")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 11
Potential protective effects of rhEGF against ultraviolet A irradiation-induced damages on human fibroblasts
Authors Kim JM, Choo JE, Kim KN, Kim YS
Received 10 April 2018
Accepted for publication 31 May 2018
Published 16 October 2018 Volume 2018:11 Pages 505—513
DOI https://doi.org/10.2147/CCID.S170697
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Ji Min Kim,1 Jung Eun Choo,1 Ki Nam Kim,1 Yang Seok Kim2
1Life Science Research Institute, Daewoong Pharmaceutical Co.,Ltd., Yongin, Korea; 2Department of Science in Korean Medicine, Kyng Hee University, Seoul, Korea
Background: Ultraviolet A (UVA) rays reach the dermal skin layer and generate oxidative stress, DNA damage, and cell inflammation, which in turn lead to photo-aging and photo-carcinogenesis. While there have been many studies about the beneficial effects of topical epidermal growth factor (EGF) treatment in the healing of wounds, the effect of EGF on UVA-induced skin irritation remains unknown. To clarify the effects of EGF on UVA-induced skin damage, it was investigated whether EGF signaling can affect intracellular reactive oxygen species (ROS) and DNA damages in UVA-irradiated human dermal fibroblasts.
Materials and methods: Fibroblasts cultured with or without rhEGF were UVA-irradiated at 40 mJ/cm2 twice per day for 5 days. After the irradiation, the intracellular ROS levels and expression of catalase and superoxide dismutase-1 (SOD-1) in the fibroblasts were ascertained. Further investigation to determine the effects of EGF on UVA-induced DNA damage, including a single cell gel electrophoresis assay and an enzyme-linked immunosorbent assay (ELISA), was carried out. Moreover, the NF-κB activity was ascertained in order to investigate the effects of EGF on UVA-irradiated fibroblasts.
Results: As a result, it was revealed that recombinant human EGF (rhEGF) inhibited UVA-increased intracellular ROS in the fibroblasts and increased the expression of catalase and SOD-1. Moreover, in UVA-irradiated fibroblasts, the longest DNA-damaged tails were observed, but this phenomenon was not detected in cells cotreated with both UVA and rhEGF. Also, it was observed that DNA damage induction, including that of cyclobutene pyrimidine dimers, pyrimidine (6-4) pyrimidone photoproducts, and 8-hydroxy-2-deoxyguanosine, was caused by UVA irradiation. Similar to previous results, it was downregulated by rhEGF. Furthermore, rhEGF also inhibited NF-κB gene expression and the NF-κB p65 protein level in the nucleus induced by UVA irradiation.
Conclusion: These results suggest that EGF might be a useful material for preventing or improving photo-aging.
Keywords: epidermal growth factor, UVA, catalase, superoxide dismutase-1, cyclobutane pyrimidine dimers, pyrimidine (6-4) pyrimidone photoproducts, 8-hydroxy-2 -deoxyguanosine
Introduction
Skin aging can be divided into endogenous aging and extrinsic aging by internal or environmental factors such as ultraviolet (UV) rays. In particular, skin – unlike most other organs – is directly affected by UV radiation.
UV radiation from the sun is classified into three types: UVA (320–400 nm), UVB (280–320 nm), and UVC (200–280 nm). While UVC is mostly absorbed by the ozone layer, UVB and UVA reach the surface of the earth and affect human skin. UVB has greater energy than UVA, so it burns the epidermis in a shorter period of time than UVA does. Conversely, UVA penetrates deeper into the dermis than UVB does and affects connective tissues such as collagen, which causes actual aging of the skin, and is known as photo-aging. Recent studies have revealed that UVA plays a key role in pigment changes that occur with age, a major sign of skin photo-aging in Asians.1 The mechanism of UVA-induced skin aging is well understood. UVA radiation directly induces major alterations in the dermal compartment through the generation of reactive oxygen species (ROS).2 UVA-induced, excess intracellular ROS activate mitogen-activated protein kinases (MAPKs) and nuclear factor-kappa B (NF-κB), which in turn induce the transcriptional regulation of matrix metalloproteinases (MMPs). Finally, this results in the degradation of collagen and elastin, subsequently leading to photo-aging.3 UVA also induces photoproducts such as pyrimidine dimers, including cyclobutane pyrimidine dimers (CPDs), and pyrimidine (6-4) pyrimidone photoproducts (6-4PP).4
Epidermal growth factor (EGF) and its receptor (EGFR) control a broad spectrum of diverse biological effects, including cellular proliferation, differentiation, wound healing, hair follicle morphogenesis, and tumorigenesis. Notably, EGF plays a key role in healing wounds through the stimulation, proliferation, and migration of keratinocytes, endothelial cells, and fibroblasts.5 Interestingly, according to Chou et al, urinary EGF started to decline in people in their 20s and was lowest in people aged 71–80 years.6 Also, aged dermal fibroblasts exhibit reduced EGFR expression, which is associated with decreased cell migration and proliferation, and the loss of flexibility and elasticity.7,8 On the skin side, EGF is a useful agent for improving chronic skin ulcers, aging, acne, and skin homeostasis.9 It is well known that topical treatment with recombinant human EGF (rhEGF) ointment encourages the healing of wounds by inducing epidermal proliferation and increasing wound contraction related to myofibroblast proliferation and collagen deposition.10 However, the effect of EGF on the production of ROS is much debated, and depends on the concentration, time, and cell state.11–14 Moreover, the effect of EGF on the production of ROS or antioxidant enzymes in skin cells is not well known.
