other hand, apoptosis has been associated with various neurological disorders, including dementia, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis (ALS).94 (Figure 2). Numerous studies have discovered neuronal death in the hippocampus of type 1 diabetic rats, which is associated with cognitive deficits and previous IGF system disturbances. Only in 8-month diabetic rats was the unique ladder configuration of genomic DNA discovered. It was associated with an increased Bax/Bcl-x ratio, L caspase-3 activity, and hippocampal neuronal death. These defects were not observed in 2-month diabetic rats, indicating that neuronal death occurs only following an extended period of insulin deficiency. Ischemia with intracytoplasmic calcium deposition and mitochondrial dysfunction,95 receptor-mediated or non-receptor-mediated processes, or decreased IGF activity are all examples of biological mechanisms that could result in neural cell apoptosis.96 Purkinje cell death in pcd mice is associated with decreased IGF-I expression.97 Reduced IGF activity may contribute to CNS disorders in streptozotocin-induced diabetic rats.98 According to a recent study, dementia patients’ vulnerable hippocampus areas have decreased IGF-I, IGF-IR, and IR activity.100 In addition, studies demonstrate that insulin resistance occurs before the onset of apoptosis.99
Cerebral blood vessel dysfunction compromises the function of capillaries, arterioles, and venules, as well as myelinated axons. Disruption of myelin and axons results in abnormalities in the white matter, which contributes to the pathophysiology of vascular dementia.100 Arterial blockade and lesions in the cortex, basal ganglia, pons, lacunar infarctions in the white matter, disrupted tight endothelial junctions, and BBB degradation appears to be significant pathologic features that impair normal cerebroarterial blood flow.101 Ependymal leaking, disturbed interstitial fluid flow, vasogenic perivenular white matter enlargement and abscesses, and A dispersion and accumulation along perivascular gaps contribute to increased apoptosis and vascularisation resistance in dementia. Ischemic infarction and vascular dementia are caused by a lack of oxygen in specific brain areas, resulting in axonal damage, apoptotic neuronal cell death, and ultimately neurodegeneration.102 Indeed, cortical neuronal apoptosis is known to occur following white matter axonal loss caused by the breakdown of afferent nerve cell synapses or regressive neuronal degradation.103 There is a strong correlation between neuronal apoptosis and pyramidal neuron death, as evidenced by the increased caspase-3 expression upon stimulation. It results in discrete subcortical protuberances in the layers III–IV of the cerebral cortex and proximal white matter metastases and neuropathies in subcortical fibres.104 Cortical apoptosis has also been associated with vascular injury, as evidenced by vacuolar atrophy, dark pyknotic nuclei, and micro-infarcts. It damaged the white matter, which eventually manifested as memory loss and dementia.105 Neurodegeneration is caused by two processes: neural apoptosis and vascular injury. They are defined by program rather than incremental cell death. Anti-apoptotic drugs were critical in masking the functionalities or progression of neuronal apoptosis and vascular damage in vascular dementia, particularly in subcortical ischemic vascular disorders, where anti-apoptotic drugs were essential in masking the functionalities or progression of neuronal apoptosis and endothelial damage.103,106
Neuronal Excitation Dysfunction
In response to an increase in extracellular glucose, a subset of neurons in the brain known as “glucose-excited” neurons depolarize and increase their activation level, potentially leading to poor insulin signaling.107 Glucose has been shown to activate POMC neurons in the hypothalamic arcuate nucleus.110 Stimulation is thought to cause cognitive deficits such as dementia and Alzheimer’s disease by ATP-induced shutdown of KATP channels in the plasma membrane.107–109 Various studies have established a critical role for neurovascular dysfunction in vascular cognitive decline and other types of dementia, including Alzheimer’s disease.111 Ischemia and neurodegenerative disease co-occurring have been shown to alter the development of clinical symptoms of dementia, implying reciprocal interactions between them.112 Cerebrovascular diseases, dementia, and Alzheimer’s disease all share common risk factors, suggesting that vascular factors may play a role in the pathogenesis of the disease.113,114 The neurovascular unit comprises endothelial cells, myocytes, neurons and their processes, astrocytes, and perivascular cells. This unit modulates cerebral blood flow, blood-brain barrier (BBB) exchange, immunological monitoring, trophic assistance, and hemostatic balance. The morphological and physiological interrelationships of the neurovascular unit are critical for brain homeostasis, as evidenced by the neurovascular unit’s severe impairment in dementia.113,115
Alterations in insulin-associated inflammation, aberrant endoplasmic reticulum-stress modulation, excitotoxicity, and lipotoxicity have all been identified as possible biochemical mechanisms of insulin resistance. The direct consequences of insulin resistance in peripheral tissues such as the brain, liver, skeletal muscle, and adipose tissue have received considerable attention.116 Numerous extra-hypothalamic nuclei, such as sensory and cognitive units in the hindbrain117,118 and autonomic, parasympathetic, and sympathetic preganglionic brainstem neurons, are also included in the control centres of the brain.119,120
Severe distortions of hormonal and nutritional signaling in pro-opiomelanocortin, neuropeptide Y, and agouti-related peptide neurons (ARPN) influence insulin metabolic activity in peripheral tissues directly through their ability to incorporate ancillary signals and adjust their electrical activity in response to resource consumption.121,122 After abruptly increasing insulin levels in the circulation using cell-specific excitatory methods, it was discovered that acute stimulation of ARPN neurons impairs systemic glucose tolerance and insulin intolerance. Following ARPN neuron excitation, transient stimulation decreased sympathetic nerve function supporting brown adipose tissues, and decreased -adrenergic activity resulted in systemic insulin resistance, which may be associated with dementia-like symptoms.123
Failure of Mitochondria Leading to Impaired Energy in Dementia
Numerous abnormalities associated with insulin resistance have been proposed to contribute to the detrimental effects of severe diabetes-related dementia.124,125 Additionally, increased renin-angiotensin system activity is associated with the development of insulin resistance, a fundamental characteristic of the cardiometabolic syndrome that is increasingly believed to be a nexus linking the syndrome.126,127 Numerous abnormalities associated with insulin resistance have been proposed to contribute to the detrimental effects of severe diabetes-related dementia.124,125 Additionally, increased renin-angiotensin system activity is associated with the development of insulin resistance, a fundamental characteristic of the cardiometabolic syndrome that is increasingly believed to be a nexus linking the syndrome.126,127 Metabolic stability predominately depends on mitochondria, which metabolises nutrients and generates ATP and heat to maintain energy balance. Mitochondrial dysfunction is defined by a decreased energy production ratio (ATP synthesis) to respiration due to an energy intake-expenditure imbalance.128,129 Exercise, food, ageing, and anxiety affect mitochondrial performance and insulin sensitivity.130,131 Significantly, mitochondrial dysfunction has been associated with insulin resistance in skeletal muscle,132–134 and other tissues such as the liver, fat, heart, blood vessels, and pancreas.135–137 Insulin resistance, caused partly by mitochondrial dysfunction, may thus contribute to the pathophysiology of a wide variety of chronic diseases. Mitochondrial function can be influenced by genetic factors, oxidative stress, mitochondrial biogenesis, and ageing, contributing to insulin resistance and other pathological conditions (Figure 2).
Mitochondrial dysfunction is frequently observed in patients suffering from dementia.138,139 Deficiencies in mitochondrial oxidative phosphorylation, specifically complex IV, have been found in brain tissue, platelets, and cell lines (cytochrome c oxidase or COX).140 Moreover, the AAβ peptide has been shown to enter the mitochondria and inhibit COX and L-3-hydroxy acyl-coenzyme Q dehydrogenase. The inhibition of these enzymes accelerates the formation of reactive oxygen species (ROS) in mitochondria.141,142 Similarly, it has been discovered that a neuron-specific protease cleaves the ApoE 4 protein, with the resulting peptides entering the mitochondrion, inhibiting oxidative phosphorylation and thus increasing ROS generation, resulting in dementia-like symptoms.143,144
Progressive Dementia Due to Microglial Overactivation
Insulin resistance is a significant risk factor for cognitive impairment and dementia in the periphery.145 Insulin affects the performance and lifespan of neurons.146 As a result, it is critical for learning and memory and regulating mechanisms associated with ageing.146 Physiological insulin contributes to the removal of beta-amyloid (A) protein, the primary cause of neurodegeneration and memory loss.147 Microglia cells, immune cells in the central nervous system, are activated by AAβ protein and cluster around or penetrate neuritic plaques, releasing neurotoxins and aggravating the neuronal injury. In particular, elevated brain insulin levels result in the formation of Aβ plaque, which stimulates microglia, produces numerous inflammatory mediators, and eventually results in memory loss and dementia.147 As a result, medications that inhibit activated microglia and Aβ protein synthesis may be potential treatments options for individuals with cognitive impairment associated with insulin resistance (Figure 2).
When advanced glycated end products (AGEs) bind to their receptors, reactive oxygen species (ROS) are generated, resulting in cell death.148 Yang et al discovered that diabetic mice with cognitive impairment had increased activation of AGEs receptors in neurons and glia.149 It has been suggested that GLP-1 analogues inhibit the expression of the AGE receptor, which could be used to mitigate the effect of AGEs on neuronal cell death.150 This study examined microglia, a critical regulator of the Aβ protein in the cell. Pioglitazone and exenatide treatment inhibited microglia activity in the hippocampus. Others validated microglia’s protective role in Alzheimer’s disease, claiming that Aβ protein stimulates microglia, causing them to release pro-inflammatory cytokines and reactive oxygen species, thereby inducing apoptosis and neuronal injury.151,152 Astrogliosis and microglial stimulation demonstrate that neuroinflammation is a pathogenic feature associated with plaque and tangle formation.153,154 In ICV-STZ-treated rats, astrocytes and microglia are activated as well.155 As a result, targeting the brain insulin signaling pathway as a potential therapeutic option for dementia treatment has emerged.156,157 Insulin intranasal administration for six weeks restored spatial memory in ICV-STZ mice, decreased tau pathology and microglial activation, and increased hippocampus neurogenesis. These findings establish a molecular basis for treating dementia with intranasal insulin. Additionally, as evidenced by astrogliosis and microglia, dementia is associated with neuroinflammation. Prolonged stimulation of these glial cells results in the production of inflammatory mediators, which contribute to the development of dementia.158 Due to impaired insulin signaling, activated microglia and astrocytes are visible in post-mortem brains, both preclinically and medically.159
Inflammation of Neural Cells in Dementia
Inflammation in the brain can result in an insulin-resistant state linked to the etiology and pathogenesis of dementia.160,161 Other common dementia symptoms include protein complexes,162 mitochondrial dysregulation,163 oxidative stress,164 neuroinflammation,165 brain cholinergic impairments,166 and adult neurogenesis disorder.167 The subventricular zone of the lateral ventricles and the subgranular zone of the hippocampus’s dentate gyrus are two neurogenic regions in the central nervous system where the resident neural stem and progenitor cells proliferate. Numerous endogenous and environmental factors can affect the adult neurogenesis process. Growth factors and neurotrophins, such as brain-derived neurotrophic factor (BDNF), stimulate neurogenesis by enhancing the differentiation, development, and lifespan of replicating NSC/NPC.
