")
Back to Journals » The Application of Clinical Genetics » Volume 11
Potential oligogenic disease of mental retardation, short stature, spastic paraparesis and osteopetrosis
Authors Alsemari A , Alsuhaibani M, Alhathlool R, Ali BM
Received 24 April 2018
Accepted for publication 1 August 2018
Published 8 November 2018 Volume 2018:11 Pages 129—134
DOI https://doi.org/10.2147/TACG.S172176
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Abdulaziz Alsemari,1 Mohanned Alsuhaibani,2 Rawabi Alhathlool,1 Bayan Mamdouh Ali1
1Department of Neurosciences, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia; 2Department of Radiology, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
Abstract: The interaction of multiple genetic factors, as opposed to monogenic inheritance, has been suspected to play a role in many diseases. This interaction has been described as an oligogenic inheritance model, which may be a useful tool in explaining certain clinical observations. The purpose of this study was to search for novel genetic defects among members of a family with traits that include mental retardation, short stature, osteopetrosis, calcification of basal ganglia, and thinning of the corpus callosum. In the index case (111-4), we identified four homozygous mutations: chromosome 8, intron2 (c.232+1G>A) at CA2 gene; chromosome 15, exon 32 (c.6100C>T) at the SPG11; chromosome 5, exon 11 (c.1015G>A) at the MCCC2; and chromosome 9, exon 9 (C.1193g>t) at the LARP gene. The mutations were confirmed by Sanger sequencing, and both parents were observed to be heterozygous for the four mutations. A moderately affected sister of the index case was homozygous for only three mutations in CA2, LARP, and Mccc2, while a nonaffected sister was heterozygous for three mutations in CA2, LARP, and MCCC2 and negative for SPG11. The clinical features of the two affected sisters can be explained distinctively by each homozygous mutation in an oligogenic pattern of inheritance. This family represents an example of an oligogenic pattern of inheritance of mental retardation, short stature, spastic paraparesis, and osteopetrosis.
Keywords: mental retardation, short stature, spastic paraparesis, CA2, SPG11, LARP, oligogenic
Introduction
Short stature, mental retardation, and facial dysmorphias represent major components of many genetic syndromes. In most of these syndromic disorders, the cause is identified at the cellular level. Growth failure and psychomotor delay are part of many syndromes and may be due to a wide variety of mechanisms, although for many of these syndromes, the underlying mechanisms are still unknown.1
There have been several reports in which molecular investigations have revealed the existence of more than one mutation in either one or several genes. Bardet–Biedl syndrome is a classic example of triallelic or oligogenic inheritance.2 This syndrome is a disorder that has helped to identify oligogenic inheritance in many diseases.3
Oligogenic inheritance refers to the concept that certain diseases are not inherited as simple single-gene Mendelian disorders nor are they classic complex traits, but rather, they fit a model in which mutations in a small number of genes may interact genetically to manifest a phenotype.4
Clinical report
Our index case is a 23-year-old female (111-4) (Figure 1). She was born normally, but she was delayed in reaching developmental milestones. She is short in stature compared with her parents. She had progressive speech and psychomotor delays. She could say only few words at the age of 2 years. She was able to stand and walk until the age of 4 but later slowly regressed and eventually lost all of her communication skills. Her neuropsychological assessment showed that she has severe cognitive delay with severe speech delay. She has micrognathia, low set ears, frontal bossing, and widely spaced teeth.
