")
Back to Archived Journals » Oncolytic Virotherapy » Volume 7
Potential of oncolytic viruses in the treatment of multiple myeloma
Authors Bartee E
Received 21 September 2017
Accepted for publication 14 December 2017
Published 23 February 2018 Volume 2018:7 Pages 1—12
DOI https://doi.org/10.2147/OV.S136644
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Eric Bartee
Department of Microbiology and Immunology, Medical University of South Carolina, Charleston, SC, USA
Abstract: Multiple myeloma (MM) is a clonal malignancy of plasma cells that is newly diagnosed in ~30,000 patients in the US each year. While recently developed therapies have improved the prognosis for MM patients, relapse rates remain unacceptably high. To overcome this challenge, researchers have begun to investigate the therapeutic potential of oncolytic viruses as a novel treatment option for MM. Preclinical work with these viruses has demonstrated that their infection can be highly specific for MM cells and results in impressive therapeutic efficacy in a variety of preclinical models. This has led to the recent initiation of several human trials. This review summarizes the current state of oncolytic therapy as a therapeutic option for MM and highlights a variety of areas that need to be addressed as the field moves forward.
Keywords: oncolytic virotherapy, multiple myeloma, review
Introduction
Multiple myeloma (MM) is a clonal malignancy of immunoglobulin-secreting plasma cells. The disease is newly diagnosed in >30,000 people in the US annually, resulting in ~12,000 deaths, making it the second-most common form of hematopoietic malignancy.1,2 The disease often evolves slowly through several progressive stages.3 If identified early, patients will often display a nonmalignant precancerous state known as monoclonal gammopathy of undetermined significance.4 Over time, this progresses to a malignant but still largely asymptomatic disease called smoldering myeloma, in which clonal plasma cells slowly increase in the bone marrow.5,6 Eventually, disease progresses to symptomatic MM, in which malignant cells proliferate rapidly in the bone marrow, causing localized sites of malignancy known as plasmacytomas. While a few patients present with single plasmacytomas,7 most develop a systemic disease characterized by multiple distinct tumors, hence the name “multiple myeloma”. Malignant cells in these lesions secrete a variety of factors that cause remodeling of local bone structure, resulting in osteolytic lesions.8 This bone remodeling, combined with malignant cells outcompeting normal bone marrow cells for proliferative space, cause the typical symptoms of MM, including elevated levels of calcium in the blood, renal failure, anemia, and bone pain or fractures (frequently referred to as CRAB).2
Historically, treatment for MM patients involved combinations of chemotherapeutic drugs, such as melphalan, cyclophosphamide, and doxorubicin, either with or without radiation. These treatments were frequently able to induce at least partial remissions; however, patients suffered extremely high rates of relapse, typically within 2–3 years. The reason for these relapses was complex and likely involved a variety of factors, including the high degree of genomic variability inherent in MM,9–11 a tendency for MM cells to display phenotypic plasticity,12,13 the relatively undefined nature of tumorigenic MM stem cells,14,15 the resistance of these cells to many chemotherapeutic agents,16,17 and the interplay between MM cells and their complex tumor microenvironment, which provides a variety of progrowth and pro-survival signals.18 Due to this poor prognosis, eligible MM patients were often treated with a more aggressive regime involving myeloablative chemotherapy combined with single or tandem autologous stem-cell transplant (ASCT).19,20 This improved prognosis; however, the myeloablative conditioning regimes used were associated with severe treatment-related morbidities, including neutropenia and increased chances of infection. This largely limited the usage of ASCT to younger, healthier patients. Additionally, even with ASCT, disease often recurred within 3–5 years. The primary cause of recurrence following ASCT was thought to be residual disease persisting within treated patients.21,22 However, studies also found that virtually all ASCT samples were contaminated with low levels of malignant MM cells that were reintroduced to the patient during transplant.23,24 The exact impact of these contaminating MM cells on disease relapse remains controversial; however, their reintroduction seems unlikely to be beneficial.
From 2000 to 2005, novel chemotherapeutic agents, including bortezomib,25 thalidomide,26 and lenalidomide,27,28 began to improve outcomes for newly diagnosed MM patients. Use of these drugs rapidly increased the median overall survival from 2.5 years in 2000 to over 6.5 years in 2010.29,30 Since 2010, a variety of new drugs have also been approved, including carfilzomib,31 pomalidomide,32 panobinostat,33 daratumumab,34 and ixazomib,35 which promise to improve diagnoses even further. Due to these advances, the long-held belief of MM being “incurable” is slowly being challenged.36 Unfortunately, even with the approval of these new drugs, the proportion of patients who achieve durable long-term remissions is still predicted to be <50%.36
Since two of the major factors contributing to therapeutic failure in MM are tumor-cell heterogeneity and plasticity,9–13 alternative therapies for MM that do not rely on the use of targeted small-molecule inhibitors are highly attractive. One such therapy is the use of live replicating viruses specifically to infect and kill malignant MM cells, a strategy known as oncolytic virotherapy (OV).37,38 OV functions through two distinct mechanisms.39,40 In the first mechanism, known as direct oncolytics, malignant cells are specifically infected with an oncolytic virus. This infection results in the specific elimination of the infected cells through a variety of potential mechanisms, including lytic viral replication, induction of apoptosis, or cellular necrosis. This specific tumor-cell killing then initiates a second OV mechanism, often referred to as oncolytic immunotherapy, in which the danger signals provided by viral infection combined with the specific killing of tumor cells result in the generation of antitumor immunoresponses. These responses are often typified by large CD8+ T-cell responses reactive against both infected and uninfected tumor cells; however, a variety of other immune cells, including CD4+ T cells and natural killer cells, have also been shown to play critical roles.41–48
Each oncolytic mechanism provides a significant benefit while also being associated with therapeutic hurdles. Each of these mechanisms provides a significant benefit, while also being associated with therapeutic hurdles. For example, direct oncolytics is extremely rapid and highly efficient in some settings; however, it only results in the elimination of cells that are directly infected with a virus. This places extremely large importance on the delivery of a virus to a high percentage of malignant cells, a significant challenge that remains largely unsolved by the field. In contrast, oncolytic immunotherapy can be completely sterilizing; however, it is much slower to initiate than direct oncolytics and frequently demonstrates clinical benefit in only a fraction of recipients. Successful OV thus requires striking a delicate balance between the rapid but incomplete direct oncolytics and the slow but sterilizing viral immunotherapy.