On the other hand, it has been demonstrated that microglial stimulation and the production of proinflammatory cytokines (eg, interleukin-1, IL-6, and tumour necrosis factor) inhibit neurogenesis by inhibiting NSC/NPC development. Prohibition results in cognitive deficits, as evidenced by difficulties with spatial and recognition memory and learning168 (Figure 2).
STZ dementia type models have shown an increase in neuroinflammation characterised by reactive gliosis and an increase in proinflammatory markers linked to cognitive impairment.155,169–171 In other studies, neurogenesis has been associated with oxidative stress172 and amyloid pathology.173 Acute and chronic neuroinflammation can impair several stages of adult neurogenesis in the mammalian brain, including proliferation, differentiation, and survival of newborn neurons. Adult neurogenesis is damaged in various neurodegenerative diseases, with neuroinflammation as a characteristic symptom.174 STZ dementia type models have shown an increase in neuroinflammation characterised by reactive gliosis and an increase in proinflammatory markers linked to cognitive impairment.155,169–171 In other studies, neurogenesis has been associated with oxidative stress172 and amyloid pathology.173 Acute and chronic neuroinflammation can impair several stages of adult neurogenesis in the mammalian brain, including proliferation, differentiation, and survival of newborn neurons. Adult neurogenesis is damaged in various neurodegenerative diseases, with neuroinflammation as a characteristic symptom.174 An investigation found a significant inflammatory response in the periventricular regions and the hippocampus of STZ-ICV rats, evidenced by increased immunoreactivity for Iba-1 (microglial marker) and GFAP (astrocytes markers). An extreme inflammatory response was found to be triggered by STZ, which is thought to be involved in inhibiting neuron growth and differentiation.175
After conducting a systematic review of the literature, another research identified 28 studies that used in vivo neuroimaging to assess one or more indicators of neuroinflammation in dementia patients. Most research used PET imaging to detect increased neuroinflammation in at least one neuroanatomical location in dementia patients, most frequently AD.176 For summary object recognition, another study employed oral extended curcumin therapy. The examined curcumin doses resulted in a slight improvement in neuroinflammation, resulting in a modest restoration of hippocampal and subventricular neurogenesis. These findings suggest that curcumin may improve object recognition recall in dementia patients. Additional research is needed to determine the efficacy of neuroinflammation treatment in dementia, particularly in models of the disease’s early stages.175
Role of Oxidative Stress in Dysregulated Insulin Linked Dementia
Dementia has been linked to insulin receptors in the brain, which are found in abundance in the hippocampus and other parts of the cerebral cortex and are crucial for memory.63 The degeneration of brain insulin receptors have been linked to the aetiology of degenerative brain diseases such as dementia. Desensitization of the insulin receptor has resulted in decreased energy metabolism and acetyl CoA production, resulting in cholinergic insufficiency.62 Increased generation of free radicals may result from neurons’ insufficient energy consumption.177,178 Reduced glucose metabolism in the brain reduces the release of acetylcholine from the cortex, which may cause cognitive decline.179 STZ treatment in the rat brain results in severe abnormalities in metabolic pathways regulated by the IR signaling cascade.180 Studies have shown that STZ injection causes low cerebral energy metabolism, which results in memory impairment, as evidenced by decreased choline acetyltransferase (ChAT) activity in the hippocampus and enhanced acetyl-CoA synthesis in the rat brain, which results in a cholinergic deficiency.181 To improve cholinergic function and thus alleviate symptoms of dementia, it is essential to inhibit the AChE enzyme. Increasing evidence has shown that free radicals can cause neuronal degeneration, emphasising the need for antioxidants to treat neurodegenerative illnesses. Poor insulin signaling has been linked to increased oxidative stress182 and mitochondrial dysfunction in brain cells, which have been linked to dementia (Figure 2).183–185
IRs have been shown to play a role in cognition and interact with the cholinergic system and oxidative stress. In a rat dementia model, the researchers used ICV STZ to estimate IR protein levels, AChE action as a marker of cholinergic activity, and oxidative stress markers (MDA and GSH) in various parts of the rat brain. Acyl CoA, glutamate, and ATP are essential for proper neuron construction and performance when brain insulin regulates glycogen breakdown via IR. Acetyl CoA is a critical component of acetylcholine synthesis and plays a role in memory and learning. The alkylating activities of STZ metabolites produce reactive oxygen species (ROS), which cause oxidative stress and DNA damage.179
Oxidative stress markers, oxidised lipids and proteins, and ROS production all contribute to neurodegenerative diseases such as dementia and Alzheimer’s disease. In the AD brain and patients with cognitive decline, enzymes involved in metabolic pathways such as glycolysis and the Krebs cycle are oxidised, implying that these changes occur early in dementia.186–188 Cerebral glucose metabolism is disrupted due to these changes, resulting in decreased ATP synthesis, which contributes to neuronal dysfunction, synaptic loss, and general neurotoxicity.186,190
The early stages of the disease have been linked to decreased mitochondrial enzymatic activity and oxidative stress.189 This could imply that the oxidative mechanisms resulting in ROS production occur before any significant accumulation of Aβ. However, in vitro studies show that the more neurotoxic Aβ oligomers can reduce cytochrome-oxidase function by increasing ROS production.190 However, in dementia, Aβ-mediated mitochondrial dysfunction and ROS generation may initiate a vicious cycle, resulting in increased insulin signaling problems. Finally, numerous evidence and studies have established that altered insulin signaling may play a role in the malfunction and progression of dementia and related neurological problems (Table 1).
 |  |  |  |  |  |
Table 1 Impaired Insulin Signaling Dysfunction in the Progression of Dementia and Related Neurological Complications |
Potential Role of GLP-1 Signaling Activators in the Protection of Various Neurodegenerative Dysfunctions Associated with Dementia
A study discovered that liraglutide, a GLP-1 analogue, protected rats’ brains from amyloid protein-induced impairments in spatial learning and memory, as well as a deficiency of late-phase long-term potentiation, and activated the cAMP signaling pathway.218 After eight weeks of systemic liraglutide treatment, a consistent study demonstrated repair of cerebral and systemic microvascular architecture in seven-month-old APP/PS1 transgenic mice.219 The clinical trial was conducted to determine whether long-acting intranasal insulin detemir, given for 21 days, improves cognition or daily functioning in individuals with amnestic mild cognitive impairment (MCI) or early-stage Alzheimer’s dementia. Daily treatment with 40 IU intranasal detemir was associated with improved verbal memory in people with MCI and AD (APOE-£4 allele carriers) and with improved visuospatial and verbal working memory in all trial participants.220
A randomised, placebo-controlled, double-blind clinical study discovered that six months of treatment with liraglutide (a GLP-1 analogue) significantly reduces the formation of amyloid β plaques and the deterioration of the brain glucose metabolism in patients with Alzheimer’s disease.221
Additionally, another study examined the effect of sitagliptin (a DPP-4 inhibitor that inhibits glucagon secretion and the degradation of GLP-1) on cognitive functions in elderly diabetic patients with and without cognitive impairment. The authors concluded that six months of sitagliptin treatment might improve cognitive function in elderly diabetic patients with or without Alzheimer’s disease.222
A case-control study conducted in Germany discovered a link between antihyperglycemic medications and the incidence of dementia in type 2 diabetes patients. The study discovered that treatment with glitazone (a PPAR-agonist that activates the GLP-1 receptor) was associated with a decreased risk of dementia (odds ratio = 0.80). Additionally, metformin monotherapy (odds ratio = 0.71) or combination treatment with sulfonylurea derivatives (odds ratio = 0.90) was associated with a decreased risk of developing dementia later.223
A subsequent population-based cohort study in Korea examined the incidence of dementia in geriatric type 2 diabetes patients treated with dipeptidyl peptidase 4 (DPP-4) inhibitors and sulfonylurea. The study found that taking a DPP-4 inhibitor reduces the risk of dementia in the elderly (hazard ratio = 0.66) compared to taking sulphonylurea. Additionally, when compared to sulphonylurea use (hazard ratio = 0.66), DPP-4 inhibitor use was associated with a significantly lower risk of Alzheimer’s disease (hazard ratio = 0.64) and a lower risk of vascular dementia (hazard ratio = 0.66).216
Moreover, a population-based longitudinal study found that pioglitazone, when used as a second-line glucose-lowering medication after metformin, may have a protective effect on the risk of dementia in type 2 diabetic study participants.224
A comparable retrospective cohort study examined the effect of metformin (a direct AMPK-dependent GLP-1 activator) use on the risk of dementia in type 2 diabetes patients using the Taiwanese National Health Insurance reimbursement database. Overall hazard ratios indicated a significantly lower risk of dementia in type 2 diabetes patients who had used metformin (especially for more than two years) in either the unmatched or matched group.225
Exendin-4 (a long-acting GLP-1 agonist) was investigated to improve cerebrovascular integrity and vascular-induced cognitive impairment and dementia (VCID) in diabetic mice when given at a dose of 30ng/kg/day for four weeks.226
All these shreds of evidence point to GLP-1 activators/analogues as a promising strategy for dementia treatment. Additionally, mechanistic research on the safety and efficacy of GLP-1 target activators may be used to develop a preventative therapy for dementia and related neurodegenerative dysfunctions. Thus, we encourage readers to understand better the cellular and molecular pathways underlying neurodegenerative diseases to establish a novel disease-modifying therapeutic intervention. GLP-1 signaling activators/analogues/agonists are neuroprotective in preventing a variety of dementia-related neurodegenerative abnormalities, as summarised in Table 2.