Emerging evidence indicates that EGFR modulates cell proliferation, differentiation, apoptosis, and DNA repair.15 The modulation of DNA repair is suggested to occur in radiation-induced DNA double-strand breaks (DSBs). Kriegs et al revealed that EGFR signaling regulates the overall DSB repair capacity by modeling nonhomologous end-joining via the MAPK pathway.16 It has been established that UVB is able to induce the formation of DSB, and it has also been suggested that it occurs following the collapse of replication forks at unrepaired CPDs, which are readily generated by UVB.17 However, the use of UVA to induce DSB formation has been debated. Furthermore, it is not well known whether EGFR signaling regulates DNA damages induced by UVA directly or indirectly.
Thus, this study sought to determine whether EGF affects events in UVA-irradiated human dermal fibroblasts by investigating the expression level of intracellular ROS, antioxidant enzymes, and DNA damage-related molecules in UVA-irradiated human dermal fibroblasts.
Materials and methods
Reagents
For the purposes of this study, rhEGF (Daewoong Pharmaceutical Co., Ltd., Seoul, Korea) was used, and ascorbic acid (Vitamin C) was purchased from Sigma-Aldrich Co. (St Louis, MI, USA).
Cell culture
Normal human dermal fibroblasts (Thermo Fisher Scientific, Waltham, MA, USA) were cultured in Medium 106 (Thermo Fisher Scientific) with a low serum growth supplement (LSGS; Thermo Fisher Scientific). Before the reagent treatment, the cells were cultured overnight in Medium 106 without LSGS (Thermo Fisher Scientific) to induce starvation. The cells were maintained in a humidified atmosphere of 5% CO2 at 37°C, and the medium was replaced every 2 days. Fibroblasts cultured with or without reagents were UVA-irradiated at 40 mJ/cm2 twice per day for a period of 5 days, totaling 400 mJ/cm2.
UVA irradiation
For the UVA irradiation, a UV Crosslinker (Ultra-Violet Product Ltd., Cambridge, UK) was used, with a UV spectrum of 365 nm. The cultured medium was removed gently and the cells were washed with PBS. Then, the PBS was also removed. After removing the PBS, the cell culture plates were placed in the UV Crosslinker and UVA-irradiated at 40 mJ/cm2. After the UVA irradiation, the cells were cultured with or without reagents. This process was repeated twice daily for a total of 5 days.
2′, 7′-Dichlorofluoresceindiacetate (DCFDA) micro-plate assay
The intracellular ROS levels were determined in fibroblasts using a DCFDA assay kit (Cell Biolabs, Inc., San Diego, CA, USA). Briefly, the cells were UVA-irradiated at 40 mJ/cm2 twice daily for 5 days, with or without reagents, and were washed with PBS three times. After that, the cells were treated with the DCFDA solution and incubated at 37°C for 1 h. Then, the cells were UVA-irradiated at 40 mJ/cm2 once more. Immediately thereafter, the fluorescence was read with a fluorometric plate reader at 480/530 nm (Molecular Devices, Sunnyvale, CA, USA).
Enzyme-linked immunosorbent assay (ELISA)
The expression of catalase and superoxide dismutase-1 (SOD-1) in the cells was determined using specific ELISA kits according to the manufacturer’s instructions. The catalase and SOD-1-specific ELISA kits were obtained from Abcam (Cambridge, UK). After treatment with UVA and/or reagents, whole-cell lysates were collected using a cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA). The expression of each enzyme was then measured according to the supplier’s protocol. In addition, a specific ELISA assay kit detected that DNA damage-related molecules – including CPD, 6-4PP, and 8-hydroxy-2′–deoxyguanosine (8-OHdG) – had been generated in the UVA-irradiated fibroblasts with or without reagents. Briefly, after UVA irradiation, the genomic DNA was extracted from the cells, using a QIAamp DNA isolation kit (Qiagen, Hilden, Germany). CPD and 6-4PP were evaluated with an OxiSelectTM UV-induced oxidative DNA damage (CPD/6-4PP) ELISA kit (Cell Biolabs., Inc.), according to the manufacturer’s protocol. Also, 8-OHdG was detected using an OxiSelectTM DNA damage ELISA kit (8-OHdG Quantitation) (Cell Biolabs., Inc.).
Single gel electrophoresis assay (comet assay)
DNA damage caused by UVA was observed using the comet assay. The UVA-irradiated cells treated with or without reagents were harvested by trypsin-EDTA. The cell pellets were washed by PBS and resuspended in 100 μL PBS. The comet assay was performed using the OxiSelectTM Comet Assay Kit (Cell Biolabs, Inc.), according to the manufacturer’s manual. The slides were imaged with a Zeiss LSM 800 confocal microscope (Carl Zeiss, Jena, Germany), and the images were analyzed using the ZEN 2012 software (Carl Zeiss).
Real-time reverse-transcription quantitative polymerase chain reaction (RT-qPCR)
To confirm the level of NF-κB gene expression, the total ribonucleic acid (RNA) was extracted from the cells using a ReliaPrep™ RNA Cell Miniprep System (Promega, Madison, WI, USA), after which 1 μg of the total RNA was converted to cDNA using a TaKaRa RNA PCR Kit v2.1 (TaKaRa Bio Inc., Shiga, Japan), under the following reaction conditions: 45°C for 45 min and 95°C for 5 min. Probes were obtained from Applied Biosystems as Assays-on-Demand™ Gene Expression Assays (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]: Hs02758991_g1, NF-κB: Hs00765730_m1, p38α: Hs01051152_m1). Reactions were carried out on an ABI StepOnePlus™ (Applied Biosystems), and the relative transcription levels were determined by GAPDH as the reference gene. The data were analyzed with the ABI StepOnePlus™ software (Applied Biosystems).