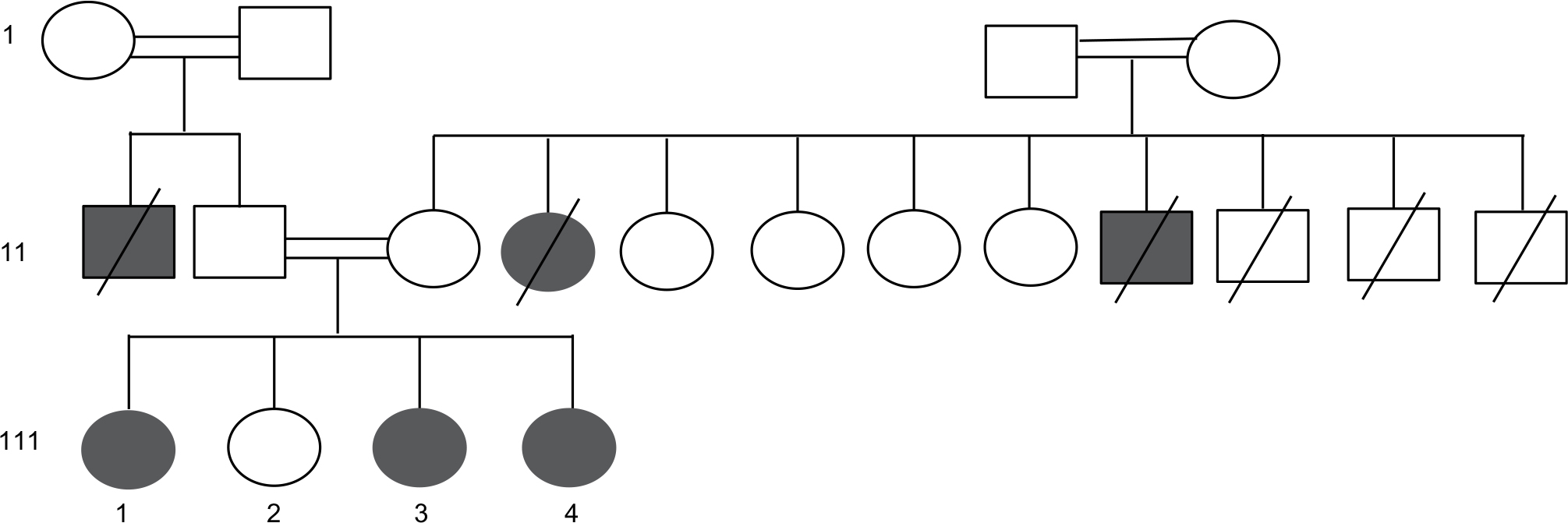 | Figure 1 Family pedigree. |
Our index case has a short stature (135 cm). Her father’s height is 185 cm, and her mother’s height is 155 cm. She exhibits spastic paraparesis with dystonic posture of the extremities. She has weakness, hyperreflexia, and increased tone. She had a normal blood count and platelets. Magnetic resonance imaging showed calcifications in the basal ganglia (Figure 2A, C), thinning of the corpus callosum, and signal changes in periventricular white matter (Figure 2B). Her skeletal survey demonstrated generalized increased sclerosis of the bony skeleton with mild metaphyseal expansion/flaring of the long tubular bones and a mild bowing deformity of the short tubular bones of the hands and feet (Table 1).
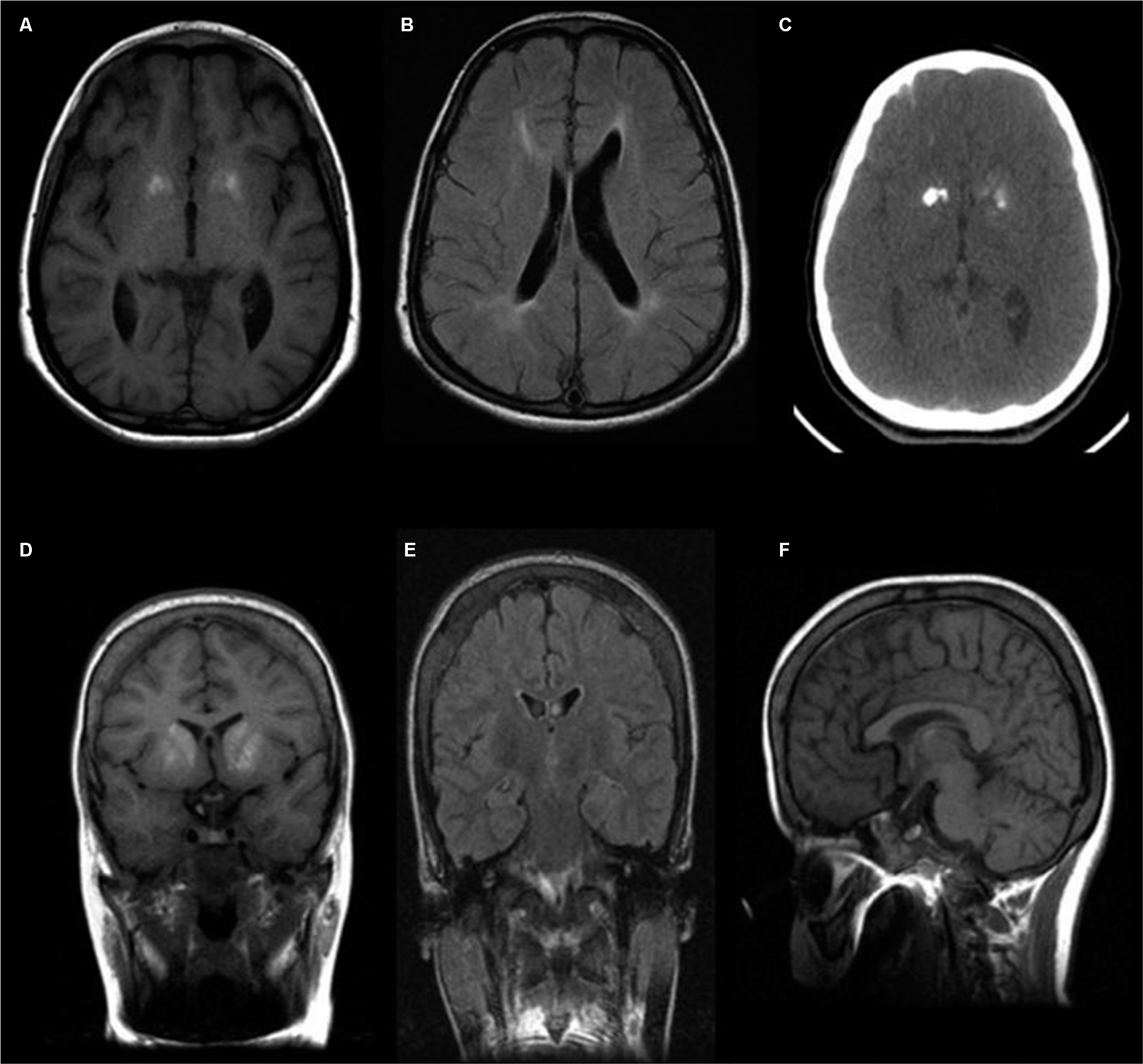 | Figure 2 Neuroimaging of the affected subjects. Notes: (A) High T1 signal in the basal ganglia. (B) Axial FLAIR sequence demonstrates bilateral periventricular