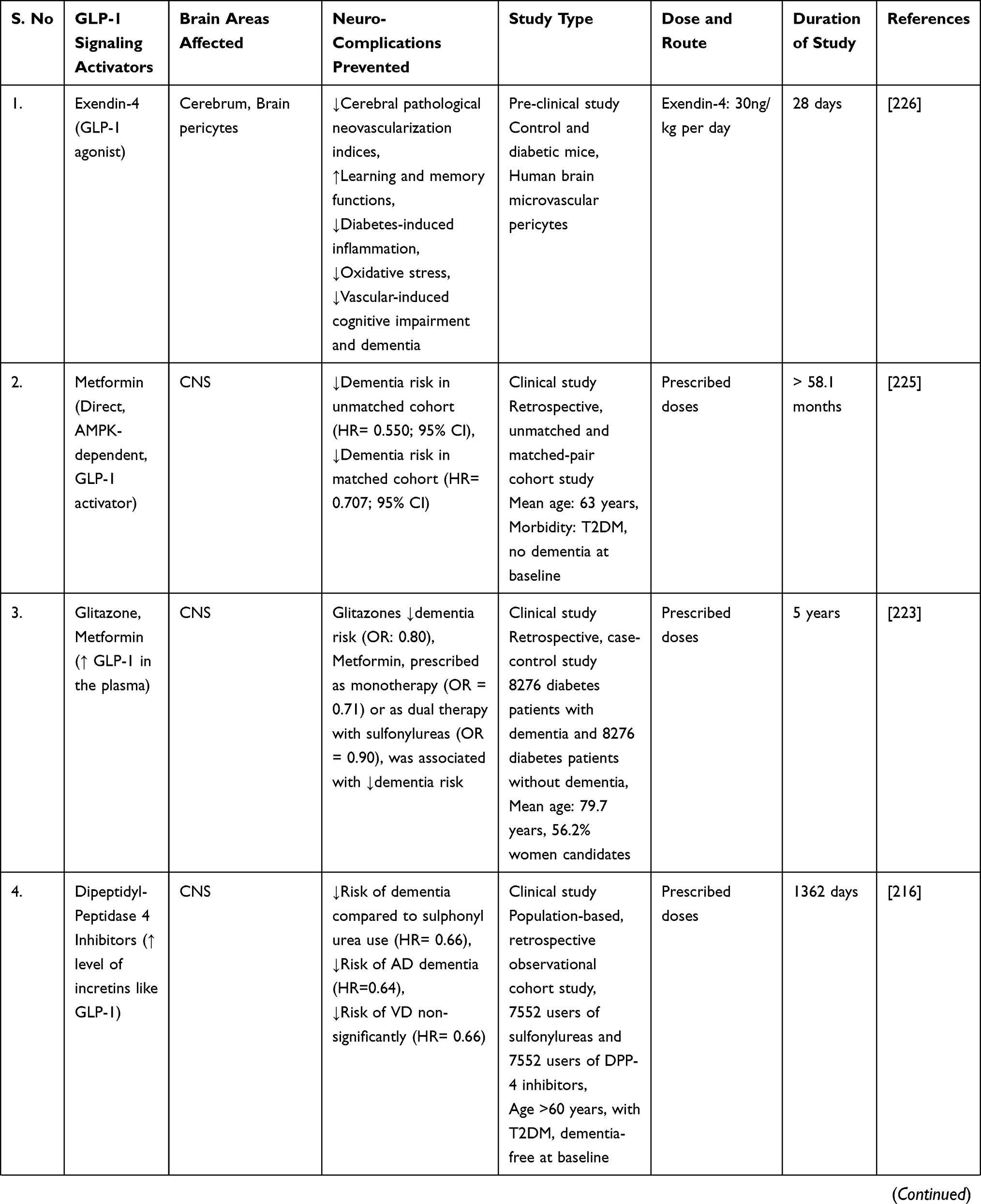 | 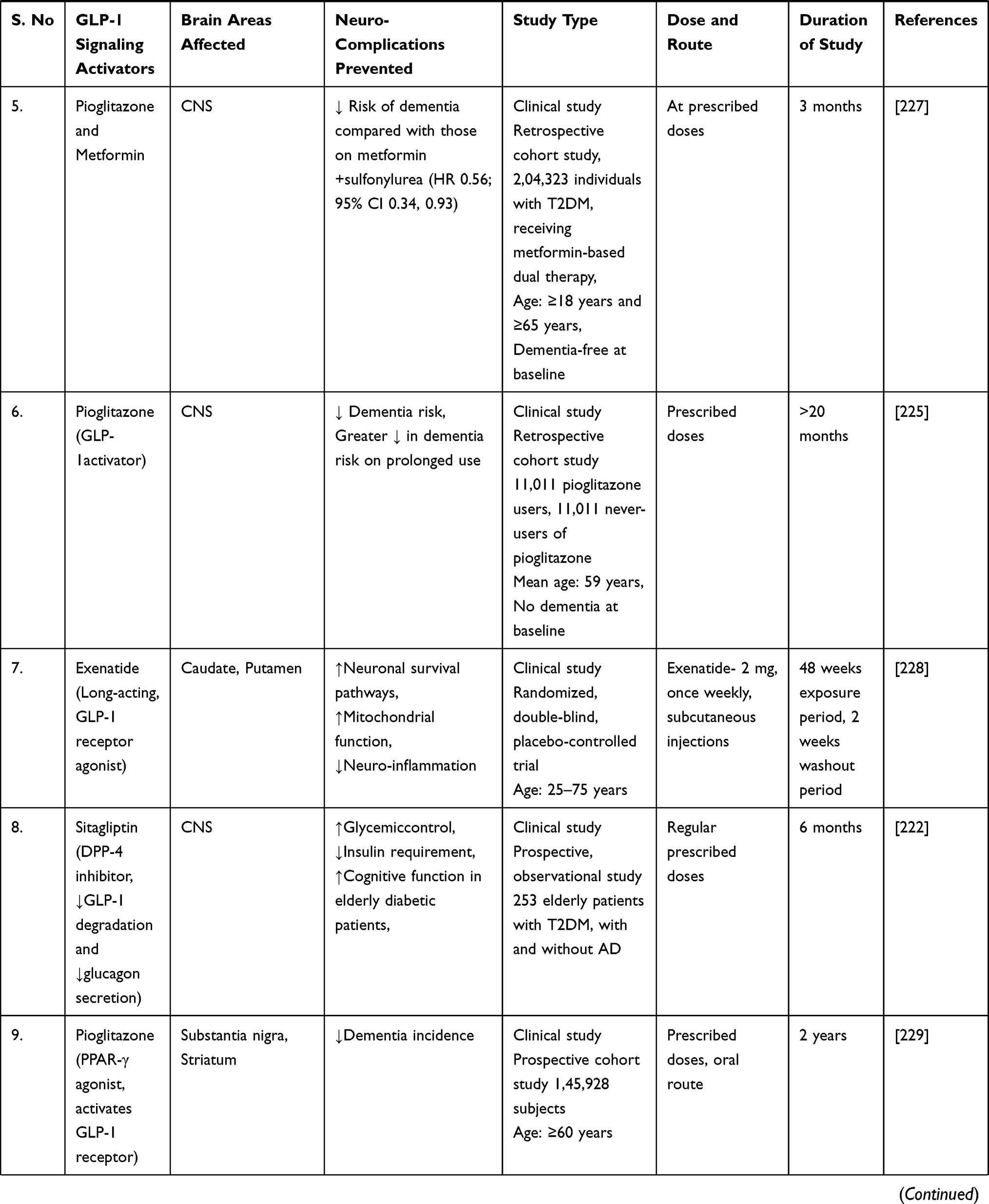 | 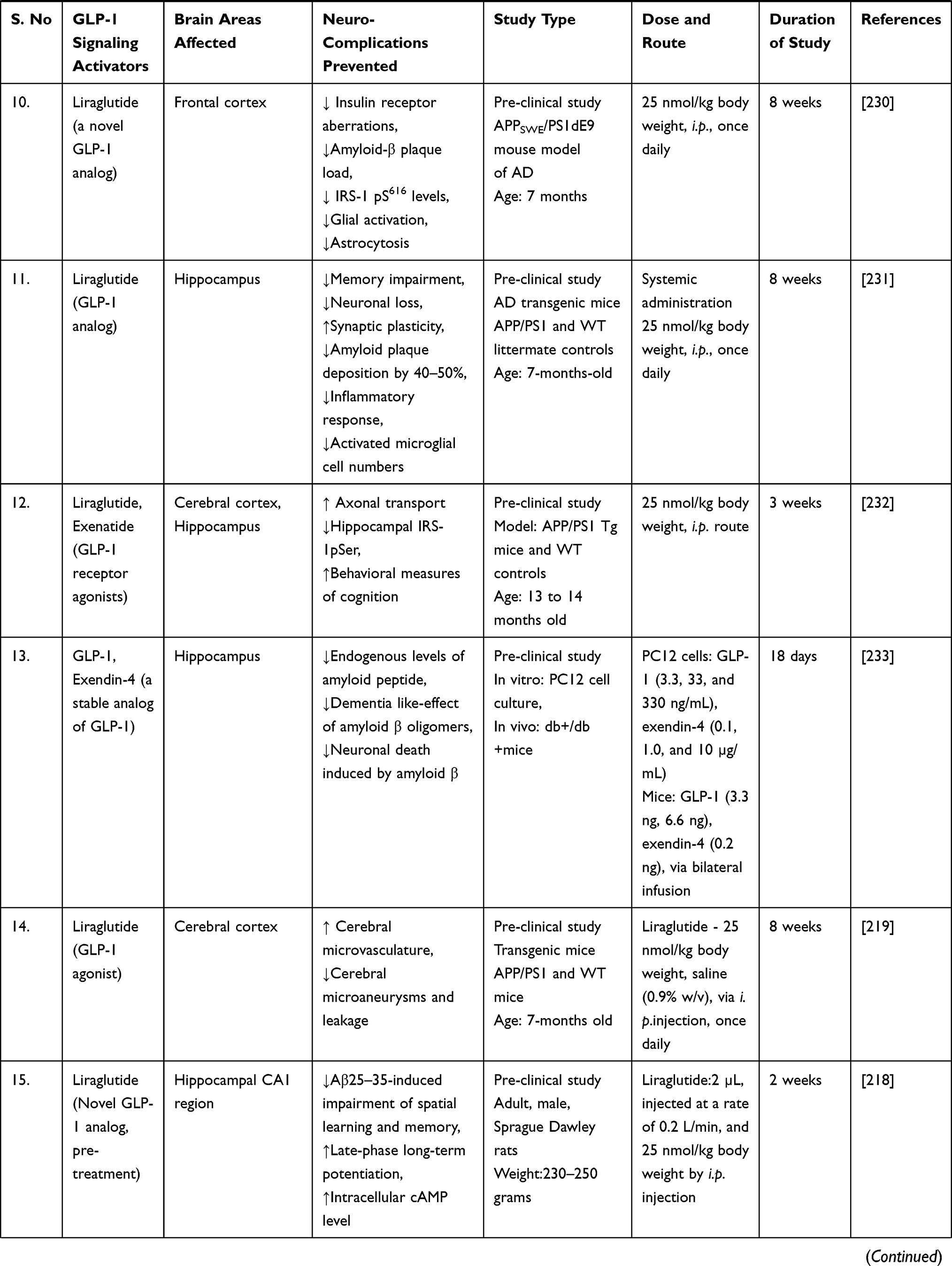 | 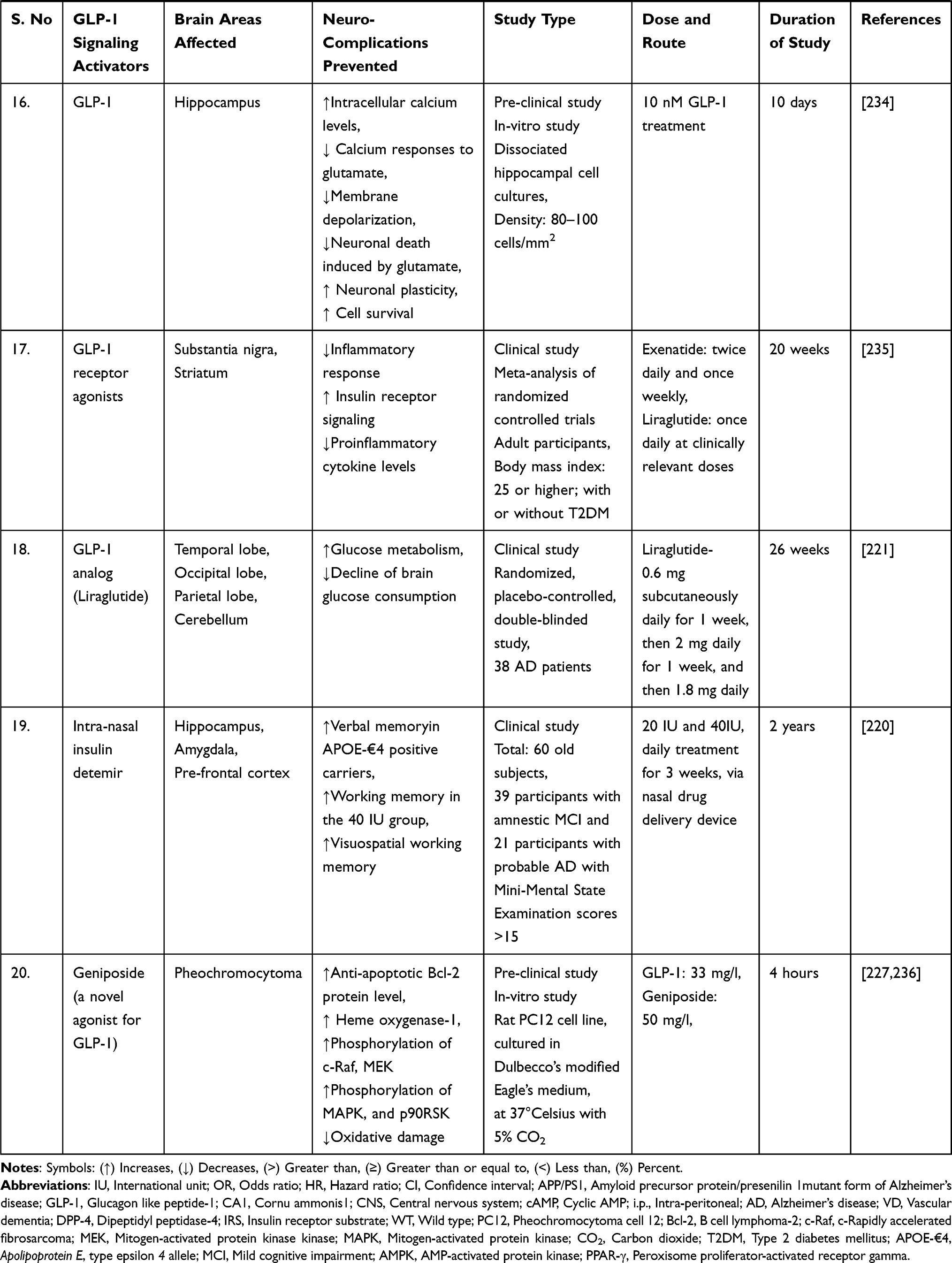 |
Table 2 Neuroprotective Role of GLP-1 Signaling Analogs in the Protection of Various Dementia Related Neurodegenerative Abnormalities |
Conclusion and Future Perspectives
This review focuses on the therapeutic potential of insulin in a variety of neurodegenerative disorders, as well as the cellular effects of insulin resistance on multiple brain cells. It summarises recent research on the roles of hypothalamic inflammation induced by obesity in neurodegeneration, impaired adult neurogenesis, and impaired neural stem cell regeneration and their relevance to obesity and related diseases. Additionally, similarities in neuroinflammation between neurodegenerative diseases and metabolic diseases caused by malnutrition are discussed. These neuro complications have been identified as promising research targets in obesity and other conditions associated with excessive food consumption. Despite strong indicators, insulin signaling and pharmacological modulators may have adverse effects, especially long-term treatments. In a nutshell, abnormalities and dysfunction of the insulin signaling pathway and its molecules may contribute to the neurodegenerative process. Numerous studies have demonstrated that altering molecules downstream in the insulin signaling pathway increases insulin sensitivity. Recently, the therapeutic effect of insulin and the detrimental impact of insulin resistance on neuronal and glial cells in neurodegenerative diseases were reviewed.
Neuroinflammation, neurodegeneration, and insulin resistance all have a strong correlation. It appears as though the outcomes vary according to the duration of the stimulus. Non-pharmacological interventions such as physical exercise and intermittent fasting may provide new information about the onset and progression of neurodegenerative diseases. Along with the impaired insulin signaling, we discussed the various agents in this class of drugs (GLP-1 receptor agonists, dual/triple receptor agonists, and DPP-4 inhibitors) and the evidence supporting their potential role in the treatment of neurodegenerative diseases.
Additional research is needed to determine the optimal insulin dose, tolerability, long-term safety, and schedule for patients with neurological disease and trauma and gain a better understanding of the role of insulin resistance in the development and progression of brain disorders.
Acknowledgments
The authors express their gratitude to Chairman, Mr. Parveen Garg, and Director, Dr. G. D. Gupta, ISF College of Pharmacy, Moga (Punjab), India, for their great vision and support.
Disclosure
The authors declared no potential conflicts of interest for this work, nor with respect to the research, authorship, and publication of this article.
References
1. Byers AL, Yaffe K. Depression and risk of developing dementia. Nat Rev Neurol. 2011;7(6):323–331. doi:10.1038/nrneurol.2011.60
2. Bierer LM, Hof PR, Purohit DP, et al. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol. 1995;52(1):81–88. doi:10.1001/archneur.1995.00540250089017
3. Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci. 2009;11(2):111.
4. Shaji S, Bose S, Verghese A. Prevalence of dementia in an urban population in Kerala, India. Br J Psychiatry. 2005;186(2):136–140. doi:10.1192/bjp.186.2.136
5. Peluso S, De Rosa A, De Lucia N, et al. Animal-assisted therapy in elderly patients: evidence and controversies in dementia and psychiatric disorders and future perspectives in other neurological diseases. J Geriatr Psychiatry Neurol. 2018;31(3):149–157. doi:10.1177/0891988718774634
6. Martinez-Horta S, Sampedro F, Horta-Barba A, et al. Structural brain correlates of dementia in Huntington’s disease. NeuroImage Clin. 2020;28:102415. doi:10.1016/j.nicl.2020.102415