A number of different oncolytic viruses have been studied for their therapeutic potential against MM. Interestingly, the findings for each virus are often unique in terms of the mechanisms through which specific infection is achieved, how this infection causes elimination of MM cells, and the therapeutic potential of the virus (Table 1). This review summarizes the existing literature for each oncolytic virus that has been studied as a therapeutic against MM, as well as attempting to provide some context for these studies and potential strengths and weaknesses of the work.
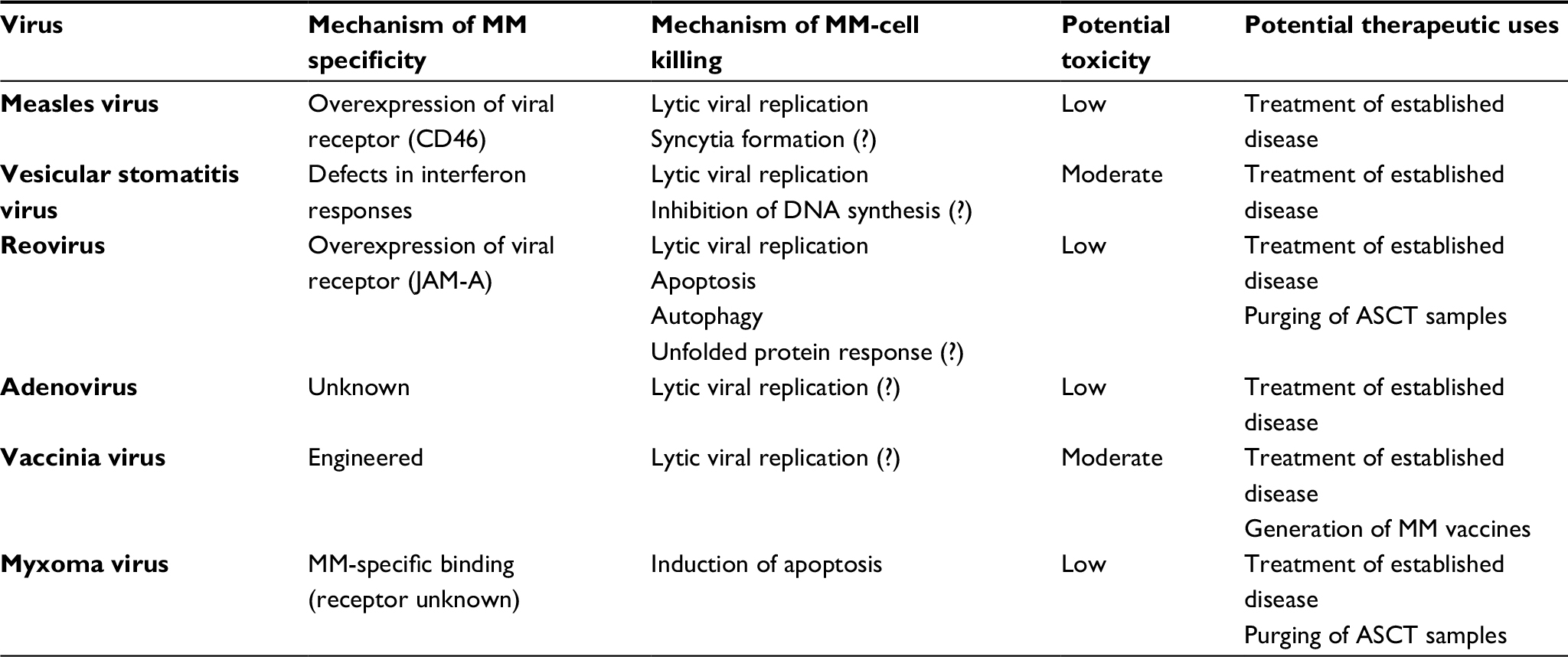 | Table 1 Treatment of MM using oncolytic viruses Notes: Overview of oncolytic viruses most commonly studied as treatments for MM, as well as mechanisms through which they achieve specific infection of MM cells, mechanisms through which they eliminate infected MM cells, potential for in vivo toxicities, and proposed therapeutic uses; (?) indicates mechanisms that are suspected, but have not yet been demonstrated. Abbreviations: MM, multiple myeloma; ASCT, autologous stem-cell transplant. |
Measles
The measles virus (MV) is a medium-sized (150 nm), negative-sense, single-stranded RNA virus from the Paramyxoviridae family. While wild-type MV is extremely pathogenic in humans, attenuated strains, including the attenuated Edmonston B strain on which oncolytic MV is based, have been used safely as vaccines for years.49 The initial oncolytic potential of MV was recognized more than 50 years ago;50 however, intensive studies into its therapeutic potential began around 2002.51
Interestingly, unlikely many oncolytic viruses, which are primarily studied as treatments for solid tumors, much of the work with oncolytic MV has been focused on hematopoietic malignancies. Therefore, while MV is far from the most commonly used oncolytic virus overall, it does represent the best-studied viral agent for the treatment of MM. Like many oncolytic viruses, MV has been shown specifically to infect both MM cell lines and CD138+ cells in MM-patient bone marrow.52,53 The specificity of this infection within the hematopoietic compartment appears to be based on an approximately sevenfold increase in expression of the endogenous MV receptor CD46 on malignant MM cells compared to nonmalignant hematopoietic cells.53 In addition, further improvements to the specificity of viral binding have been obtained by fusing the viral glycoprotein H to single-chain variable-fragment antibodies specific to either CD3854 or the Wue1 epitope.55 Both studies demonstrated improvements in the specificity of MV infection for malignant MM cells in vitro; however, neither strategy appears to have been adopted in other MV studies. Interestingly, while MV has been well studied as a therapeutic agent against MM, the mechanisms responsible for MM elimination following MV infection appear largely unknown. Presumably, lytic viral replication plays a major role; however, MV is also highly fusogenic, and infection results in the formation of large syncytia.52,53,56 The relative impact of each of these processes on elimination of infected MM cells thus remains unclear.