confluent high FLAIR signal intensity. (C) Axial CT scan shows bilateral basal ganglia calcifications. (D) Coronal T1-WI shows bilateral symmetric high T1 signal of the basal ganglia compatible with calcification. (E) Coronal FLAIR demonstrates no significant abnormal white matter signal. (F) Midsagittal T1-WI demonstrates normal appearance of the midline corpus callosum. Abbreviations: CT, computed tomography; FLAIR, fluid-attenuated inversion recover. |
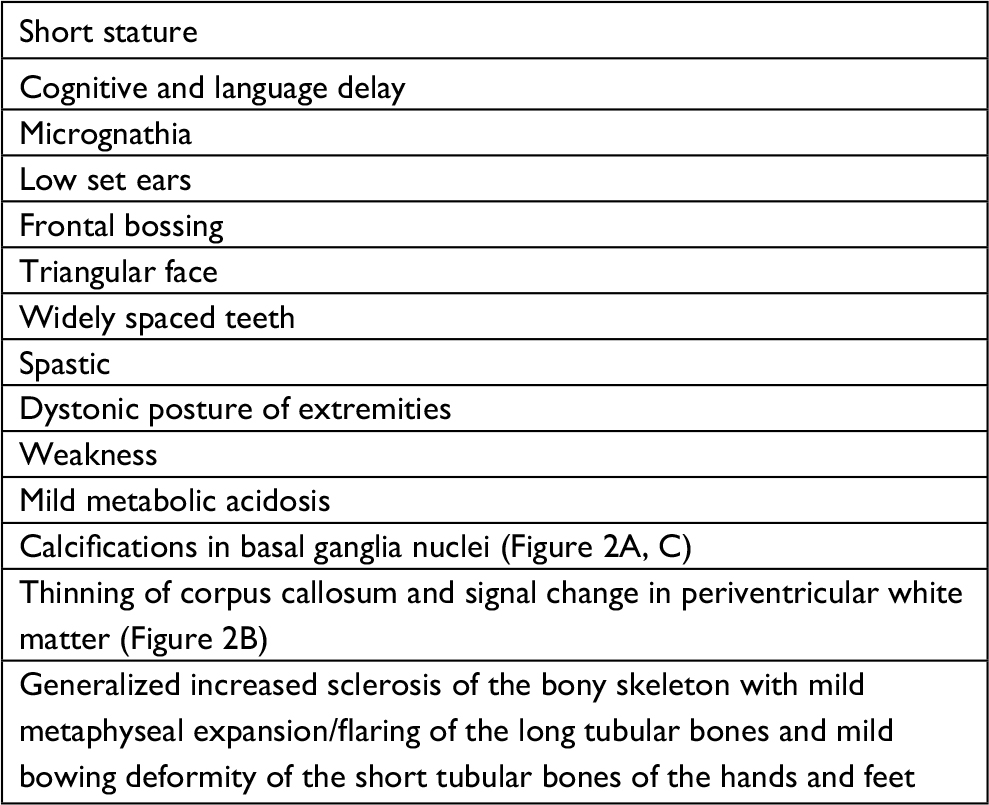 | Table 1 Clinical features of the severely affected sister (111-4) |
She has three sisters, of whom the eldest sister (111-1) is severely affected with a similar syndrome. The eldest sister is in a vegetative state and resides in a nursing home. A second sister, who is 29 years old, is affected with a similar syndrome but with a milder course. She is ambulatory and able to communicate and has no signs of spastic paraparesis (111-2). She is short in stature with mild cognitive