7. Tschampa HJ, Neumann M, Zerr I, et al. Patients with Alzheimer’s disease and dementia with Lewy bodies mistaken for Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2001;71(1):33–39. doi:10.1136/jnnp.71.1.33
8. Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386(10004):1672–1682. doi:10.1016/S0140-6736(15)00461-4
9. Pervin F, Edwards C, Lippa CF. Dementia with Lew body: impacts of surgery. Am J Alzheimers Dis Other Dement. 2016;31(1):5–17. doi:10.1177/1533317515581704
10. Mazucanti CH, Liu QR, Lang D, et al. Release of insulin produced by the choroid plexis is regulated by serotonergic signaling. JCI Insight. 2019;4(23). doi:10.1172/jci.insight.131682
11. Siso S, Jeffrey M, Gonzalez L. Sensory circumventricular organs in health and disease. Acta Neuropathol. 2010;120:689–705. doi:10.1007/s00401-010-0743-5
12. García-Cáceres C, Quarta C, Varela L, et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell. 2016;166:867–880. doi:10.1016/j.cell.2016.07.028
13. Reno CM, Puente EC, Sheng Z, et al. Brain GLUT4 knockout mice have impaired glucose tolerance, decreased insulin sensitivity, and impaired hypoglycemiccounterregulation. Diabetes. 2017;66:587–597. doi:10.2337/db16-0917
14. Rojas FA, Hirata AE, Saad MJ. Regulation of insulin receptor substrate-2 tyrosine phosphorylation in animal models of insulin resistance. Endocrine. 2003;21:115–122. doi:10.1385/endo:21:2:115
15. Qiu J, Zhang C, Borgquist A, et al. Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab. 2014;19:682–693. doi:10.1016/j.cmet.2014.03.004
16. Ferreira LS, Fernandes CS, Vieira MN, De Felice FG. Insulin resistance in Alzheimer’s disease. Front Neurosci. 2018;12:830. doi:10.3389/fnins.2018.00830
17. Yun SP, Kam TI, Panicker N, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med. 2018;24(7):931–938. doi:10.1038/s41591-018-0051-5
18. Athauda D, Foltynie T. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: mechanisms of action. Drug Discov Today. 2016;21(5):802–818. doi:10.1016/j.drudis.2016.01.013
19. Hunot S, Brugg B, Ricard D, et al. Nuclear translocation of NF-κB is increased in dopaminergic neurons of patients with Parkinson disease. Proc Natl Acad Sci. 1997;94(14):7531–7536. doi:10.1073/pnas.94.14.7531
20. Perry T, Lahiri DK, Chen D, et al. A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J Pharmacol Exp Ther. 2002;300(3):958–966. doi:10.1124/jpet.300.3.958
21. Querfurth HW, LaFerla FM. Mechanisms of disease. N Engl J Med. 2010;362(4):329–344. doi:10.1056/NEJMra0909142
22. Hainfellner JA, Wanschitz J, Jellinger K, Liberski PP, Gullotta F, Budka H. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol. 1998;96(2):116–122. doi:10.1007/s004010050870
23. Bateman RJ, Aisen PS, De Strooper B, et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res Ther. 2011;3(1):1–3.
24. Chen JX, Yan SS. Role of mitochondrial amyloid-β in Alzheimer’s disease. J Alzheimers Dis. 2010;20(s2):S569–78. doi:10.3233/JAD-2010-100357
25. Crews L, Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum Mol Genet. 2010;19(R1):R12–20. doi:10.1093/hmg/ddq160
26. Tiwari S, Atluri V, Kaushik A, Yndart A, Nair M. Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int J Nanomedicine. 2019;14:5541. doi:10.2147/IJN.S200490
27. Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286(17):15317–15331. doi:10.1074/jbc.M110.209296
28. Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci. 2013;33(3):1024–1037. doi:10.1523/JNEUROSCI.2642-12.2013
29. Medina M, Avila J. The role of extracellular Tau in the spreading of neurofibrillary pathology. Front Cell Neurosci. 2014;8:113. doi:10.3389/fncel.2014.00113
30. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging‐Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–269. doi:10.1016/j.jalz.2011.03.005
31. Markowitsch HJ, Staniloiu A. Amnesic disorders. Lancet. 2012;380(9851):1429–1440. doi:10.1016/S0140-6736(11)61304-4
32. Aljanabi NM, Mamtani S, Al-Ghuraibawi MM, Yadav S, Nasr L. Alzheimer’s and hyperglycemia: role of the insulin signaling pathway and GSK-3 inhibition in paving a path to dementia. Cureus. 2020;12(2):e6885.