In terms of therapy, studies on MV have focused on the treatment of established disease, and efficacy has been shown in both systemically disseminated and localized plasmacytoma models.52,57–63 In these models, MV has shown exceptionally high efficacy, including the ability to induce potentially curative responses in some settings.52,57,58 Unfortunately, MV does not naturally infect mice, due to an inability to bind to murine MV receptors.64 This has limited the study of oncolytic MV to models in which human MM cells are xenografted into immunodeficient mice. This inherent limitation has resulted in two major translational hurdles that remain to be adequately addressed. First is that MV therapy, particularly following intravenous injection, is highly restricted by existing αMV humoral immunity. Although MM patients do present with slightly decreased αMV-antibody titers compared to healthy controls,65 virtually 100% of patients remain αMV-antibody-positive, and these antibodies can potently neutralize MV infection.57 In an oncolytic setting, studies have demonstrated that passive transfer of these αMV antibodies into tumor-bearing mice negatively impacts the resulting efficacy of MV therapy;57,59 however, whether this passive transfer fully recapitulates a functional humoral response seems questionable. Several methods to overcome this challenge have been attempted, primarily focusing on the use of different carrier cells, including T cells,57 macrophages,61 and irradiated tumor cells,59 to deliver the virus to sites of residual MM. Of these, irradiated tumor cells seem to be the most promising; however, this strategy has not yet been adopted clinically, and MV therapy remains largely restricted to patients who present with low αMV titers.66 The second major challenge involves the potential impact of T cells on MV therapy. These cells could either restrict viral therapy by eliminating infected cells prior to viral replication or be induced to generate potent αMM immunoresponses.39 Unfortunately, the use of xenografted, immunodeficient animal models largely eliminates the possible study of how T-cell responses might impact MV therapy.
Despite these unresolved issues, MV represents one of only two oncolytic viruses that have progressed into clinical trials for the treatment of MM.66–68 The virus used in these trials is a derivative of the Edmonston B vaccine strain that was modified to include the sodium–iodine symporter (NIS). Inclusion of the nis gene causes infected cells to import high levels of radioactive iodine, which allows for both live imaging of infected cells during therapy and improvement of clinical efficacy by increasing accumulation of toxic auger-emitting radioisotopes. Comprehensive preclinical toxicology studies carried out in either MV-susceptible squirrel monkeys or human CD46 transgenic mice68 have demonstrated that MV-NIS is safe for use at high doses. Similarly, while a recently completed Phase I trial did identify some potential grade III and grade IV hematological toxicities, including neutropenia, lymphopenia, anemia, and thrombocytopenia, no dose-limiting toxicities following therapy were identified. While this study was not powered for analysis of efficacy, some impressive results, including one complete disease regression, were observed.66,67 Overall, the response rate at the highest dose was 36% (4 or 11 patients). MV was detected in the blood following treatment; however, patients in this study rapidly seroconverted, suggesting that αMV humoral immunity could be a major limitation even in patients who initially present as MV-naïve. Unpublished reports have suggested that responding patients in this trial can develop long-term αMM immunological memory, implicating the potential impact of T-cell immunotherapy; however, this possibility has not yet been thoroughly studied. Despite a number of remaining hurdles, however, its established clinical successes currently make MV the most advanced oncolytic candidate for the treatment of MM.
Vesicular stomatitis virus
The vesicular stomatitis virus (VSV) is a small (75–120 nm), enveloped, negative-sense, single-stranded RNA virus from the Rhabdoviridae family. VSV is a severe animal pathogen, particularly in cattle, where it causes pathology virtually indistinguishable from foot-and-mouth disease. Adsorption of VSV to target cells is accomplished through the viral glycoprotein G, which binds to the ubiquitously expressed low-density-lipoprotein receptor. After entry, VSV displays an extremely rapid replication cycle in which new viral progeny can be generated in as little as 1–2 hours. This replication, however, is rapidly blocked by the presence of functional innate immunoresponses, particularly interferon. This naturally limits VSV replication to cells in which these responses are not present, including a wide array of transformed malignant cells. Due to its extremely rapid replication cycle and natural restriction to cancerous cells, VSV represents one of the more potent direct oncolytic viruses being studied.
With regard to MM, VSV has been shown directly to infect and kill both MM cell lines and CD138+ cells in MM-patient bone-marrow samples.69,70 This is likely due to lytic viral replication; however, the virus has also been shown to inhibit DNA and RNA synthesis rapidly in infected MM cells, which could represent a second potential mechanism of killing.71 Viral replication is predominantly MM-cell-specific, since signs of infection are typically not observed in most peripheral blood cells, including T cells, B cells, and natural killer cells. However, low-level infection can be seen in some neutrophils, and the virus appears fully infectious to normal monocyte.69 Additionally, toxicity studies done in mice using an oncolytic VSV armed with IFNβ and NIS demonstrated that in addition to malignant cells, viral RNA could be recovered from the liver and spleen72 and fully infectious virus found in the spleen. These studies also demonstrated the potential for intravenous injection to cause systemic inflammatory responses and liver toxicity (measured by ALT and AST). In addition to these “off-target” toxicities, “on-target” infection of meningeal MM deposits has been hypothesized to cause potentially lethal inflammation in the central nervous system.73 Therefore, while unmodified VSV appears naturally oncotropic for MM cells, additional work remains needed to identify ways to reduce toxicity that might be associated with systemic therapy.