delay. She also has micrognathia, low set ears, frontal bossing, and widely spaced teeth (Figure 3A, B). MRI brain of patient (111-2) showed calcifications of the basal ganglia (Figure 2D–F) and evidence of generalized increased sclerosis of the bony skeleton (Table 2).
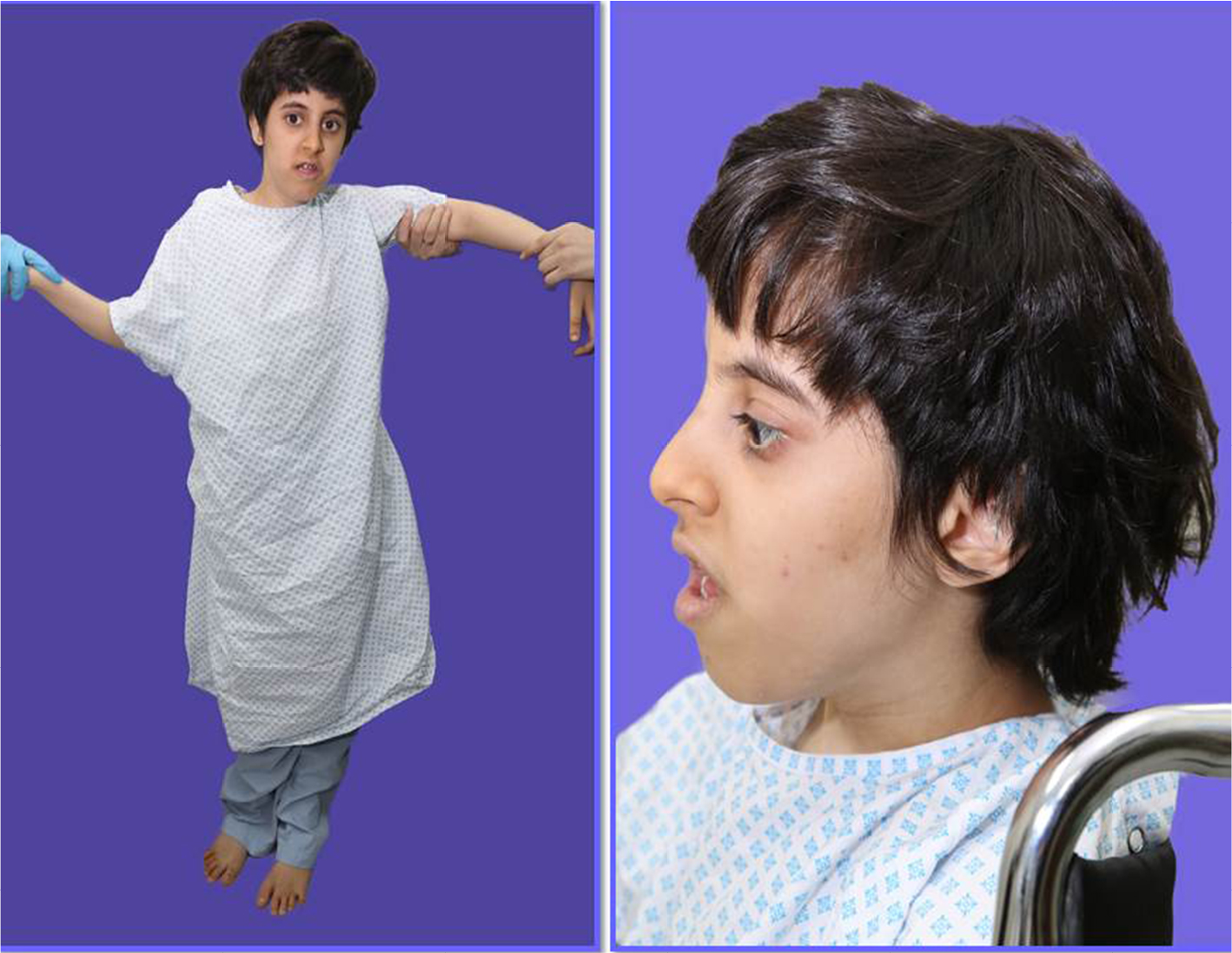 | Figure 3 Photographs of affected subject (111-2). Notes: (A, B) (111-2) is an affected sister with short stature, mild cognitive delay, micrognathia, low set ears, frontal bossing, and widely spaced teeth. |
 | Table 2 Clinical features of the moderately affected sister (111-2) |
Whole exome sequencing
Genome DNA was fragmented by sonication of the genomic DNA and ligating the fragments to the Illumina multiplexing PE adapters. The adapter-ligated DNA was later amplified by PCR using primers with sequencing barcodes (indexes). For the target enrichment/exome capture procedure, the precapture library was enriched by hybridization to biotin-labeled VC Rome 2.1 in-solution exome probes3 at 47°C for 64–72 hours. Additional probes for over 3,600 Mendelian disease genes were included in the capture in order to improve the exome coverage. For massively parallel sequencing, the postcapture library DNA was subjected to sequence analysis on the Illumina HiSeq platform for 100 bp paired-end reads.
Results
Synonymous variants, intronic variants not affecting the splicing site, and common benign variants were excluded from the interpretation unless they were previously reported as pathogenic variants. The variants were interpreted according to American College of Medical Genetics and Genomics guidelines and patient phenotypes. Variants related to patient phenotypes were confirmed by Sanger sequencing for patients and parents. The pathogenic variants in genes related to the clinical phenotype of the index case are summarized in Table 3. And the summary of the whole exome sequencing of the family members is charted in Table 4.