33. Zola-Morgan S, Squire LR, Amaral DG. Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci. 1986;6(10):2950–2967. doi:10.1523/JNEUROSCI.06-10-02950.1986
34. Peters F, Collette F, Degueldre C, Sterpenich V, Majerus S, Salmon E. The neural correlates of verbal short-term memory in Alzheimer’s disease: an fMRI study. Brain. 2009;132(7):1833–1846. doi:10.1093/brain/awp075
35. Abbott MA, Wells DG, Fallon JR. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J Neurosci. 1999;19(17):7300–7308. doi:10.1523/JNEUROSCI.19-17-07300.1999
36. Skeberdis VA, Lan JY, Zheng X, Zukin RS, Bennett MV. Insulin promotes rapid delivery of N-methyl-D-aspartate receptors to the cell surface by exocytosis. Proc Natl Acad Sci. 2001;98(6):3561–3566. doi:10.1073/pnas.051634698
37. Bingham EM, Hopkins D, Smith D, et al. The role of insulin in human brain glucose metabolism: an 18fluoro-deoxyglucose positron emission tomography study. Diabetes. 2002;51(12):3384–3390. doi:10.2337/diabetes.51.12.3384
38. Craft S, Asthana S, Cook DG, et al. Insulin dose–response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: interactions with apolipoprotein E genotype. Psychoneuroendocrinology. 2003;28(6):809–822. doi:10.1016/S0306-4530(02)00087-2
39. Townsend M, Mehta T, Selkoe DJ. Soluble Aβ inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007;282(46):33305–33312. doi:10.1074/jbc.M610390200
40. De Felice FG, Vieira MN, Bomfim TR, et al. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc Natl Acad Sci. 2009;106(6):1971–1976. doi:10.1073/pnas.0809158106
41. Lee CC, Kuo YM, Huang CC, Hsu KS. Insulin rescues amyloid β-induced impairment of hippocampal long-term potentiation. Neurobiol Aging. 2009;30(3):377–387. doi:10.1016/j.neurobiolaging.2007.06.014
42. Sato S, Tatebayashi Y, Akagi T, et al. Aberrant tau phosphorylation by glycogen synthase kinase-3β and JNK3 induces oligomeric tau fibrils in COS-7 cells. J Biol Chem. 2002;277(44):42060–42065. doi:10.1074/jbc.M202241200
43. Harman D. Alzheimer’s disease: role of aging in pathogenesis. Ann N Y Acad Sci. 2002;959(1):384–395. doi:10.1111/j.1749-6632.2002.tb02109.x
44. Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimers Dis. 2005;8(3):247–268. doi:10.3233/JAD-2005-8304
45. Lesort M, Johnson GV. Insulin-like growth factor-1 and insulin mediate transient site-selective increases in tau phosphorylation in primary cortical neurons. Neuroscience. 2000;99(2):305–316. doi:10.1016/S0306-4522(00)00200-1
46. Planel E, Richter KE, Nolan CE, et al. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007;27(12):3090–3097. doi:10.1523/JNEUROSCI.4854-06.2007
47. Mi K, Johnson GV. The role of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2006;3(5):449–463. doi:10.2174/156720506779025279
48. Alonso AD, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci. 2001;98(12):6923–6928. doi:10.1073/pnas.121119298
49. Planel E, Tatebayashi Y, Miyasaka T, et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J Neurosci. 2007;27(50):13635–13648. doi:10.1523/JNEUROSCI.3949-07.2007
50. Schubert M, Brazil DP, Burks DJ, et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci. 2003;23(18):7084–7092. doi:10.1523/JNEUROSCI.23-18-07084.2003
51. Schubert M, Gautam D, Surjo D, et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci. 2004;101(9):3100–3105. doi:10.1073/pnas.0308724101
52. Long C, Grueter CE, Song K, et al. Ataxia and Purkinje cell degeneration in mice lacking the CAMTA1 transcription factor. Proc Natl Acad Sci. 2014;111(31):11521–11526. doi:10.1073/pnas.1411251111
53. Manczak M, Park BS, Jung Y, Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease. Neuromolecular Med. 2004;5(2):147–162. doi:10.1385/NMM:5:2:147
54. Zhu X, Smith MA, Perry G, Aliev G. Mitochondrial failures in Alzheimer’s disease. Am J Alzheimers Dis Other Dement. 2004;19(6):345–352. doi:10.1177/153331750401900611
55. Hirai K, Aliev G, Nunomura A, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21(9):3017–3023. doi:10.1523/JNEUROSCI.21-09-03017.2001
56. Rademakers R, Cruts M, Van Broeckhoven C. Genetics of early-onset Alzheimer dementia. Sci World J. 2003;3:497–519. doi:10.1100/tsw.2003.39
57. German DC, Eisch AJ. Mouse models of Alzheimer’s disease: insight into treatment. Rev Neurosci. 2004;15(5):353–370. doi:10.1515/REVNEURO.2004.15.5.353
58. Selkoe DJ. Alzheimer disease: mechanistic understanding predicts novel therapies. Ann Intern Med. 2004;140(8):627–638. doi:10.7326/0003-4819-140-8-200404200-00047
59. Bhutada P, Mundhada Y, Humane V, et al. Agmatine, an endogenous ligand of imidazoline receptor protects against memory impairment and biochemical alterations in streptozotocin-induced diabetic rats. Prog Neuropsychopharmacol Biol Psychiatry. 2012;37(1):96–105. doi:10.1016/j.pnpbp.2012.01.009
60. Richard F, Pasquier F. Can the treatment of vascular risk factors slow cognitive decline in Alzheimer’s disease patients? J Alzheimers Dis. 2012;32(3):765–772. doi:10.3233/JAD-2012-121012
61. Cao Q, Jiang K, Zhang M, et al. Brain glucose metabolism and neuropsychological test in patients with mild cognitive impairment. Chin Med J. 2003;116(8):1235–1238.
62. Biessels GJ, van der Heide LP, Kamal A, Bleys RL, Gispen WH. Ageing and diabetes: implications for brain function. Eur J Pharmacol. 2002;441(1–2):1–4. doi:10.1016/S0014-2999(02)01486-3
63. Zhao WQ, Chen H, Quon MJ, Alkon DL. Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol. 2004;490(1–3):71–81. doi:10.1016/j.ejphar.2004.02.045
64. Kamal A, Biessels GJ, Gispen WH, Ramakers GM. Synaptic transmission changes in the pyramidal cells of the hippocampus in streptozotocin-induced diabetes mellitus in rats. Brain Res. 2006;1073:276–280. doi:10.1016/j.brainres.2005.12.070
65. Cardoso S, Santos MS, Seiça R, Moreira PI. Cortical and hippocampal mitochondria bioenergetics and oxidative status during hyperglycemia and/or insulin-induced hypoglycemia. Biochim Biophys Acta Mol Basis Dis. 2010;1802(11):942–951. doi:10.1016/j.bbadis.2010.07.001
66. Zhao WQ, Townsend M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis. 2009;1792(5):482–496.
67. Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. doi:10.1038/35008121
68. Park HR, Park M, Choi J, Park KY, Chung HY, Lee J. A high-fat diet impairs neurogenesis: involvement of lipid peroxidation and brain-derived neurotrophic factor. Neurosci Lett. 2010;482(3):235–239. doi:10.1016/j.neulet.2010.07.046
69. Parikh V, Kozak R, Martinez V, Sarter M. Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron. 2007;56(1):141–154. doi:10.1016/j.neuron.2007.08.025
70. Hasselmo ME, Sarter M. Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology. 2011;36(1):52–73. doi:10.1038/npp.2010.104
71. Pakiam AS, Bergeron C, Lang AE. Diffuse Lewy body disease presenting as multiple system atrophy. Can J Neurol Sci. 1999;26(2):127–131.
72. Mark MH, Sage JI, Dickson DW, et al. Meige syndrome in the spectrum of Lewy body disease. Neurology. 1994;44(8):1432. doi:10.1212/WNL.44.8.1432
73. Davies P. Neurotransmitter-related enzymes in senile dementia of the Alzheimer type. Brain Res. 1979;171(2):319–327. doi:10.1016/0006-8993(79)90336-6
74. Court JA, Perry EK. Dementia: the neurochemical basis of putative transmitter orientated therapy. Pharmacol Ther. 1991;52(3):423–443. doi:10.1016/0163-7258(91)90035-K
75. Kodl CT, Seaquist ER. Cognitive dysfunction and diabetes mellitus. Endocr Rev. 2008;29(4):494–511. doi:10.1210/er.2007-0034