Therapeutically, VSV has demonstrated efficacy in MM models following both localized and systemic injections.70,74–76 Critically, much of this work has been done in syngeneic models of both localized plasmacytomas and systemically disseminated MM. This work thus allows for analysis of both direct and immunotherapeutic OV mechanisms. Interestingly, while oncolytically-induced immunotherapy is often essential in solid tumor models, the work in MM suggests that this process is largely inhibitory to VSV therapy. Two studies comparing the efficacy of VSV in immunodeficient and immunocompetent models found better therapeutic efficacy in the absence of an immune system.70,76 Additionally, in two other models with clear immunotherapeutic involvement, depletion of CD4+ and CD8+ T cells resulted in only minor loss of efficacy.73,75 These data suggest that VSV-based treatment of MM might be predominantly through a direct oncolytic mechanism. Alternatively, it has been shown that systemic VSV therapy is limited by the presence of αVSV antibodies in the blood,77 which cause rapid clearance of free virus by the liver. This concern might be addressable, since chemical modifications of the viral virions using polyethylene glycol (PEG) have been shown to slow viral clearance. The effects of this PEGylation during actual OV of MM, however, have not been demonstrated. Therefore, while the potential efficacy of VSV against MM appears well established, more studies into the exact mechanisms involved remain needed.
Reovirus
Reovirus (ReoV) is an extremely large (600–1,000 nm), nonenveloped, double-stranded RNA virus from the Reoviridae family. While this virus can infect humans, in either the gastrointestinal or respiratory tract, it typically causes only subclinical pathology and is considered safe for clinical use. The oncolytic potential of ReoV in models of both solid tumors and hematological malignancies has been studied for many years,78,79 and a clinical grade ReoV (derived from the Dearing strain) is currently available under the brand Reolysin.80–83
In vitro, ReoV has been shown specifically to infect and kill both established MM cell lines84–88 and CD138+ cells in primary MM bone-marrow samples.85,86 The specificity of this infection is likely due to high expression of the ReoV receptor JAM-A on malignant MM cells, since the sensitivity of MM cells to ReoV treatment correlates with their JAM-A expression,87,88 elimination of JAM-A from MM cell lines prevents their infection with ReoV, and overexpression of JAM-A significantly increases in the sensitivity of MM cells to ReoV.87 In contrast, the mechanism through which MM cells are killed following ReoV infection remains somewhat unclear. Killing is clearly dependent on lytic viral replication;85,86 however, viral treatment has also been shown to induce cell death through both apoptotic and autophagic pathways.85 Additionally, viral infection significantly impacts the unfolded protein response, which is known to play a critical role in MM-cell survival.89 It thus remains to be determined which of these potential mechanisms truly mediates ReoV elimination of infected MM cells.
Therapeutically, ReoV has been shown to be efficacious in a variety of preclinical MM models. Thirukkumaran et al demonstrated that ReoV is unable to infect normal CD34+ bone-marrow progenitor cells and that the virus can specifically identify low levels of CD138+ MM cells contained in mixtures of normal bone-marrow aspirates.84,86 This allows the virus to be used as an ex vivo purging agent to prevent MM reintroduction during ASCT while not negatively impacting hematopoietic reconstitution.84–86 Additionally, direct viral injection has been shown to be somewhat therapeutically effective against MM in both systemic85,88–90 and localized plasmacytoma89 models. Unfortunately, the results of viral monotherapy in these studies have frequently been modest, and complete regression has not often been observed. Similarly, while a Phase I clinical trial of Reolysin monotherapy in relapsed MM patients identified no dose-limiting toxicities, only modest efficacy was seen, with the best clinical outcomes being stable disease (observed in 25% of patients).83 These data suggest that the use of ReoV in combination treatments might be needed to increase therapeutic efficacy.
In this context, it is interesting to note that several groups have shown that existing MM therapies, including histone deacetylase inhibitors or the proteasome inhibitor bortezomib, can sensitize MM cells to ReoV infection by increasing the expression of the viral receptor JAM-A.87,88 This suggests that ReoV might be more effective in MM patients who have already failed one or more existing therapies. Alternatively, ReoV and histone deacetylase inhibitors have been shown to act synergistically when used together in vivo, suggesting the possible development of rational combination therapies.88 Finally, a recently published work has suggested the possibility of using ReoV to improve response rates of MM patients to additional immunotherapeutic treatments, such as PDL1 blockade.90 More work, however, is clearly needed to identify and optimize potential combinatorial therapies involving ReoV.
Adenovirus
Adenovirus (AdV) is a medium-sized (90–100 nm), non-enveloped, double-stranded DNA virus from the Adenoviridae family. The term “adenovirus” actually refers to a large number of distinct viral serotypes, many of which display highly distinct pathologies and infectious characteristics. Taken together, AdV infections in humans are extremely common and can account for almost 10% of all respiratory infections. These infections typically present as a “common cold”, although some serotypes are associated with more severe symptoms, including pneumonia. As therapeutic agents, AdVs are one of the best-studied viral families. They have been used in a variety of therapeutic modalities, including lytically replicating oncolytic studies, vaccine studies, and gene-therapy studies. Due to the substantial breadth of this work, this review focuses on lytically replicating oncolytic studies using AdV.
In the context of MM, AdV has been shown to be highly infectious to both established MM cell lines and CD138+ cells from MM-patient bone-marrow samples.91,92 Interestingly, while much of the oncolytic work in solid tumors has focused on a single AdV serotype (type 5), work in MM has directly compared the efficacy of a wide range of AdV serotypes, with interesting results. Senac et al found that serotypes 6, 26, and 48 killed MM cells while having only minimal effects of normal peripheral blood mononuclear cells. In contrast, serotypes 11, 35, 40, and 41 displayed the opposite specificity, killing normal peripheral blood mononuclear cells while largely sparing malignant MM cells.91 This observation was supported by additional research demonstrating that species D AdV, including the previously identified serotypes 26 and 48, was a highly efficient MM killer, while species B AdV, including serotypes 11 and 35, was a highly inefficient killer. This work identified the most commonly used oncolytic serotype (AdV type 5) as only a modest killer of MM cells. This serotype hierarchy appeared to be maintained during in vivo therapy, since serotypes 26 and 45 displayed improved efficacy against established MM tumors in vivo.92 Interestingly, unlike many oncolytic viruses, whose preferential infection of MM cells appears to be mediated by receptor specificity, the preferential killing of MM by different AdV serotypes correlated better with replication kinetics than with viral adsorption.