NF-κB p65 transcription factor assay (ELISA)
To examine the NF-κB p65 transcription factor protein level, nuclear extracts were collected using a nuclear extraction kit (Cayman, Ann Arbor, MI, USA) in the UVA-irradiated fibroblasts with or without reagents. The protein concentrations were determined with a Bio-Rad reagent (Bio-Rad Laboratories, Hercules, CA, USA). The activation of the NF-κB (p65) transcription factor was detected using an NF-κB (p65) transcription factor assay kit (Cayman). Briefly, 10 μg of nuclear proteins were added to the wells with a complete transcription factor buffer and incubated overnight at 4°C. Blank wells, a positive control, and nonspecific binding samples were also included on the plate. After that, NF-κB binding was detected by incubating monoclonal and secondary antibodies against the NF-κB p65 subunit. The reaction was quantified at 450 nm. The percentage change in the activity of each test sample relative to the average of the untreated samples was then determined.
Statistical analysis
All of the experiments were performed in triplicate, and the results are expressed as the mean ± standard deviation (STDEV). P-values <0.05 were considered statistically significant. A one-way analysis of variance with Dunnett’s test was conducted using a GraphPad Prism v 7 (GraphPad Software, San Diego, CA, USA).
Results
UVA-induced intracellular ROS were inhibited by rhEGF
To confirm the UVA dosage, fibroblasts were UVA-irradiated at 10, 20, or 40 mJ/cm2 twice daily for 5 days, for a total of 100, 200, or 400 mJ/cm2, respectively. The intracellular ROS level was increased by UVA in a dose-dependent manner (Figure 1A). Thus, we set the dose of UVA irradiation at 40 mJ/cm2. Then, fibroblasts with or without reagents – including several concentrations of rhEGF and 250 μM vitamin C used as the positive control – were UVA-irradiated at 40 mJ/cm2 twice daily for 5 days, for a total of 400 mJ/cm2. Intracellular ROS increased by UVA were inhibited by rhEGF in a concentration-dependent manner, and vitamin C also inhibited the increment of intracellular ROS in the UVA-treated cells (Figure 1B). To determine the reason for the decrease of intracellular ROS production by rhEGF, we measured the expression of antioxidative enzymes, including catalase and SOD-1. As a result, rhEGF increased the catalase and SOD-1 expression in the UVA-treated human dermal fibroblasts (Figure 1C and D). Moreover, 1 and 10 ng/mL rhEGF increased the expression of catalase and SOD-1 more than that increased by 250 μM vitamin C (Figure 1C and D).