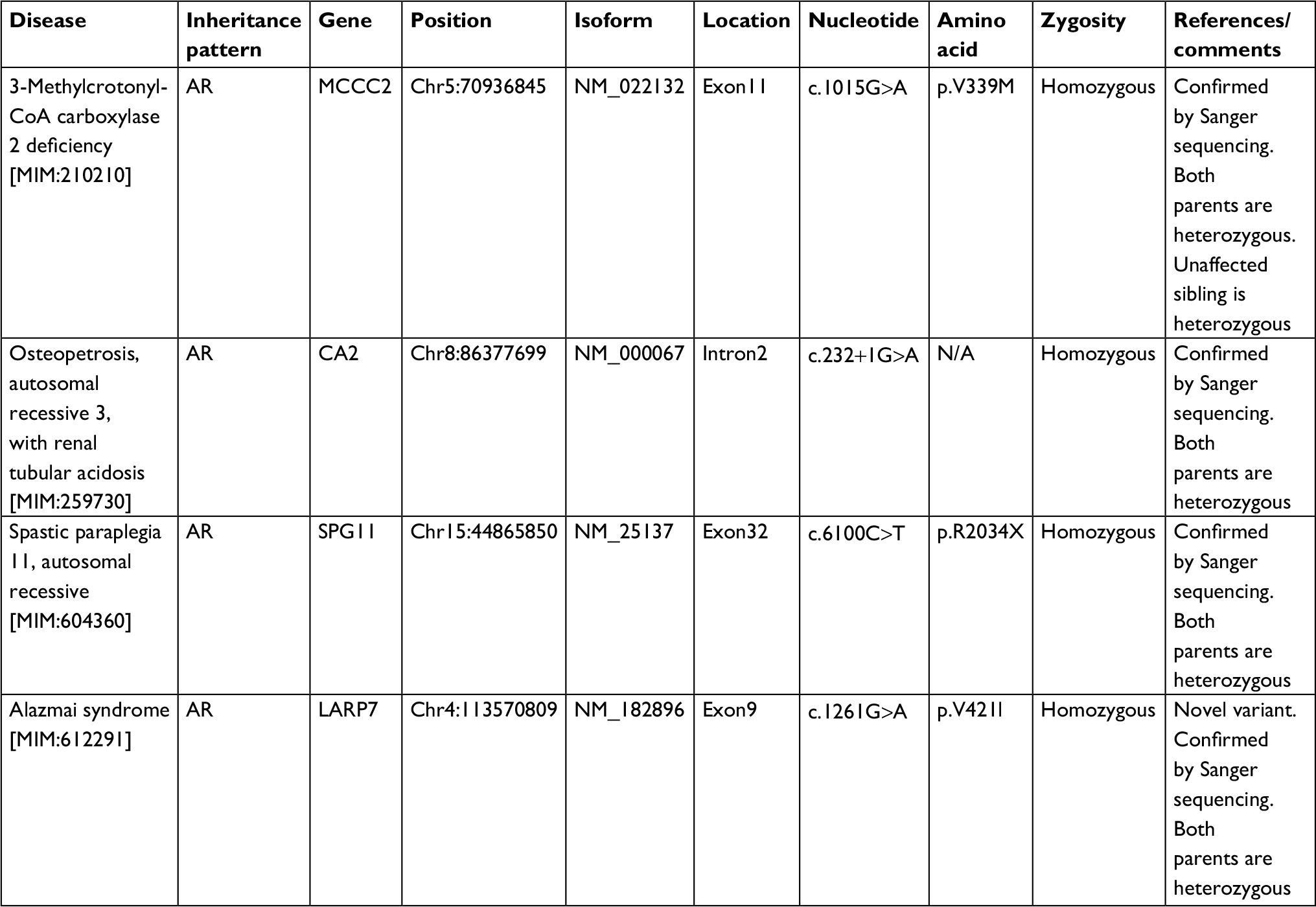 | Table 3 Pathogenic variants in disease genes related to clinical phenotype of the index case Abbreviation: AR, autosomal recessive. |
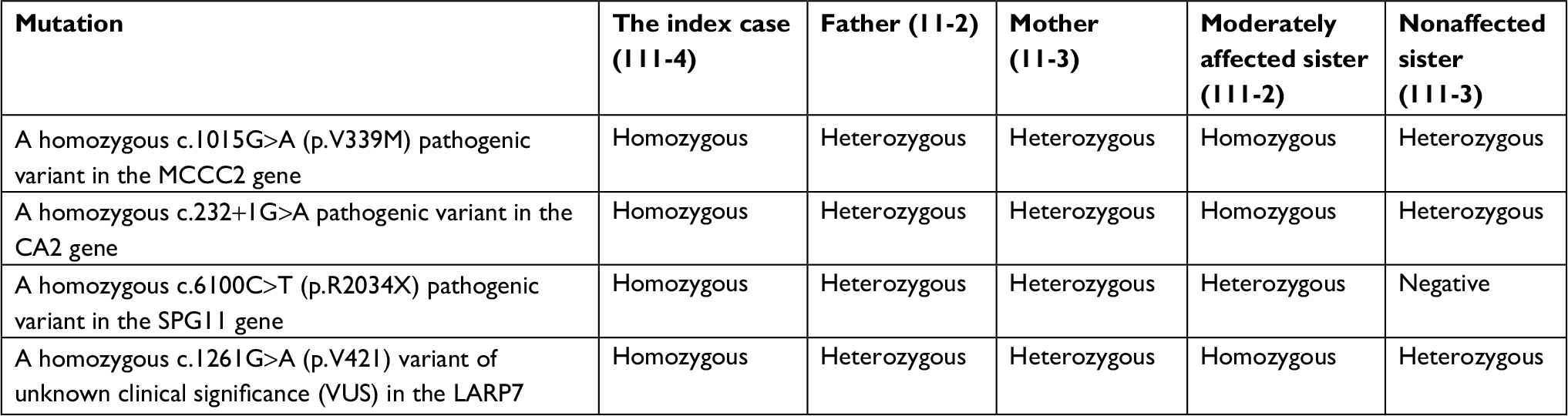 | Table 4 Summary of the whole exome sequencing of the family members |
Functional studies of the linked pathogenic genes
Carbonic anhydrase II is expressed at high levels in osteoclasts during bone resorption. It was demonstrated in two in vitro culture systems that antisense RNA and DNA block expression of CA II and lead to decreased bone resorption. In the choroid plexus, carbonic anhydrase II supports the transport of bicarbonate ions, sodium ions, and water from the blood to the cerebrospinal fluid.5
He et al identified human LARP7, which they called PIP7S. He et al identified PIP7S as a component of the 7SK snRNP. Nearly all nuclear 7SK was associated with PIP7S, and knockdown of PIP7S with short hairpin RNA (shRNA) reduced the levels of 7SK, indicating that PIP7S is required for 7SK stability.6
The SPG11 gene encodes spatacsin, a protein with a role in neuronal axonal growth, function, and intracellular cargo trafficking. By immunoprecipitation analysis of HeLa cells, Słabicki et al showed that KIAA0415 exists in a core protein complex containing SPG11, SPG15, C20ORF29, and DKFZp761E198.7
Discussion
Methodological and conceptual advances in human genetics have led to the identification of a large number of genes associated with human disease. Studies have shown that a substantial amount of the phenotypic variation in “Mendelian” disorders is due to genetic interaction of several genes.8,9 This research has also revealed that the traditional distinction between Mendelian and complex disorders might be blurred.8