76. Auer RN. Hypoglycemic brain damage. In: Acute Neuronal Injury; 2018:175–188.
77. Sonnewald U, McKenna M. Metabolic compartmentation in cortical synaptosomes: influence of glucose and preferential incorporation of endogenous glutamate into GABA. Neurochem Res. 2002;27(1):43–50. doi:10.1023/A:1014846404492
78. Frigerio F, Casimir M, Carobbio S, Maechler P. Tissue specificity of mitochondrial glutamate pathways and the control of metabolic homeostasis. Biochim Biophys Acta Bioenerg. 2008;1777(7–8):965–972. doi:10.1016/j.bbabio.2008.04.031
79. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA‐glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. 2006;98(3):641–653. doi:10.1111/j.1471-4159.2006.03913.x
80. Alberdi E, Sánchez-Gómez MV, Cavaliere F, et al. Amyloid β oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium. 2010;47(3):264–272. doi:10.1016/j.ceca.2009.12.010
81. Gundersen V, Fonnum F, Ottersen OP, Storm-Mathisen J. Redistribution of neuroactive amino acids in hippocampus and striatum during hypoglycemia: a quantitative immunogold study. J Cereb Blood Flow Metab. 2001;21(1):41–51. doi:10.1097/00004647-200101000-00006
82. Clementi F. Distribution of nicotinic subtypes in human brain. Alzheimer Dis Assoc Disord. 1995;9 Suppl 2:6–14. doi:10.1097/00002093-199501002-00003
83. Bartus RT, Dean R, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217(4558):408–414. doi:10.1126/science.7046051
84. Court JA, Piggott MA, Lloyd S, et al. Nicotine binding in human striatum: elevation in schizophrenia and reductions in dementia with Lewy bodies, Parkinson’s disease and Alzheimer’s disease and in relation to neuroleptic medication. Neuroscience. 2000;98(1):79–87. doi:10.1016/S0306-4522(00)00071-3
85. Piggott MA, Marshall EF, Thomas N, et al. Striatal dopaminergic markers in dementia with Lewy bodies, Alzheimer’s and Parkinson’s diseases: rostrocaudal distribution. Brain. 1999;122(8):1449–1468. doi:10.1093/brain/122.8.1449
86. Jones GM, Sahakian BJ, Levy R, Warburton DM, Gray JA. Effects of acute subcutaneous nicotine on attention, information processing and short-term memory in Alzheimer’s disease. Psychopharmacology. 1992;108(4):485–494. doi:10.1007/BF02247426
87. Levin ED, Briggs SJ, Christopher NC, Rose JE. Persistence of chronic nicotine-induced cognitive facilitation. Behav Neural Biol. 1992;58(2):152–158. doi:10.1016/0163-1047(92)90399-O
88. Levin ED, Bettegowda C, Blosser J, Gordon J. AR-R17779, and alpha7 nicotinic agonist, improves learning and memory in rats. Behav Pharmacol. 1999;10(6–7):675–680. doi:10.1097/00008877-199911000-00014
89. Alkondon M, Rocha ES, Maelicke A, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat brain. V. alpha-Bungarotoxin-sensitive nicotinic receptors in olfactory bulb neurons and presynaptic modulation of glutamate release. J Pharmacol Exp Ther. 1996;278(3):1460–1471.
90. Lippa CF, Smith TW, Perry E. Dementia with Lewy bodies: choline acetyltransferase parallels nucleus basalis pathology. J Neural Transm. 1999;106(5):525–535. doi:10.1007/s007020050176
91. Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267(5203):1456–1462. doi:10.1126/science.7878464
92. Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102(4):783–791. doi:10.1172/JCI2425
93. Zhang W, Khanna P, Chan LL, Campbell G, Ansari NH. Diabetes-induced apoptosis in rat kidney. Biochem Mol Med. 1997;61(1):58–62. doi:10.1006/bmme.1997.2592
94. Stefani L, Burke RE, Greene LA. Apoptosis in neurodegenerative disorders. Curr Opin Neurol. 1997;10:299–305. doi:10.1097/00019052-199708000-00004
95. White BC, Sullivan JM, DeGracia DJ, et al. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci. 2000;179(1–2):1–33. doi:10.1016/s0022-510x(00)00386-5
96. Resnicoff M, Burgaud JL, Rotman HL, Abraham D, Baserga R. Correlation between apoptosis, tumorigenesis, and levels of insulin-like growth factor I receptors. Cancer Res. 1995;55(17):3739–3741.
97. Zhang W, Ghetti B, Lee WH. Decreased IGF-I gene expression during the apoptosis of Purkinje cells in pcd mice. Dev Brain Res. 1997;98(2):164–176. doi:10.1016/S0165-3806(96)00168-X
98. Li ZG, Zhang W, Grunberger G, Sima AA. Hippocampal neuronal apoptosis in type 1 diabetes. Brain Res. 2002;946(2):221–231. doi:10.1016/S0006-8993(02)02887-1
99. Folch J, Olloquequi J, Ettcheto M, et al. The involvement of peripheral and brain insulin resistance in late onset Alzheimer’s dementia. Front Aging Neurosci. 2019;11:236. doi:10.3389/fnagi.2019.00236
100. Kalaria RN. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol. 2016;131(5):659–685. doi:10.1007/s00401-016-1571-z
101. Xiong Y, Zhou H, Zhang L. Influences of hyperthermia-induced seizures on learning, memory and phosphorylative state of CaMKIIα in rat hippocampus. Brain Res. 2014;1557:190–200. doi:10.1016/j.brainres.2014.02.026
102. Sairanen T, Karjalainen-Lindsberg ML, Paetau A, Ijäs P, Lindsberg PJ. Apoptosis dominant in the periinfarct area of human ischaemic stroke—a possible target of antiapoptotic treatments. Brain. 2006;129(1):189–199. doi:10.1093/brain/awh645
103. Viswanathan A, Gray F, Bousser MG, Baudrimont M, Chabriat H. Cortical neuronal apoptosis in CADASIL. Stroke. 2006;37(11):2690–2695. doi:10.1161/01.STR.0000245091.28429.6a
104. Wang XX, Zhang B, Xia R, Jia QY. Inflammation, apoptosis and autophagy as critical players in vascular dementia. Eur Rev Med Pharmacol Sci. 2020;24(18):9601–9614. doi:10.26355/eurrev_202009_23048
105. Kalaria RN. Cerebrovascular disease and mechanisms of cognitive impairment: evidence from clinicopathological studies in humans. Stroke. 2012;43(9):2526–2534. doi:10.1161/STROKEAHA.112.655803
106. Mungas D, Jagust WJ, Reed BR, et al. MRI predictors of cognition in subcortical ischemic vascular disease and Alzheimer’s disease. Neurology. 2001;57(12):2229–2235. doi:10.1212/WNL.57.12.2229
107. Ashford ML, Boden PR, Treherne JM. Glucose-induced excitation of hypothalamic neurones is mediated by ATP-sensitive K+ channels. Pflügers Archiv. 1990;415(4):479–483. doi:10.1007/BF00373626
108. Miki T, Liss B, Minami K, et al. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci. 2001;4(5):507–512. doi:10.1038/87455
109. Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes. 2004;53(3):549–559. doi:10.2337/diabetes.53.3.549
110. Ibrahim N, Bosch MA, Smart JL, et al. Hypothalamic proopiomelanocortin neurons are glucose responsive and express KATP channels. Endocrinology. 2003;144(4):1331–1340. doi:10.1210/en.2002-221033
111. Popa-Wagner A, Buga AM, Popescu B, Muresanu D. Vascular cognitive impairment, dementia, aging and energy demand. A vicious cycle. J Neural Transm. 2015;122(1):47–54. doi:10.1007/s00702-013-1129-3
112. Jellinger KA, Attems J. Prevalence of dementia disorders in the oldest-old: an autopsy study. Acta Neuropathol. 2010;119(4):421–433. doi:10.1007/s00401-010-0654-5
113. Iadecola C, Gorelick PB. Converging pathogenic mechanisms in vascular and neurodegenerative dementia. Stroke. 2003;34(2):335–337. doi:10.1161/01.STR.0000054050.51530.76
114. Pluta R, Jolkkonen J, Cuzzocrea S, Pedata F, Cechetto D, Popa-Wagner A. Cognitive impairment with vascular impairment and degeneration. Curr Neurovasc Res. 2011;8(4):342–350. doi:10.2174/156720211798120981
115. Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57(2):178–201. doi:10.1016/j.neuron.2008.01.003
116. Kasuga M, Karlsson FA, Kahn CR. Insulin stimulates the phosphorylation of the 95,000-dalton subunit of its own receptor. Science. 1982;215(4529):185–187. doi:10.1126/science.7031900
117. Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005;1(1):53–61. doi:10.1016/j.cmet.2004.11.001
118. Filippi BM, Yang CS, Tang C, Lam TK. Insulin activates Erk1/2 signaling in the dorsal vagal complex to inhibit glucose production. Cell Metab. 2012;16(4):500–510. doi:10.1016/j.cmet.2012.09.005
119. Rossi J, Balthasar N, Olson D, et al. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab. 2011;13(2):195–204. doi:10.1016/j.cmet.2011.01.010
120. Berglund ED, Liu T, Kong X, et al. Melanocortin 4 receptors in autonomic neurons regulate thermogenesis and glycemia. Nat Neurosci. 2014;17(7):911–913. doi:10.1038/nn.3737
121. Hill JW, Elias CF, Fukuda M, et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010;11(4):286–297. doi:10.1016/j.cmet.2010.03.002
122. Könner AC, Janoschek R, Plum L, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5(6):438–449. doi:10.1016/j.cmet.2007.05.004
123. Ruud J, Steculorum SM, Brüning JC. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat Commun. 2017;8(1):1–2. doi:10.1038/ncomms15259