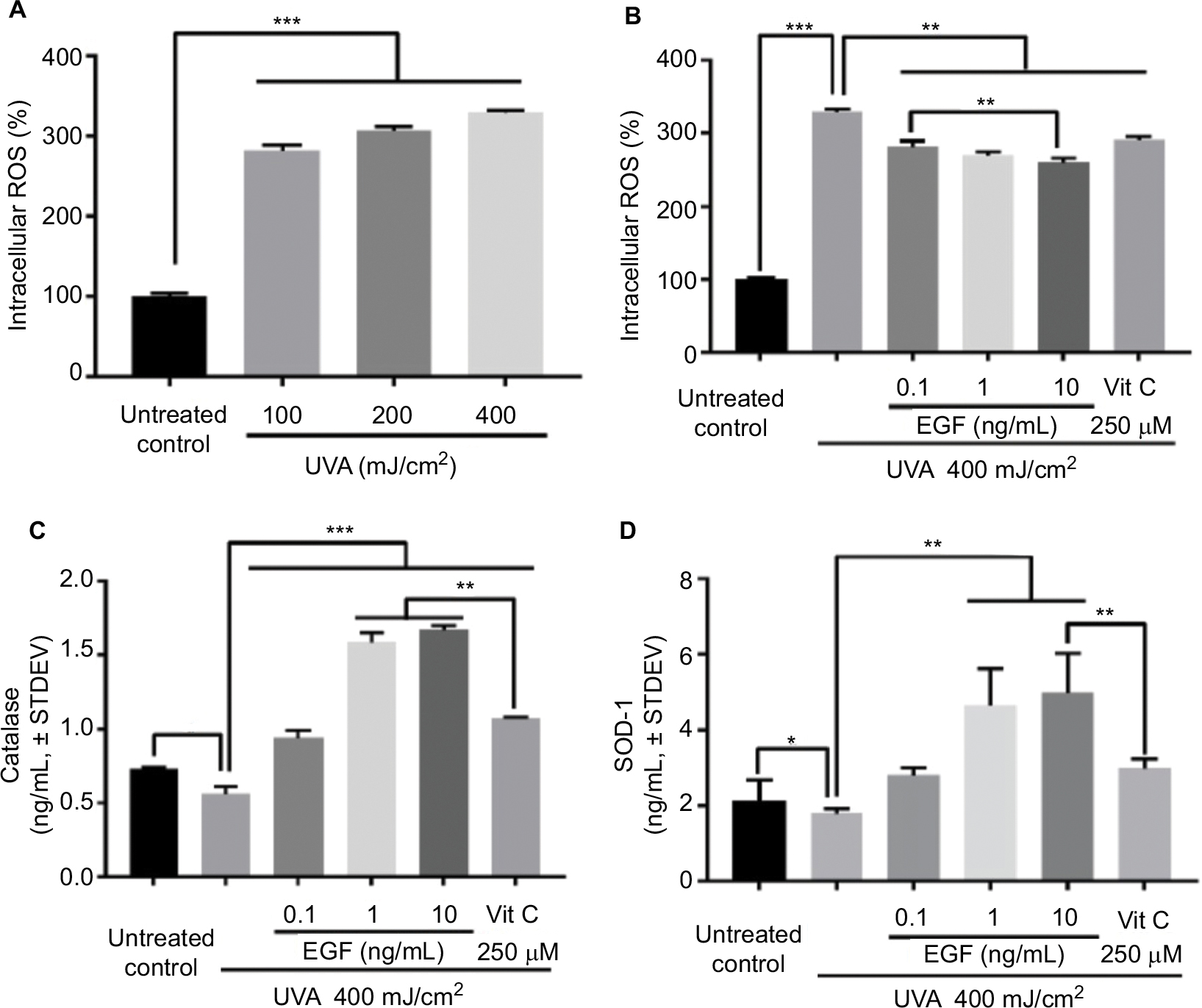 | Figure 1 rhEGF inhibited UVA-increased intracellular ROS in fibroblasts in a concentration-dependent manner: (A) Fibroblasts were UVA-irradiated at 10, 20, or 40 mJ/cm2 twice daily for 5 days (for a total of 100, 200, or 400 mJ/cm2). The intracellular ROS level was increased by UVA in a dose-dependent manner. (B) Fibroblasts cultured with or without reagents, including rhEGF and vitamin C, were UVA-irradiated at 40 mJ/cm2 twice daily for 5 days, for a total of 400 mJ/cm2. The UVA-induced intracellular ROS level was inhibited by rhEGF and vitamin C. (C, D) rhEGF increased the expression of catalase and SOD-1 in UVA-irradiated fibroblasts. Vitamin C also upregulated the expression of catalase and SOD-1 in UVA-irradiated fibroblasts (* P<0.05, **P<0.01, ***P<0.001). Abbreviations: ROS, reactive oxygen species; EGF, epidermal growth factor; UVA, ultraviolet A; STDEV, standard deviation; rhEGF, recombinant human EGF; SOD-1, superoxide dismutase-1; Vit, vitamin. |
rhEGF protects against DNA damage induced by UVA irradiation
Next, we assessed the UVA-induced DNA damage using a single cell gel electrophoresis assay known as the comet assay. As a result, in the untreated control group, the presence of a tail signifying DNA damage was not observed (Figure 2A). However, a tail was observed in the UVA-irradiated cells (Figure 2B). On the other hand, no tail was observed in the cells cotreated with UVA and various concentrations of rhEGF (Figure 2C–E). Similarly, no tail was observed in the cells cotreated with vitamin C and UVA (Figure 2F). After that, we measured the length of the tail using the ZEN 2012 blue edition software. As a result, the tail was observed to be longest in the UVA-irradiated cells, while the tail length in the UVA and rhEGF or vitamin C cotreated cells was similar to that of the untreated control (Figure 2G). Next, we quantified the DNA damage using CPD, 6-4PP, and 8-OHdG specific ELISA kits. In the UVA-irradiated fibroblasts, three kinds of DNA damage molecules increased; and the detection expression level of 8-OHdG was the highest while that of 6-4PP was the lowest (Figure 2H). Interestingly, 1 and 10 ng/mL rhEGF inhibited the increment, but 0.1 ng/mL rhEGF did not (Figure 2H).