Therapeutically, unmodified AdVs have been shown to be effective against established MM in several studies.91–93 Unfortunately, these studies have focused on the treatment of localized human plasmacytomas in immunodeficient animals. Efficacy in more clinically relevant systemic, immunocompetent models has yet to be tested. This is particularly important for AdV-based therapy, given that virtually all patients are exposed to AdV throughout life and many carry potent neutralizing humoral responses against the more common AdV serotypes. In addition to this caveat, it must be noted that the overall efficacy of AdV therapy, even in localized models, has typically been modest, and very few long-term cures have been demonstrated. In order to improve this efficacy, several groups have studied the possibility of “arming” AdV to promote therapy. AdV armed with TRAIL has been shown to display enhanced killing of MM cells in vitro while also demonstrating therapeutic synergy with PI3K or proteasome inhibitors.94 Experiments arming AdV with CD40L have also displayed improved killing of MM cells both in vitro and in vivo.93 Interestingly, while CD40L is a known immunomodulator, the improved efficacy in the latter studies appeared to be mediated by direct induction of apoptosis, and not through enhanced T-cell responses (a possibility that was never examined). Additionally, it is important to note that both experiments arming oncolytic AdV used serotype 5 as their viral backbone. Utilizing similar arming strategies on more potent αMM serotypes has yet to be explored.
Vaccinia virus
The vaccinia virus (VacV) is a large (200–300 nm), enveloped, double-stranded DNA virus from the Poxviridae family. While the evolutionary host for VacV remains somewhat controversial, it is clear that the wild-type virus is fully infectious in a variety of mammals, including humans, and that systemic infection can result in severe disease symptoms, including death. Due to their use as vaccine platforms to eradicate smallpox, however, attenuated VacV strains have been extensively studied in humans and display relatively good safety profiles. As an oncolytic agent, VacV has been well studied in the context of solid tumors in both preclinical95,96 and clinical97–102 settings. In particular, recombinant VacV encoding either GM-CSF (known as JX594) or PSA (known as Prostvac) has advanced to large Phase III clinical trials.
While oncolytic VacV is well established as a potential treatment for solid tumors, its use in a hematopoietic setting is much less well studied. This is likely due to the fact that while VacV is extremely lytic, the virus is not naturally oncotropic and requires additional genetic modification to restrict viral replication to malignant cells. In solid tumors, this can be accomplished through a single deletion of the viral tk gene. This restricts viral replication to cells with an abundance of thymidine, a state predominately found in rapidly dividing cancer cells. Unfortunately, this restriction is not absolute, and tk–/– VacV clones can infect normal tissue, causing severe pathology following system injection.103 Therefore, the use of VacV as an oncolytic agent against systemic malignancies, such as MM, requires additional genetic engineering further to restrict viral replication and limit toxicity. A variety of methods to enhance viral specificity have been attempted. One of the most commons methods is to delete the vgf gene in tk–/– clones. VGF stimulates cellular proliferation through activation of EGFR. Its removal in so-called double-deleted VacV further restricts viral replication to rapidly dividing cells, resulting in improved safety profiles.104 A second approach that has been attempted is actively to restrict viral replication by placing essential viral genes under miRNA restriction. In this approach, several Let7a miRNA-binding sites are placed within the promoter of the essential viral gene B5R. This causes an inhibition of B5R expression in normal cells (which express high levels of Let7a). In contrast, in MM cells (which fail to express Let7a), B5R expression occurs at normal levels, thus promoting viral replication only in malignant MM cells.103 Attempts have also been made to alter viral binding specifically to increase adsorption of virions in malignant cells.105 While this is an attractive theoretical approach, the binding determinants of VacV are complex and remain poorly understood,106 making successful retargeting difficult. In constructs that display successful targeting, elimination of infected MM cells appears to be dependent on lytic replication.104 Interestingly, the efficacy of this elimination seems somewhat controversial. Several groups have suggested that VacV is extremely lytic toward infected MM cells, with massive reductions in cellular viability shown within 24 hours after infection.103,104,106 In contrast, Lei et al saw only minimal reductions in MM-cell viability following infection with VacV.107 In this work, elimination of infected cells occurred through the induction of apoptosis, and efficient killing required “arming” the oncolytic VacV with additional apoptotic modulators, including miR34a and Smac.
Therapeutically, VacV has been used as a treatment for MM in two distinct ways. The first way is the direct treatment of established disease in vivo.103,104,107 This work has demonstrated that VacV has the potential to treat established MM. Unfortunately, it has primarily utilized models of human plasmacytomas established in immunodeficient mice. Anecdotal evidence from a single case report suggests that VacV can (at least transiently) reduce systemic MM-tumor burden in a human patient;108 however, the efficacy of viral treatment against disseminated MM remains largely unclear. The second therapeutic modality in which VacV has been studied is the use of the virus to generate αMM-tumor vaccines. In this regard, it has been demonstrated that VacV can be used as an adjuvant to increase the magnitude of αMM immunoresponses following vaccination. Animals vaccinated with tumor cells infected ex vivo with VacV and then lethally irradiated can completely reject secondary challenge with uninfected MM cells,109–113 and vaccination of tumor-bearing animals can result in remission of established MM.114 This effect appears to be mediated by cytotoxic T cells, although the exact subset has not been identified.111,113 Unfortunately, this methodology requires prevaccination of tumor-naïve individuals with VacV to achieve maximal efficacy, which could severely limit its translational potential. Nevertheless, further investigation into this approach might be warranted.