Our index case (111-4, Figure 1) is homozygous for four mutations (Tables 3 and 4). The mutations have been previously reported and are associated with mental retardation, short stature, and craniofacial dysmorphic features.10–13 Nevertheless, 57% of MCCC2 mutations are asymptomatic,12 and the negative tests for organic aciduria in the affected patients make it unlikely that this is directly related to the clinical features in these patients.
The moderately affected sister of the index case is heterozygous for SPG11 mutation (111-2, Figure 1, Table 2). Consequently, she did not exhibit spasticity of her lower limbs and her corpus callosum was normal. Her clinical features are thought to be mainly related to the CA2 mutation.
The cognitive delay, osteopetrosis, and calcifications of the basal ganglia in the index case are linked to the pathogenic CA2 mutation.14 However, her spastic paraparesis and thinning of the corpus callosum can only be explained by the SPG11 mutation.11 The patient’s osteopetrosis is not severe; no bone fractures or compressive neuropathies were identified.
In 2012, Alazami et al15 described a novel syndrome of primordial dwarfism with distinct facial features and severe intellectual disability. A homozygous frameshift mutation in LARP7, a chaperone of the noncoding RNA 7SK, was discovered in patients from a single consanguineous Saudi family.
Hollink et al13 have broadened the phenotypic spectrum of pathogenic LARP7 variants through a report of two cases with intellectual disability, variable growth retardation, and distinct facial features that have been linked to two homozygous pathogenic LARP7 variants: c.1091_1094delCGGT in a Dutch case and c.1045_1051dupAAGGATA in a Saudi Arabian case. The LARP7 mutation is another probable gene in the network related to the phenomena of oligogenic pattern of inheritance of mental retardation, short stature, and craniofacial dysmorphic features in this family.