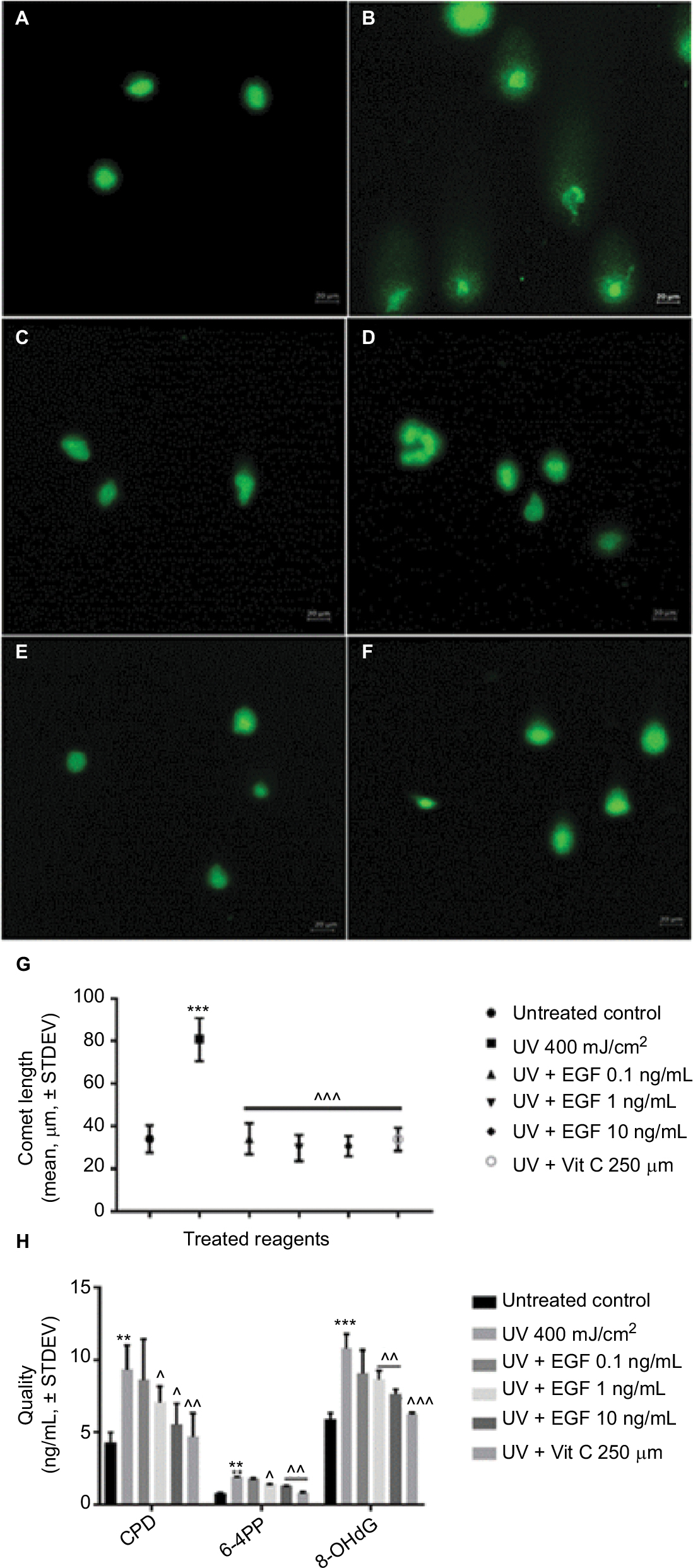 | Figure 2 rhEGF inhibited DNA damage induced by UVA irradiation in fibroblasts. (A–F) UVA-induced DNA damage was detected by a single cell gel electrophoresis assay: (A) untreated control, (B) UVA 400 mJ/cm2, (C) UVA 400 mJ/cm2 + EGF 0.1 ng/mL, (D) UVA 400 mJ/cm2 + EGF 1 ng/mL, (E) UVA 400 mJ/cm2 + EGF 10 ng/mL, (F) UVA 400 mJ/cm2 + vitamin C 250 μM, (G) Tail length was measured by ZEN 2012 blue edition software. rhEGF and vitamin C had protective effect on DNA damage in UVA-irradiated fibroblasts. (H) Directly or indirectly induced DNA damaged molecules by UVA irradiation were quantified using specific ELSIA kits. CPD, 6-4PP, and 8-OHdG expressions were increased by UVA, but this was inhibited by rhEGF and vitamin C. (** P<0.01, *** P<0.001 compared with the untreated control; ^P<0.05, ^^P<0.01, ^^^P<0.001 compared with UVA 400 mJ/cm2). Abbreviations: EGF, epidermal growth factor; UVA, ultraviolet A; STDEV, standard deviation; rhEGF, recombinant human EGF; Vit, vitamin; CPD, cyclobutene pyrimidine dimmers; 6-4PP, pyrimidine (6-4) pyrimidone photoproduct; 8-OHdG, 8-hydroxy-2-deoxyguanosine; ELISA, enzyme-linked immunosorbent assay. |
rhEGF inhibited NF-κB gene expression and the NF-κB p65 protein levels in the nucleus induced by UVA irradiation
Finally, we examined the changes of NF-κB expression in the UVA-irradiated cells with or without reagents. The mRNA expression of NF-κB was induced by the UVA-irradiated cells, but that increment was downregulated by 1 and 10 ng/mL rhEGF and vitamin C (Figure 3A). The NF-κB p65 protein levels in the nucleus also increased in the UVA-irradiated fibroblasts (Figure 3B). Interestingly, unlike the mRNA expression result, only the 10 ng/mL rhEGF and vitamin C decreased the NF-κB p65 protein levels in the nucleus of the UVA-irradiated fibroblasts (Figure 3B).
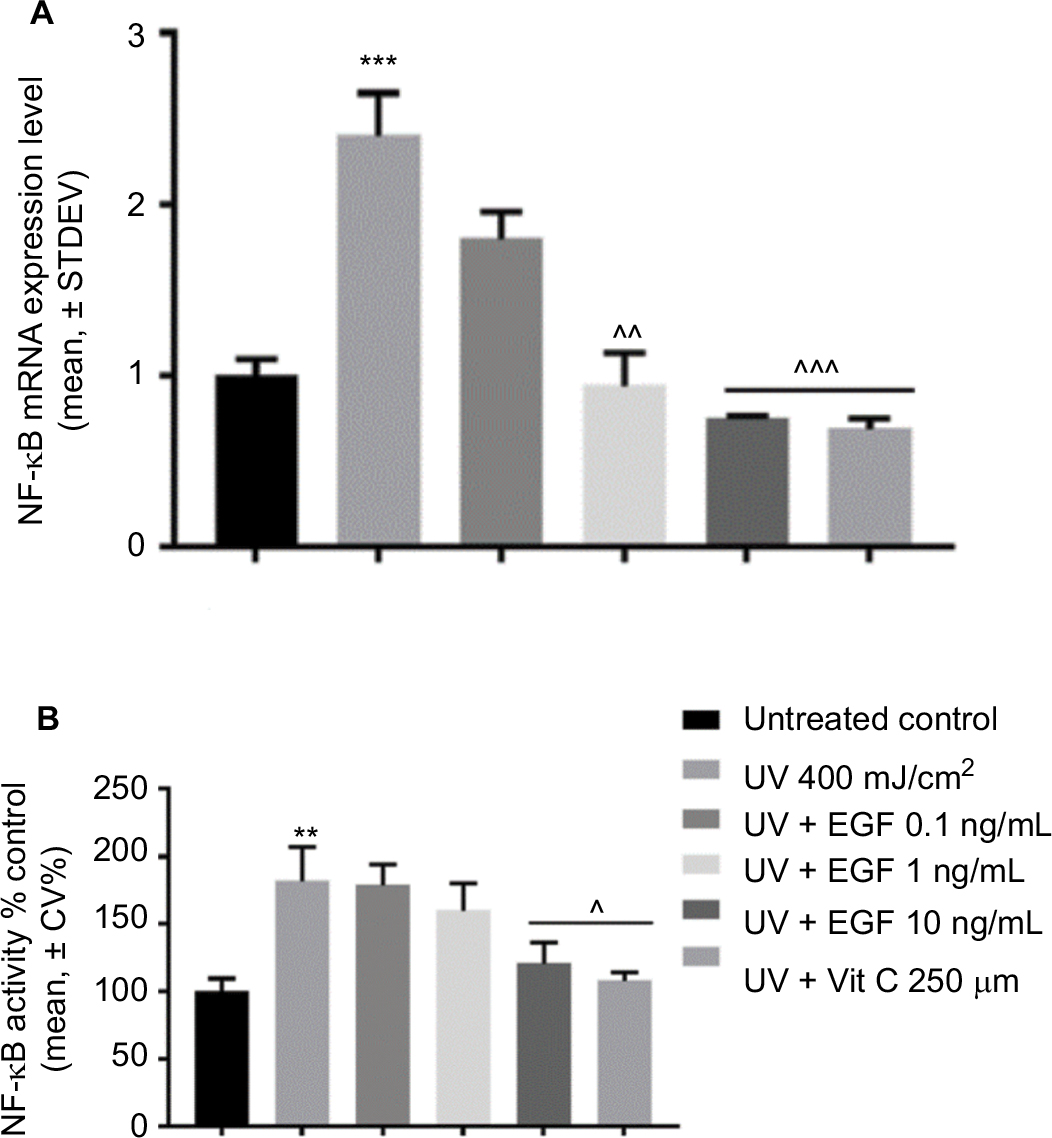 | Figure 3 rhEGF inhibited NF-kB mRNA expression and activity. (A) The mRNA expression level of NF-κB was increased by UVA, but this increment was downregulated by rhEGF and vitamin C. (B) The activity of NF-κB was also increased by UVA, but this was inhibited by 10 ng/mL EGF and 250 μM vitamin C. (** P<0.01, *** P<0.001 compared with the untreated control, ^P<0.05, ^^P<0.01, ^^^P<0.001 compared with UV 400 mJ/cm2). Abbreviations: EGF, epidermal growth factor; UVA, ultraviolet A; STDEV, standard deviation; rhEGF, recombinant human EGF; Vit, vitamin. |
Discussion