Myxoma virus
The myxoma virus (MyxV) is a large (200–300 nm), enveloped, double-stranded DNA virus from the Poxviridae family that is somewhat related to VacV.115 Unlike VacV, however, MyxV displays a tightly restricted host range specific to lagomorphs (rabbits). No instance of natural MyxV infection has ever been documented in any nonrabbit species, and even direct injection of large amounts of the virus does not elicit noticeable pathology in either humans or mice.116–118 MyxV is a relatively novel oncolytic virus whose therapeutic potential was recognized <10 years ago.119 Since then, it has been investigated in preclinical models of a number of different malignancies, including melanoma,42,119,120 rhabdoid tumors,121,122 pancreatic cancer,123,124 glioma,125,126 and gallbladder cancer.127
Like many oncolytic viruses, MyxV has been shown preferentially to infect and kill both established MM cell lines and CD138+ cells found in MM-patient bone-marrow samples.117 This killing appears selective for MM cells, based on a strong preferential binding of the virus to CD138+ cells compared to nonmalignant CD138– cells found in MM-patient bone marrow.117,118 Like ReoV, this preferential binding also excludes absorption of MyxV to CD34+ bone-marrow progenitors, allowing the virus to be used as an ex vivo purging agent during ASCT.117,118,128,129 Interestingly, unlike the vast majority of OVs, killing of MM cells by MyxV appears to be independent of lytic viral replication, since no new viral progeny are produced during treatment and replication-incompetent virus is still fully capable to kill infected cells.117 Instead, elimination of MyxV-infected MM cells appears to be mediated by the induction of extrinsic apoptosis caused by depletion of cellular apoptotic inhibitors, Bcl2, Mcl1, XlAP, and survivin. This depletion results from the rapid decapping of cellular mRNA early in infection.130,131 Interestingly, while a truly comprehensive study has never been published, 100% of the primary MM-patient samples studied to date have responded to MyxV therapy, suggesting that the unique mechanism through which MyxV kills MM cells might overcome some of the challenges associated with MM-cell heterogeneity and resistance.106,117
Therapeutically, many of the studies into MyxV’s potential to treat MM have focused on the virus’s ability to improve the treatment for MM patients receiving ASCT.117,128,129 Bartee et al showed that MyxV treatment of MM-patient bone-marrow biopsies resulted in the rapid and specific infection of CD138+ cells while sparing the CD34+ bone-marrow progenitors.117,118 Additionally, treatment of mixtures of human MM cells and bone-marrow progenitors with MyxV prior to transplant could specifically prevent establishment of MM tumors, while still allowing for stem-cell engraftment and hematopoietic reconstitution.117 Due to these results, it has been suggested that MyxV could be used as an ex vivo purging agent to eliminate contaminating MM cells from ASCT samples. It has also been shown that direct intravenous injection of MyxV into immunocompetent mice bearing established, systemic MM can result in both a rapid reduction in tumor burden and potentially curative induction of αMM immunoresponses.43 Unfortunately, while the results of these experiments were striking in some animals, the overall response rates achieved were very low. In order to improve therapy, several groups have studied the possibility of combining ex vivo purging with treatment of established disease.128,129 These groups have demonstrated that treatment of ASCT samples with MyxV ex vivo results in the loading of viral particles onto either T cells128 or neutrophils,129 which has beneficial effects through improving the delivery of the virus in vivo or activating immune cells to improve αMM immunity.
Picornavirus
In addition to the more commonly used viruses listed already, a small number of studies have also demonstrated the oncolytic potential of the Picornaviridae family against MM. For example, it has been shown that MM cells overexpress Coxsackie virus receptors ICAM1 and DAF, rendering them susceptible to infection with the oncolytic Coxsackie virus A21.132 Additional work has shown that the efficacy of A21 infection in killing MM cells was high and the virus could be used to treat solid plasmacytomas.132,133 However, viral treatment also had a negative impact on bone-marrow progenitor differentiation, suggesting a possible hurdle to translation. Mengovirus (another member of Picornaviridae) has also been shown to infect and kill MM cells; however, in vivo studies with this virus demonstrated only modest efficacy along with relatively high toxicity.134 Coxsackie viruses are positive-sense, single-stranded RNA viruses, which means their nucleic acid is directly infectious. In an interesting study, Hadac et al were able to demonstrate that injection of infectious A21 nucleic acid into solid MM plasmacytomas initiated an oncolytic infection with efficacy similar to that seen using intact virus.133 While this is a strategy with somewhat restricted usage, it represents an interesting approach to overcoming some of the challenges that appear to be associated with using intact picornaviruses in the clinic.
Final words
Review of the existing literature clearly demonstrates the potential for OV to be used as a viable therapy for MM patients. A large number of viruses have been shown specifically to infect MM cells, with infection resulting in the rapid elimination of these cells through a variety of mechanisms. Preclinical studies conducted with most of these viruses have typically resulted in at least stable disease or partial disease remission, and early results from clinical studies in human patients appear to be promising. However, the field also suffers from several obvious issues, which must be addressed. First and foremost is the use of appropriate preclinical models to study the mechanisms and efficacies of OV. With the notable exception of VSV, the majority of work studying OV treatment of MM has been conducted in immunodeficient models of single subcutaneous plasmacytoma. While these models represent a technically easy starting point, they largely fail to recapitulate the clinical realities of MM in two major ways. First, due to the nature of oncolytic virus particles, the challenges associated with treatment of systemic versus localized disease are very different. In particular, delivery of a virus through the bloodstream to sites of systemic disease is known to be a major translational hurdle. Since MM typically presents as a systemic disease, this hurdle should be addressed in any preclinical study. Unfortunately, the use of subcutaneous plasmacytoma models largely bypasses this issue, possibly skewing results in a more favorable manner. Second, the immune system is known to impact OV efficacy significantly in a variety of both positive and negative ways. For example, serum complement and existing antiviral humoral responses can inhibit viral infectivity.135–137 In contrast, much of the efficacy of OV is now thought to be mediated through the induction of antitumor T-cell-mediated immunotherapy.39 The use of immunodeficient animal models largely precludes studies into how these issues might affect MM therapy. In particular, as immunotherapy becomes more clinically prevalent, the propensity of OV to induce α-tumor T-cell responses raises the possibility that OV can be used to improve the response of MM to other immunotherapeutic agents, such as PD1- or CTLA4-blocking antibodies. To date, these antibodies have proven only modestly successful as single agents against MM;138–140 however, given their success in other malignancies, this line of study still appears conceptually attractive. Importantly, a variety of immunocompetent MM models have been developed in recent years that can be used to address these issues.141,142 While none of these models perfectly recapitulates the clinical realties of MM, they are all clearly better suited to preclinical studies on OV than subcutaneous immunodeficient models.