Oligogenic disorders remain primarily genetic in origin, in contrast to polygenic traits, which are thought to occur as a result of complex interactions between genes and the environment. The synergistic action of a small number of mutant alleles at a small number of loci is necessary in order for these disorders to occur. The severity of the disorder depends on three main variables: whether a major locus is involved, the number of loci involved, and the extent of environmental involvement.8
Modifier genes form the basis of oligogenic disease. These genes act to vary the expression of traits that are inherited from one gene. Modifier genes are defined as inherited genetic variations that lead to a qualitative or quantitative difference in any aspect of the disease phenotype.16
Based on these results, our family may represent another example of oligogenic disease with a major locus of the CA2 homozygous mutation and modifier pathogenic mutations of SPG11 and LARP genes in this phenotype.
Informed consent
Written informed consent has been provided by the parents of the family members to have the case details and any accompanying images published.
Disclosure
The authors report no conflicts of interest in this work.
References
Şıklar Z, Berberoğlu M. Syndromic disorders with short stature. J Clin Res Pediatr Endocrinol. 2014;6(1):1–8. | ||
Katsanis N, Ansley SJ, Badano JL, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293(5538):2256–2259. | ||
Penchev V, Boueva A, Kamenarova K, et al. A familial case of severe infantile nephronophthisis explained by oligogenic inheritance. Eur J Med Genet. 2017;60(6):321–325. | ||
Eichers ER, Lewis RA, Katsanis N, Lupski JR. Triallelic inheritance: a bridge between Mendelian and multifactorial traits. Ann Med. 2004;36(4):262–272. | ||
Laitala T, Väänänen HK. Inhibition of bone resorption in vitro by antisense RNA and DNA molecules targeted against carbonic anhydrase II or two subunits of vacuolar H(+)-ATPase. J Clin Invest. 1994;93(6):2311–2318. | ||
He N, Jahchan NS, Hong E, et al. A La-related protein modulates 7SK snRNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis. Mol Cell. 2008;29(5):588–599. | ||
Słabicki M, Theis M, Krastev DB, et al. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol. 2010;8(6):e1000408. | ||
Badano JL, Katsanis N. Beyond Mendel: an evolving view of human genetic disease transmission. Nat Rev Genet. 2002;3(10):779–789. | ||
Wagstaff J. Genetics beyond Mendel. Understanding nontraditional inheritance patterns. Postgrad Med. 2000;108(3):131137–133138. | ||
Ohlsson A, Cumming WA, Paul A, Sly WS. Carbonic anhydrase II deficiency syndrome: recessive osteopetrosis with renal tubular acidosis and cerebral calcification. Pediatrics. 1986;77(3):371–381. | ||
Boukhris A, Stevanin G, Feki I, et al. Hereditary spastic paraplegia with mental impairment and thin corpus callosum in Tunisia: SPG11, SPG15, and further genetic heterogeneity. Arch Neurol. 2008;65(3):393–402. | ||
Grünert SC, Stucki M, Morscher RJ, et al. 3-methylcrotonyl-CoA carboxylase deficiency: clinical, biochemical, enzymatic and molecular studies in 88 individuals. Orphanet J Rare Dis. 2012;7:31. | ||
Hollink IH, Alfadhel M, Al-Wakeel AS, et al. Broadening the phenotypic spectrum of pathogenic LARP7 variants: two cases with intellectual disability, variable growth retardation and distinct facial features. J Hum Genet. 2016;61(3):229–233. | ||
Funderburk SJ. Osteopetrosis in two brothers with severe mental retardation. Birth Defects Orig Artic Ser. 1975;11(6):91–98. | ||
Alazami AM, et al. Loss of function mutation in LARP7, chaperone of 7SK ncRNA, causes a syndrome of facial dysmorphism, intellectual disability, and primordial dwarfism. Hum Mutat, 2012;33(10):1429–1434. | ||
Houlston RS, Tomlinson IP. Modifier genes in humans: strategies for identification. Eur J Hum Genet. 1998;6(1):80–88. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.