ROS are an important factor in several chronic human diseases, including atherosclerosis, vascular diseases, mutagenesis and cancer, neurodegeneration, immunologic disorders, and even aging.18 In the skin, about 1.5–5% of the consumed oxygen is converted into ROS by intrinsic processes.19 ROS are continuously produced in the mitochondria via the aerobic metabolism, and are regarded as the main cause of intrinsic aging.20 UV radiation, and even visible wavelengths of radiation from the sun, can accelerate ROS generation.21 An increase of ROS by UV leads to the activation of MAPK p38, c-Jun amino-terminal kinase, and extracellular signal-regulated kinase, and this in turn provokes the expression of the transcription factor activator protein 1, resulting in the expression of MMP1, 3, and 9 in fibroblasts and keratinocytes.22–24 This process is reinforced by the production of ROS, which also results in the activation of MAPK and leads to the expression of NF-κB. In this study, we confirmed the induction of the intracellular ROS level in UVA-irradiated human dermal fibroblasts, which was inhibited by rhEGF in a dose-dependent manner. A previous study has revealed that rhEGF treatment increased the contractility of old human fibroblasts,9 and also showed that topical treatment with rhEGF induced collagen synthesis in rats.10 It discussed the possibility that EGF has potential for use as a regenerative agent for aging skin. The present study confirmed the effects of ROS inhibition and DNA breaks on the recovery of rhEGF. Therefore, we considered that these results may suggest a new mechanism for improving the aging skin by EGF.
SOD is one of the most important enzymes that can regulate ROS. In mammals, there are three isoforms of SOD, including SOD-1, -2, and -3. All three human SODs have a huge impact on aging skin. Normally, a deletion of SOD is lethal, as demonstrated in mice, but the lifespan of mice with SOD mimetics can be prolonged for several weeks.25 Sod1–/– mice show clear skin atrophy that is also observed in aged human beings.26,27 Catalase is also a very important enzyme in protecting cells from damage due to oxidation by ROS. The catalase enzyme is very prominently expressed in the skin, especially in the stratum corneum, where the amount of catalase exceeds the amount of SOD. Moreover, it was observed that the activity of this enzyme decreased as it penetrated further into the skin layer. 28,29 It has been revealed that UVB as well as UVA inhibit the expression and activity of these enzymes. 30–32 In the present research, rhEGF increased the expression of antioxidant-related enzymes, including catalase and SOD-1, in UVA-irradiated fibroblasts. As a result, the amount of SOD-1 expression was higher than that of catalase, which is thought to be due to the cell line.