The second issue that arises during a review of the literature is the need to examine the impact of disease heterogeneity on OV. A number of studies have shown that oncolytic viruses can infect and typically kill a high percentage of CD138+ cells in MM-patient bone marrow. However, the number of samples used in these studies is often small (typically data from only one to three patients is shown), and the methodologies used (often flow cytometry or immunofluorescence) are typically unable to detect small numbers of resistant cells. One of the major challenges in treating MM is the heterogeneity of disease, both between patients and within single patients, which increases the likelihood of resistant clones developing following therapy. While OV theoretically has the potential to overcome these challenges, a demonstration of this ability is notably lacking in the current literature. The field would thus benefit from a comprehensive study of the efficacy of OV therapy on a large number of patient samples using sensitive techniques to study efficacy versus the development of resistance.
Finally, as is often the case in the field of OV, the literature reveals a set of studies that are often disjointed and unconnected. Multiple lines of research that appear promising within a single virus are never combined, nor are these lines of research adopted in other viral platforms. Additionally, the results of combining OV with more typical MM standards of care, such as chemotherapy, radiation, or monoclonal antibodies, are generally lacking. This latter point is especially critical, in order to place OV in the context of other MM therapies. For example, should OV treatment be used as frontline therapy, or is it more suited for relapsed or refractory patients? Does OV synergize with existing MM treatments? If so, which one? At some point, if the field is to advance from preclinical work to meaningful human trials, the “optimal” genetic modifications must be identified, synergistic combinatorial modifications must be introduced, and therapy must be integrated into current standards of care. In conclusion, a number of significant issues must still be resolved before OV for MM is to become truly clinically viable. However, for a field that should probably be considered in its infancy, the results appear extremely promising, and more work is clearly warranted.
Disclosure
The author reports no conflicts of interest in this work.
References
Surveillance, Epidemiology, and End Results Program. SEER data, 1973–2014. 2017. Available from: http://seer.cancer.gov/data. Accessed December 21, 2017. | ||
Soiety Ll. Disease information and support: myeloma. 2012. Available from: https://www.lls.org/facts-and-statistics/facts-and-statistics-overview. Accessed February 15, 2018. | ||
Kumar SK, Rajkumar V, Kyle RA, et al. Multiple myeloma. Nat Rev Dis Primers. 2017;3:17046. | ||
Kyle RA. Monoclonal gammopathy of undetermined significance: natural history in 241 cases. Am J Med. 1978;64(5):814–826. | ||
Kyle RA, Greipp PR. Smoldering multiple myeloma. N Engl J Med. 1980;302(24):1347–1349. | ||
Crowley JP. Smoldering multiple myeloma. N Engl J Med. 1980; 303(16):941. | ||
Grammatico S, Scalzulli E, Petrucci MT. Solitary plasmacytoma. Mediterr J Hematol Infect Dis. 2017;9(1):e2017052. | ||
Panaroni C, Yee AJ, Raje NS. Myeloma and bone disease. Curr Osteoporos Rep. 2017;15(5):483–498. | ||
Zhan F, Huang Y, Colla S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–2028. | ||
Bolli N, Avet-Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. | ||
Lohr JG, Stojanov P, Carter SL, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25(1):91–101. | ||
Chaidos A, Barnes CP, Cowan G, et al. Clinical drug resistance linked to interconvertible phenotypic and functional states of tumor-propagating cells in multiple myeloma. Blood. 2013;121(2):318–328. | ||
Hajek R, Okubote SA, Svachova H. Myeloma stem cell concepts, heterogeneity and plasticity of multiple myeloma. Br J Haematol. 2013;163(5):551–564. | ||
Matsui W, Huff CA, Wang Q, et al. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103(6):2332–2336. | ||
Kim D, Park CY, Medeiros BC, Weissman IL. CD19-CD45 low/- CD38 high/CD138+ plasma cells enrich for human tumorigenic myeloma cells. Leukemia. 2012;26(12):2530–2537. | ||
Matsui W, Wang Q, Barber JP, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008;68(1):190–197. | ||
Boucher K, Parquet N, Widen R, et al. Stemness of B-cell progenitors in multiple myeloma bone marrow. Clin Cancer Res. 2012;18(22):6155–6168. | ||
McDonald MM, Fairfield H, Falank C, Reagan MR. Adipose, bone, and myeloma: contributions from the microenvironment. Calcif Tissue Int. 2017;100(5):433–448. | ||
McElwain TJ, Selby PJ, Gore ME, et al. High-dose chemotherapy and autologous bone marrow transplantation for myeloma. Eur J Haematol Suppl. 1989;51:152–156. | ||
Selby P, Zulian G, Forgeson G, et al. The development of high dose melphalan and of autologous bone marrow transplantation in the treatment of multiple myeloma: Royal Marsden and St Bartholomew’s Hospital studies. Hematol Oncol. 1988;6(2):173–179. | ||
Bourhis JH, Bouko Y, Koscielny S, et al. Relapse risk after autologous transplantation in patients with newly diagnosed myeloma is not related with infused tumor cell load and the outcome is not improved by CD34+ cell selection: long term follow-up of an EBMT phase III randomized study. Haematologica. 2007;92(8):1083–1090. | ||