124. Freeman AM, Pennings N. Insulin resistance. In: StatPearls [Internet]; 2021.
125. Akter K, Lanza EA, Martin SA, Myronyuk N, Rua M, Raffa RB. Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment? Br J Clin Pharmacol. 2011;71(3):365–376. doi:10.1111/j.1365-2125.2010.03830.x
126. Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102(4):401–414. doi:10.1161/CIRCRESAHA.107.165472
127. Cooper SA, Whaley-Connell A, Habibi J, et al. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. Am J Physiol Heart Circ Physiol. 2007;293(4):H2009–23. doi:10.1152/ajpheart.00522.2007
128. Sowers JR, Stump CS. Insights into the biology of diabetic vascular disease: what’s new? Am J Hypertens. 2004;17(S2):2S–6S. doi:10.1016/j.amjhyper.2003.08.015
129. Brehm A, Krssak M, Schmid AI, Nowotny P, Waldhäusl W, Roden M. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes. 2006;55(1):136–140. doi:10.2337/diabetes.55.01.06.db05-1286
130. Ritz P, Berrut G. Mitochondrial function, energy expenditure, aging and insulin resistance. Diabetes Metab. 2005;31:5S67–73. doi:10.1016/S1262-3636(05)73654-5
131. Frisard M, Ravussin E. Energy metabolism and oxidative stress: impact on the metabolic syndrome and the aging process. Endocrine. 2006;29(1):27–32. doi:10.1385/ENDO:29:1:27
132. Befroy DE, Petersen KF, Dufour S, et al. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56(5):1376–1381. doi:10.2337/db06-0783
133. Krssak MF, Petersen KF, Dresner A, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42(1):113–116. doi:10.1007/s001250051123
134. Mogensen M, Sahlin K, Fernström M, et al. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2007;56(6):1592–1599. doi:10.2337/db06-0981
135. Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation. 2007;116(4):434–448. doi:10.1161/CIRCULATIONAHA.107.702795
136. Nisoli E, Clementi E, Carruba MO, Moncada S. Defective mitochondrial biogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome? Circ Res. 2007;100(6):795–806. doi:10.1161/01.RES.0000259591.97107.6c
137. Wiederkehr A, Wollheim CB. Minireview: implication of mitochondria in insulin secretion and action. Endocrinology. 2006;147(6):2643–2649. doi:10.1210/en.2006-0057
138. Aliev G, Gasimov E, Obrenovich ME, et al. Atherosclerotic lesions and mitochondria DNA deletions in brain microvessels: implication in the pathogenesis of Alzheimer’s disease. Vasc Health Risk Manag. 2008;4(3):721. doi:10.2147/VHRM.S2608
139. Abdul HM, Sultana R, Clair DK, Markesbery WR, Butterfield DA. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic Biol Med. 2008;45(10):1420–1425. doi:10.1016/j.freeradbiomed.2008.08.012
140. Coskun PE, Wyrembak J, Derbereva O, et al. Systemic mitochondrial dysfunction and the etiology of Alzheimer’s disease and down syndrome dementia. J Alzheimers Dis. 2010;20(s2):S293–310. doi:10.3233/JAD-2010-100351
141. Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116(3):615–622. doi:10.1172/JCI27794
142. Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54(1):8–14. doi:10.2337/diabetes.54.1.8
143. Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator–activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106(7):847–856. doi:10.1172/JCI10268
144. Baar K, Wende AR, Jones TE, et al. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC‐1. FASEB J. 2002;16(14):1879–1886. doi:10.1096/fj.02-0367com
145. Yaffe K, Blackwell T, Kanaya AM, Davidowitz N, Barrett-Connor E, Krueger K. Diabetes, impaired fasting glucose, and development of cognitive impairment in older women. Neurology. 2004;63(4):658–663. doi:10.1212/01.WNL.0000134666.64593.BA
146. Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3(3):169–178. doi:10.1016/S1474-4422(04)00681-7
147. Gad ES, Zaitone SA, Moustafa YM. Pioglitazone and exenatide enhance cognition and downregulate hippocampal beta amyloid oligomer and microglia expression in insulin-resistant rats. Can J Physiol Pharmacol. 2016;94(8):819–828. doi:10.1139/cjpp-2015-0242
148. Carvalho C, Correia SC, Santos RX, et al. Role of mitochondrial-mediated signaling pathways in Alzheimer disease and hypoxia. J Bioenerg Biomembr. 2009;41(5):433. doi:10.1007/s10863-009-9247-1
149. Yang C, DeVisser A, Martinez JA, et al. Retracted: differential impact of diabetes and hypertension in the brain: adverse effects in white matter. Neurobiol Dis. 2011;42(3):446–458. doi:10.1016/j.nbd.2011.02.007
150. Puddu A, Mach F, Nencioni A, Viviani GL, Montecucco F. An emerging role of glucagon-like peptide-1 in preventing advanced-glycation-end-product-mediated damages in diabetes. Mediators Inflamm. 2013;2013:1–9. doi:10.1155/2013/591056
151. Mao H, Liu B. Synergistic microglial reactive oxygen species generation induced by pesticides lindane and dieldrin. Neuroreport. 2008;19(13):1317–1320. doi:10.1097/WNR.0b013e32830b3677
152. Kemp K, Griffiths J, Campbell S, Lovell K. An exploration of the follow-up up needs of patients with inflammatory bowel disease. J Crohns Colitis. 2013;7(9):e386–95. doi:10.1016/j.crohns.2013.03.001
153. Mrak RE, Griffin WS. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol Aging. 2001;22(6):903–908. doi:10.1016/S0197-4580(01)00287-1
154. Zilka N, Ferencik M, Hulin I. Neuroinflammation in Alzheimer’s disease: protector or promoter? Bratisl Lek Listy. 2006;107(9–10):374–383.
155. Chen Y, Liang Z, Blanchard J, et al. A non-transgenic mouse model (icv-STZ mouse) of Alzheimer’s disease: similarities to and differences from the transgenic model (3xTg-AD mouse). Mol Neurobiol. 2013;47(2):711–725. doi:10.1007/s12035-012-8375-5
156. Banks WA, Owen JB, Erickson MA. Insulin in the brain: there and back again. Pharmacol Ther. 2012;136(1):82–93. doi:10.1016/j.pharmthera.2012.07.006
157. Chen Y, Zhang J, Zhang B, Gong CX. Targeting insulin signaling for the treatment of Alzheimer’s disease. Curr Top Med Chem. 2016;16(5):485–492. doi:10.2174/1568026615666150813142423
158. Bouvier DS, Murai KK. Synergistic actions of microglia and astrocytes in the progression of Alzheimer’s disease. J Alzheimers Dis. 2015;45(4):1001–1014. doi:10.3233/JAD-143156
159. Guo Z, Chen Y, Mao YF, et al. Long-term treatment with intranasal insulin ameliorates cognitive impairment, tau hyperphosphorylation, and microglial activation in a streptozotocin-induced Alzheimer’s rat model. Sci Rep. 2017;7(1):1–2. doi:10.1038/s41598-016-0028-x
160. Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA. Insulin-resistant brain state: the culprit in sporadic Alzheimer’s disease? Ageing Res Rev. 2011;10(2):264–273. doi:10.1016/j.arr.2011.01.001
161. Correia SC, Santos RX, Carvalho C, et al. Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012;1441:64–78. doi:10.1016/j.brainres.2011.12.063
162. Suh YH, Checler F. Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer’s disease (vol 54, pg 469, 2002). Pharmacol Rev. 2006;58(2):280.
163. Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta Mol Basis Dis. 2010;1802(1):2–10. doi:10.1016/j.bbadis.2009.10.006
164. Bonda DJ, Wang X, Perry G, et al. Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology. 2010;59(4–5):290–294. doi:10.1016/j.neuropharm.2010.04.005
165. Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer’s disease. Neurobiol Dis. 2010;37(3):503–509. doi:10.1016/j.nbd.2009.10.006
166. Auld DS, Kornecook TJ, Bastianetto S, Quirion R. Alzheimer’s disease and the basal forebrain cholinergic system: relations to β-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol. 2002;68(3):209–245. doi:10.1016/s0301-0082(02)00079-5
167. Jin K, Peel AL, Mao XO, et al. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci. 2004;101(1):343–347. doi:10.1073/pnas.2634794100
168. Vadodaria KC, Gage FH. SnapShot: adult hippocampal neurogenesis. Cell. 2014;156(5):1114. doi:10.1016/j.cell.2014.02.029
169. Djurfeldt M, Hjorth J, Eppler JM, et al. Run-time interoperability between neuronal network simulators based on the MUSIC framework. Neuroinformatics. 2010;8(1):43–60. doi:10.1007/s12021-010-9064-z
170. Kraska A, Santin MD, Dorieux O, et al. In vivo cross-sectional characterization of cerebral alterations induced by intracerebroventricular administration of streptozotocin. PLoS One. 2012;7(9):e46196. doi:10.1371/journal.pone.0046196
171. Javed H, Khan MM, Ahmad A, et al. Rutin prevents cognitive impairments by ameliorating oxidative stress and neuroinflammation in rat model of sporadic dementia of Alzheimer type. Neuroscience. 2012;210:340–352. doi:10.1016/j.neuroscience.2012.02.046
172. Qu ZQ, Zhou Y, Zeng YS, et al. Protective effects of a Rhodiola crenulata extract and salidroside on hippocampal neurogenesis against streptozotocin-induced neural injury in the rat. PLoS One. 2012;7(1):e29641. doi:10.1371/journal.pone.0029641
173. Sun P, Knezovic A, Parlak M, et al. Long-term effects of intracerebroventricular streptozotocin treatment on adult neurogenesis in the rat hippocampus. Curr Alzheimer Res. 2015;12(8):772–784. doi:10.2174/1567205012666150710112147
174. Wang B, Jin K. Current perspectives on the link between neuroinflammation and neurogenesis. Metab Brain Dis. 2015;30(2):355–365. doi:10.1007/s11011-014-9523-6
175. Bassani TB, Turnes JM, Moura EL, et al. Effects of curcumin on short-term spatial and recognition memory, adult neurogenesis and neuroinflammation in a streptozotocin-induced rat model of dementia of Alzheimer’s type. Behav Brain Res. 2017;335:41–54. doi:10.1016/j.bbr.2017.08.014
176. Stefaniak J, O’Brien J. Imaging of neuroinflammation in dementia: a review. J Neurol Neurosurg Psychiatry. 2016;87(1):21–28.
177. Byrne E. Does mitochondrial respiratory chain dysfunction have a role in common neurodegenerative disorders? J Clin Neurosci. 2002;9(5):497–501. doi:10.1054/jocn.2001.0991
178. Vajda FJ. Neuroprotection and neurodegenerative disease. J Clin Neurosci. 2002;9(1):4–8. doi:10.1054/jocn.2001.1027
179. Agrawal R, Tyagi E, Shukla R, Nath C. A study of brain insulin receptors, AChE activity and oxidative stress in rat model of ICV STZ induced dementia. Neuropharmacology. 2009;56(4):779–787. doi:10.1016/j.neuropharm.2009.01.005
180. Grünblatt E, Salkovic‐Petrisic M, Osmanovic J, Riederer P, Hoyer S. Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J Neurochem. 2007;101(3):757–770. doi:10.1111/j.1471-4159.2006.04368.x
181. Sonkusare S, Srinivasan K, Kaul C, Ramarao P. Effect of donepezil and lercanidipine on memory impairment induced by intracerebroventricular streptozotocin in rats. Life Sci. 2005;77(1):1–4. doi:10.1016/j.lfs.2004.10.036
182. Droege W, Kinscherf R. Aberrant insulin receptor signaling and amino acid homeostasis as a major cause of oxidative stress in aging. Antioxid Redox Signal. 2008;10(4):661–678. doi:10.1089/ars.2007.1953
183. Seo JH, Ahn Y, Lee SR, Yeo CY, Hur KC. The major target of the endogenously generated reactive oxygen species in response to insulin stimulation is phosphatase and tensin homolog and not phosphoinositide-3 kinase (PI-3 kinase) in the PI-3 kinase/Akt pathway. Mol Biol Cell. 2005;16(1):348–357. doi:10.1091/mbc.e04-05-0369
184. Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem. 2001;276(24):21938–21942. doi:10.1074/jbc.C100109200
185. Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9(2):387–399. doi:10.1016/S1097-2765(02)00445-8
186. Butterfield DA, Di Domenico F, Barone E. Elevated risk of type 2 diabetes for development of Alzheimer disease: a key role for oxidative stress in brain. Biochim Biophys Acta Mol Basis Dis. 2014;1842(9):1693–1706.