It is well known that UV radiation can alter DNA strand breaks. Direct UV rays induce two main photochemical reactions in DNA, including CPD and 6-4PP.33,34 Recently, it was revealed that UVA induced CPD via a direct photochemical mechanism.35 Moreover, UVA-induced DNA DSBs can be generated from the repair of clustered oxidative DNA damages.36 On the other hand, UV radiation indirectly affects DNA damage by increasing the ROS level.37 Among the numerous types of oxidative DNA damage, the formation of 8-OHdG is a ubiquitous marker of oxidative stress. According to Cadet et al, UV radiation also targets the DNA base guanine, giving rise to 8-OHdG in the strand DNA.38 In this study, rhEGF appears to have a protective effect against UVA-induced DNA damages. It was observed that the length of the tail, which signifies DNA damage, was shorter in the groups cotreated with UVA and rhEGF than in the group treated with UVA alone. Also, the UVA-induced expression of CPD, 6-4PP, and 8-OHdG was decreased by rhEGF in a dose-dependent manner. These results suggest that rhEGF might play a role in protecting against DNA damage whether induced directly or indirectly by UVA, leading us to conclude that further studies of the related mechanisms are required. One possibility is that the reduction in ROS by rhEGF may be associated with a decrease in indirect lesion breaks and 8-OHdG by rhEGF. In addition, a possible mechanism involved in the reduction of CPD and 6-4PP by rhEGF could be that EGF may be helping cells to increase the function of DNA repair proteins which were affected by ROS after UVA irradiation.
As previously described, UVA increases MMPs expression through MAPK or NF-κB signaling. We have previously reported that rhEGF regulates the signaling of NF-κB to inhibit the expression of pro-inflammatory cytokines in inflammatory milieu in vitro and in vivo.39 Therefore, in this study, we investigated whether rhEGF affects the NF-κB signaling induced by UVA. As a result, it was confirmed that not only the gene expression of NF-κB by UVA, but also its activity was inhibited by rhEGF.
In conclusion, our results indicate that EGF plays an important role in regulating UVA-induced skin damage through the suppression of ROS expression and on DNA damage in relation with NF-κB signaling. Therefore, these findings suggest that EGF might have a beneficial effect on skin aging, and more particularly photo-aging.
Acknowledgment
An earlier version of this work was presented as an abstract in Growth Factors, Cell Adhesion and Matrix Biology, Journal of Investigative Dermatology, 2017.
Author contributions
JMK performed the experiments, analyzed the data, and wrote the manuscript. JEC and KNK critically reviewed the study proposal and discussed the data. YSK contributed to the study design, and assisted in drafting and editing the manuscript. All of the authors read and approved the final version of the manuscript. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Battie C, Jitsukawa S, Bernerd F, Del Bino S, Marionnet C, Verschoore M. New insights in photoaging, UVA induced damage and skin types. Exp Dermatol. 2014;23(Suppl 1):7–12. | ||
Murai M, Tsuji G, Hashimoto-Hachiya A, Kawakami Y, Furue M, Mitoma C. An endogenous tryptophan photo-product, FICZ, is potentially involved in photo-aging by reducing TGF-beta-regulated collagen homeostasis. J Dermatol Sci. 2017;89(1):19–26. | ||
Pittayapruek P, Meephansan J, Prapapan O, Komine M, Ohtsuki M. Role of matrix metalloproteinases in photoaging and photocarcinogenesis. Int J Mol Sci. 2016;17(6). pii: E868. | ||
Douki T, Reynaud-Angelin A, Cadet J, Sage E. Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation. Biochemistry. 2003;42(30):9221–9226. | ||
Zhang Z, Xiao C, Gibson AM, Bass SA, Khurana Hershey GK. EGFR signaling blunts allergen-induced IL-6 production and Th17 responses in the skin and attenuates development and relapse of atopic dermatitis. J Immunol. 2014;192(3):859–866. | ||
Chou JS, Reiser IW, Porush JG. Aging and urinary excretion of epidermal growth factor. Ann Clin Lab Sci. 1997;27(2):116–122. | ||
Liu Y, Guyton KZ, Gorospe M, et al. Age-related decline in mitogen-activated protein kinase activity in epidermal growth factor-stimulated rat hepatocytes. J Biol Chem. 1996;271(7):3604–3607. | ||
Reenstra WR, Yaar M, Gilchrest BA. Aging affects epidermal growth factor receptor phosphorylation and traffic kinetics. Exp Cell Res. 1996;227(2):252–255. | ||
Kim D, Kim SY, Mun SK, Rhee S, Kim BJ. Epidermal growth factor improves the migration and contractility of aged fibroblasts cultured on 3D collagen matrices. Int J Mol Med. 2015;35(4):1017–1025. | ||
Kwon YB, Kim HW, Roh DH, et al. Topical application of epidermal growth factor accelerates wound healing by myofibroblast proliferation and collagen synthesis in rat. J Vet Sci. 2006;7(2):105–109. | ||
Price LT, Chen Y, Frank L. Epidermal growth factor increases antioxidant enzyme and surfactant system development during hyperoxia and protects fetal rat lungs in vitro from hyperoxic toxicity. Pediatr Res. 1993;34(5):577–585. | ||
Koken T, Erkasap N, Serteser M, Kahraman A. Epidermal growth factor increases tissue antioxidant enzyme activities in ethanol-induced gastric injury in rat. J Physiol Biochem. 2006;62(4):237–243. | ||
Coskun S, Gulec EG, Balabanli B, Acarturk F. Effects of epidermal growth factor on lipid peroxidation and nitric oxide levels in oral mucosal ulcer healing: a time-course study. Surg Today. 2007;37(7):570–574. | ||
Peng H, Wen TC, Tanaka J, et al. Epidermal growth factor protects neuronal cells in vivo and in vitro against transient forebrain ischemia- and free radical-induced injuries. J Cereb Blood Flow Metab. 1998;18(4):349–360. | ||
Szumiel I. Epidermal growth factor receptor and DNA double strand break repair: the cell’s self-defence. Cell Signal. 2006;18(10):1537–1548. | ||
Kriegs M, Kasten-Pisula U, Rieckmann T, et al. The epidermal growth factor receptor modulates DNA double-strand break repair by regulating non-homologous end-joining. DNA Repair (Amst). 2010;9(8):889–897. | ||
Garinis GA, Mitchell JR, Moorhouse MJ, et al. Transcriptome analysis reveals cyclobutane pyrimidine dimers as a major source of UV-induced DNA breaks. EMBO J. 2005;24(22):3952–3962. | ||
Castro L, Freeman BA. Reactive oxygen species in human health and disease. Nutrition. 2001;17(2):161, 163–165. | ||
Poljsak B, Dahmane RG, Godic A. Intrinsic skin aging: the role of oxidative stress. Acta Dermatovenerol Alp Pannonica Adriat. 2012;21(2):33–36. | ||
Farage MA, Miller KW, Elsner P, Maibach HI. Intrinsic and extrinsic factors in skin ageing: a review. Int J Cosmet Sci. 2008;30(2):87–95. | ||
Pattison DI, Davies MJ. Actions of ultraviolet light on cellular structures. EXS. 2006(96):131–157. | ||
Kohl E, Steinbauer J, Landthaler M, Szeimies RM. Skin ageing. J Eur Acad Dermatol Venereol. 2011;25(8):873–884. | ||
Lavker RM, Veres DA, Irwin CJ, Kaidbey KH. Quantitative assessment of cumulative damage from repetitive exposures to suberythemogenic doses of UVA in human skin. Photochem Photobiol. 1995;62(2):348–352. | ||
Fisher GJ, Datta SC, Talwar HS, et al. Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature. 1996;379(6563):335–339. | ||
Rinnerthaler M, Bischof J, Streubel MK, Trost A, Richter K. Oxidative stress in aging human skin. Biomolecules. 2015;5(2):545–589. | ||
Murakami K, Inagaki J, Saito M, et al. Skin atrophy in cytoplasmic SOD-deficient mice and its complete recovery using a vitamin C derivative. Biochem Biophys Res Commun. 2009;382(2):457–461. | ||
Shibuya S, Ozawa Y, Watanabe K, et al. Palladium and platinum nanoparticles attenuate aging-like skin atrophy via antioxidant activity in mice. PLoS One. 2014;9(10):e109288. | ||
Giacomoni PU, Declercq L, Hellemans L, Maes D. Aging of human skin: review of a mechanistic model and first experimental data. IUBMB Life. 2000;49(4):259–263. | ||
Hellemans L, Corstjens H, Neven A, Declercq L, Maes D. Antioxidant enzyme activity in human stratum corneum shows seasonal variation with an age-dependent recovery. J Invest Dermatol. 2003;120(3):434–439. | ||
Gegotek A, Rybaltowska-Kawalko P, Skrzydlewska E. Rutin as a Mediator of Lipid Metabolism and Cellular Signaling Pathways Interactions in Fibroblasts Altered by UVA and UVB Radiation. Oxid Med Cell Longev. 2017;2017:4721352. | ||
Hahn HJ, Kim KB, Bae S, et al. Pretreatment of Ferulic Acid Protects Human Dermal Fibroblasts against Ultraviolet A Irradiation. Ann Dermatol. 2016;28(6):740–748. | ||
Min W, Liu X, Qian Q, et al. Effects of baicalin against UVA-induced photoaging in skin fibroblasts. Am J Chin Med. 2014;42(3):709–727. | ||
Batista LF, Kaina B, Meneghini R, Menck CF. How DNA lesions are turned into powerful killing structures: insights from UV-induced apoptosis. Mutat Res. 2009;681(2-3):197–208. | ||
Setlow RB, Carrier WL. Pyrimidine dimers in ultraviolet-irradiated DNA’s. J Mol Biol. 1966;17(1):237–254. | ||
Mouret S, Philippe C, Gracia-Chantegrel J, et al. UVA-induced cyclobutane pyrimidine dimers in DNA: a direct photochemical mechanism? Org Biomol Chem. 2010;8(7):1706–1711. | ||
Greinert R, Volkmer B, Henning S, et al. UVA-induced DNA double-strand breaks result from the repair of clustered oxidative DNA damages. Nucleic Acids Res. 2012;40(20):10263–10273. | ||
Zhang X, Rosenstein BS, Wang Y, Lebwohl M, Wei H. Identification of possible reactive oxygen species involved in ultraviolet radiation-induced oxidative DNA damage. Free Radic Biol Med. 1997;23(7):980–985. | ||
Cadet J, Douki T, Pouget JP, Ravanat JL. Singlet oxygen DNA damage products: formation and measurement. Methods Enzymol. 2000;319:143–153. | ||
Choi SY, Lee YJ, Kim JM, Kang HJ, Cho SH, Chang SE. Epidermal Growth Factor Relieves Inflammatory Signals in Staphylococcus aureus-Treated Human Epidermal Keratinocytes and Atopic Dermatitis-Like Skin Lesions in Nc/Nga Mice. BioMed Res Int. 2018;2018:9. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.