187. Aluise CD, Robinson RA, Cai J, Pierce WM, Markesbery WR, Butterfield DA. Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical Alzheimer’s disease: insights into memory loss in MCI. J Alzheimers Dis. 2011;23(2):257–269. doi:10.3233/JAD-2010-101083
188. Chen Z, Zhong C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol. 2013;108:21–43. doi:10.1016/j.pneurobio.2013.06.004
189. Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci. 2009;106(34):14670–14675. doi:10.1073/pnas.0903563106
190. Reddy PH, Tripathi R, Troung Q, et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta Mol Basis Dis. 2012;1822(5):639–649. doi:10.1016/j.bbadis.2011.10.011
191. Velazquez R, Tran A, Ishimwe E, et al. Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer’s disease. Neurobiol Aging. 2017;58:1–13. doi:10.1016/j.neurobiolaging.2017.06.003
192. Kim YG, Jeon JY, Kim HJ, et al. Risk of dementia in older patients with type 2 diabetes on dipeptidyl-peptidase IV inhibitors versus sulfonylureas: a real-world population-based cohort study. J Clin Med. 2018;8(1):28. doi:10.3390/jcm8010028
193. Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61:661–666. doi:10.1001/archneur.61.5.661
194. Peila R, Rodriguez BL, Launer LJ, Honolulu-Asia AS. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–1262. doi:10.2337/diabetes.51.4.1256
195. Arvanitakis Z, Schneider JA, Wilson RS, et al. Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology. 2006;67(11):1960–1965. doi:10.1212/01.wnl.0000247053.45483.4e
196. Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53(9):1937–1942. doi:10.1212/WNL.53.9.1937
197. Luchsinger JA, Tang M-X, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology. 2004;63(7):1187–1192. doi:10.1212/01.WNL.0000140292.04932.87
198. MacKnight C, Rockwood K, Awalt E, McDowell I. Diabetes mellitus and the risk of dementia, Alzheimer’s disease and vascular cognitive impairment in the Canadian Study of Health and Aging. Dement Geriatr Cogn Disord. 2002;14(2):77–83. doi:10.1159/000064928
199. Craft S, Peskind E, Schwartz MW, Schellenberg GD, Raskind M, Porte D
200. Xu WL, Qiu CX, Wahlin A, Winblad B, Fratiglioni L. Diabetes mellitus and risk of dementia in the Kungsholmen project: a 6-year follow-up study. Neurology. 2004;63(7):1181–1186. doi:10.1212/01.WNL.0000140291.86406.D1
201. Schnaider Beeri M, Goldbourt U, Silverman JM, et al. Diabetes mellitus in midlife and the risk of dementia three decades later. Neurology. 2004;63(10):1902–1907. doi:10.1212/01.WNL.0000144278.79488.DD
202. Bruce DG, Davis WA, Casey GP, et al. Severe hypoglycaemia and cognitive impairment in older patients with diabetes: the Fremantle Diabetes Study. Diabetologia. 2009;52:1808–1815. doi:10.1007/s00125-009-1437-1
203. Rönnemaa E, Zethelius B, Sundelöf J, et al. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology. 2008;71(14):1065–1071. doi:10.1212/01.wnl.0000310646.32212.3a
204. Whitmer RA, Karter AJ, Yaffe K, Quesenberry CP
205. Lin Z, Tian H, Lam KS, et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013;17(5):779–789. doi:10.1016/j.cmet.2013.04.005
206. Yaffe K, Falvey CM, Hamilton N, et al. Association between hypoglycemia and dementia in a biracial cohort of older adults with diabetes mellitus. JAMA Intern Med. 2013;173(14):1300–1306. doi:10.1001/jamainternmed.2013.6176
207. Chin SO, Rhee SY, Chon S, et al. Hypoglycemia is associated with dementia in elderly patients with type 2 diabetes mellitus: an analysis based on the Korea National Diabetes Program Cohort. Diabetes Res Clin Pract. 2016;122:54–61. doi:10.1016/j.diabres.2016.09.027
208. Mehta HB, Mehta V, Goodwin JS. Association of hypoglycemia with subsequent dementia in older patients with type 2 diabetes mellitus. J Gerontol Series A Biomed Sci Med Sci. 2017;72(8):1110–1116.
209. Cheng G, Huang C, Deng H, Wang H. Diabetes as a risk factor for dementia and mild cognitive impairment: a meta‐analysis of longitudinal studies. Intern Med J. 2012;42:484–491. doi:10.1111/j.1445-5994.2012.02758.x
210. Feinkohl I, Aung PP, Keller M, et al.; Edinburgh Type 2 Diabetes Study (ET2DS) Investigators. Severe hypoglycemia and cognitive decline in older people with type 2 diabetes: the Edinburgh type 2 diabetes study. Diabetes Care. 2014;37(2):507–515. doi:10.2337/dc13-1384
211. Smolina K, Wotton CJ, Goldacre MJ. Risk of dementia in patients hospitalized with type 1 and type 2 diabetes in England, 1998–2011: A retrospective national record linkage cohort study. Diabetologia. 2015;58:942–950. doi:10.1007/s00125-015-3515-x
212. Aung PP, Strachan MW, Frier BM, Butcher I, Deary IJ, Price JF. Severe hypoglycaemia and late-life cognitive ability in older people with Type 2 diabetes: the Edinburgh Type 2 Diabetes Study. Diabet Med. 2012;29(3):328–336. doi:10.1111/j.1464-5491.2011.03505.x
213. Matsuzaki T, Sasaki K, Tanizaki Y, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology. 2010;75(9):764–770. doi:10.1212/WNL.0b013e3181eee25f
214. Gudala K, Bansal D, Schifano F, Bhansali A. Diabetes mellitus and risk of dementia: a meta‐analysis of prospective observational studies. J Diabetes Investig. 2013;4:640–650. doi:10.1111/jdi.12087
215. Cukierman T, Gerstein H, Williamson J. Cognitive decline and dementia in diabetes—systematic overview of prospective observational studies. Diabetologia. 2005;48(12):2460–2469. doi:10.1007/s00125-005-0023-4
216. Kim YG, Park DG, Moon SY, et al. Hypoglycemia and dementia risk in older patients with type 2 diabetes mellitus: a propensity-score matched analysis of a population-based cohort study. Diabetes Metab J. 2020;44(1):125–133. doi:10.4093/dmj.2018.0260
217. Lin CH, Sheu WH. Hypoglycaemic episodes and risk of dementia in diabetes mellitus: 7-year follow-up study. J Intern Med. 2013;273(1):102–110. doi:10.1111/joim.12000
218. Han WN, Hölscher C, Yuan L, et al. Liraglutide protects against amyloid-beta protein-induced impairment of spatial learning and memory in rats. Neurobiol Aging. 2013;34(2):576–588. doi:10.1016/j.neurobiolaging.2012.04.009
219. Kelly P, McClean PL, Ackermann M, Konerding MA, Hölscher C, Mitchell CA. Restoration of cerebral and systemic microvascular architecture in APP/PS1 transgenic mice following treatment with liraglutide. Microcirculation. 2015;22(2):133–145. doi:10.1111/micc.12186
220. Claxton A, Baker LD, Hanson A, et al. Long acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J Alzheimers Dis. 2015;45(4):1269–1270. doi:10.3233/JAD-159002
221. Gejl M, Gjedde A, Egefjord L, et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled Double-Blind Clinical Trial. Front Aging Neurosci. 2016;8:108. doi:10.3389/fnagi.2016.00108
222. Isik AT, Soysal P, Yay A, Usarel C. The effects of sitagliptin, a DPP4 inhibitor, on cognitive functions in elderly diabetic patients with or without Alzheimer’s disease. Diabetes Res Clin Pract. 2017;123:192–198.
223. Bohlken J, Jacob L, Kostev K. Association between the use of antihyperglycemic drugs and dementia risk: a case-control study. J Alzheimers Dis. 2018;66(2):725–732. doi:10.3233/JAD-180808
224. Lu CH, Yang CY, Li CY, Hsieh CY, Ou HT. Lower risk of dementia with pioglitazone, compared with other second-line treatments, in metformin-based dual therapy: a population-based longitudinal study. Diabetologia. 2018;61(3):562–573. doi:10.1007/s00125-017-4499-5
225. Tseng CH. Pioglitazone reduces dementia risk in patients with type 2 diabetes mellitus: a retrospective cohort analysis. J Clin Med. 2018;7(10):306. doi:10.3390/jcm7100306
226. Bailey JM, Ghaly M, Barrett AC, Hafez S, Coucha MM, Abdelsaid MA. GLP-1 Agonists Improve Cerebrovascular Integrity and Vascular Induced Cognitive Impairment and Dementia Beyond Glycemic Control via Restoration of Brain Pericytes Functions in Diabetic Mice. Stroke. 2020;51(Suppl_1):A39–A39. doi:10.1161/str.51.suppl_1.39
227. Liu J, Yin F, Zheng X, Jing J, Hu Y. Geniposide, a novel agonist for GLP-1 receptor, prevents PC12 cells from oxidative damage via MAP kinase pathway. Neurochem Int. 2007;51(6–7):361–369. doi:10.1016/j.neuint.2007.04.021
228. Athauda D, Maclagan K, Skene SS, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10103):1664–1675. PMID: 28781108. doi:10.1016/S0140-6736(17)31585-4
229. Heneka MT, Fink A, Doblhammer G. Effect of pioglitazone medication on the incidence of dementia. Ann Neurol. 2015;78(2):284–294. doi:10.1002/ana.24439
230. Long-Smith CM, Manning S, McClean PL, et al. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signaling in parallel with decreasing both amyloid-β plaque and glial pathology in a mouse model of Alzheimer’s disease. Neuromolecular Med. 2013;15(1):102–114. doi:10.1007/s12017-012-8199-5
231. McClean PL, Parthsarathy V, Faivre E, Hölscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci. 2011;31(17):6587–6594. doi:10.1523/JNEUROSCI.0529-11.2011
232. Bomfim TR, Forny-Germano L, Sathler LB, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Ab oligomers. J Clin Invest. 2012;122:1339–1353. doi:10.1172/JCI57256
233. Perry T, Lahiri DK, Sambamurti K, et al. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J Neurosci Res. 2003;72(5):603–612. doi:10.1002/jnr.10611
234. Gilman CP, Perry T, Furukawa K, Grieg NH, Egan JM, Mattson MP. Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J Neurochem. 2003;87(5):1137–1144. doi:10.1046/j.1471-4159.2003.02073.x
235. Vilsbøll T, Christensen M, Junker AE, Knop FK, Gluud LL. Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. BMJ. 2012;344:7771. doi:10.1136/bmj.d7771
236. Chin-Hsiao T. Metformin and the risk of dementia in type 2 diabetes patients. Aging Dis. 2019;10(1):37–48. 27. doi:10.14336/AD.2017.1202
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.