Dyson PG, Horvath N, Joshua D, et al. CD34+ selection of autologous peripheral blood stem cells for transplantation following sequential cycles of high-dose therapy and mobilization in multiple myeloma. Bone Marrow Transplant. 2000;25(11):1175–1184. | ||
Kopp HG, Yildirim S, Weisel KC, Kanz L, Vogel W. Contamination of autologous peripheral blood progenitor cell grafts predicts overall survival after high-dose chemotherapy in multiple myeloma. J Cancer Res Clin Oncol. 2009;135(4):637–642. | ||
Vogel W, Kopp HG, Kanz L, Einsele H. Myeloma cell contamination of peripheral blood stem-cell grafts can predict the outcome in multiple myeloma patients after high-dose chemotherapy and autologous stem-cell transplantation. J Cancer Res Clin Oncol. 2005;131(4):214–218. | ||
Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352(24):2487–2498. | ||
Singhal S, Mehta J, Desikan R, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341(21):1565–1571. | ||
Rajkumar SV, Hayman SR, Lacy MQ, et al. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood. 2005;106(13):4050–4053. | ||
Richardson PG, Blood E, Mitsiades CS, et al. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood. 2006;108(10):3458–3464. | ||
Rajkumar SV. Multiple myeloma: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol. 2014;89(10):999–1009. | ||
Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28(5):1122–1128. | ||
Avet-Loiseau H, Fonseca R, Siegel D, et al. Carfilzomib significantly improves the progression-free survival of high-risk patients in multiple myeloma. Blood. 2016;128(9):1174–1180. | ||
San Miguel J, Weisel K, Moreau P, et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14(11):1055–1066. | ||
San Miguel JF, Hungria VT, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15(11):1195–1206. | ||
Chari A, Suvannasankha A, Fay JW, et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood. 2017;130(8):974–981. | ||
Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;374(17):1621–1634. | ||
Barlogie B, Mitchell A, van Rhee F, Epstein J, Morgan GJ, Crowley J. Curing myeloma at last: defining criteria and providing the evidence. Blood. 2014;124(20):3043–3051. | ||
Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658–670. | ||
Lawler SE, Speranza MC, Cho CF, Chiocca EA. Oncolytic viruses in cancer treatment: a review. JAMA Oncol. 2017;3(6):841–849. | ||
Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. 2011;19(6):1008–1016. | ||
Choi AH, O’Leary MP, Fong Y, Chen NG. From benchtop to bedside: a review of oncolytic virotherapy. Biomedicines. 2016;4(3):E18. | ||
Chen A, Zhang Y, Meng G, et al. Oncolytic measles virus enhances antitumour responses of adoptive CD8+NKG2D+ cells in hepatocellular carcinoma treatment. Sci Rep. 2017;7(1):5170. | ||
Bartee MY, Dunlap KM, Bartee E. Tumor-localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res. 2017;77(11):2952–2963. | ||
Bartee E, Bartee MY, Bogen B, Yu XZ. Systemic therapy with oncolytic myxoma virus cures established residual multiple myeloma in mice. Mol Ther Oncolytics. 2016;3:16032. | ||
Ishihara M, Seo N, Mitsui J, et al. Systemic CD8+ T cell-mediated tumoricidal effects by intratumoral treatment of oncolytic herpes simplex virus with the agonistic monoclonal antibody for murine glucocorticoid-induced tumor necrosis factor receptor. PloS One. 2014;9(8):e104669. | ||
Bridle BW, Clouthier D, Zhang L, et al. Oncolytic vesicular stomatitis virus quantitatively and qualitatively improves primary CD8+ T-cell responses to anticancer vaccines. Oncoimmunology. 2013;2(8):e26013. | ||
Morales O, Richard A, Martin N, et al. Activation of a helper and not regulatory human CD4+ T cell response by oncolytic H-1 parvovirus. PloS One. 2012;7(2):e32197. | ||
Ye JF, Qi WX, Liu MY, Li Y. The combination of NK and CD8+ T cells with CCL20/IL15-armed oncolytic adenoviruses enhances the growth suppression of TERT-positive tumor cells. Cell Immunol. 2017;318:35–41. | ||
Yoo JY, Jaime-Ramirez AC, Bolyard C, et al. Bortezomib treatment sensitizes oncolytic HSV-1-treated tumors to NK cell immunotherapy. Clin Cancer Res. 2016;22(21):5265–5276. | ||
Katz SL, Kempe CH, Black FL, et al. Studies on an attenuated measles-virus vaccine – VIII: general summary and evaluation of the results of vaccination. Am J Dis Child. 1960;100:942–946. | ||
Albano A. [Adaptation of rat myeloma cells to continuous culture and their susceptibility to enteroviruses, adenoviruses and measles virus]. Boll Ist Sieroter Milan. 1962;41:599–607. Italian. | ||
Grote D, Russell SJ, Cornu TI, et al. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood. 2001;97(12):3746–3754. | ||
Peng KW, Ahmann GJ, Pham L, Greipp PR, Cattaneo R, Russell SJ. Systemic therapy of myeloma xenografts by an attenuated measles virus. Blood. 2001;98(7):2002–2007. | ||
Ong HT, Timm MM, Greipp PR, et al. Oncolytic measles virus targets high CD46 expression on multiple myeloma cells. Exp Hematol. 2006;34(6):713–720. | ||
Peng KW, Donovan KA, Schneider U, Cattaneo R, Lust JA, Russell SJ. Oncolytic measles viruses displaying a single-chain antibody against CD38, a myeloma cell marker. Blood. 2003;101(7):2557–2562. | ||
Hummel HD, Kuntz G, Russell SJ, et al. Genetically engineered attenuated measles virus specifically infects and kills primary multiple myeloma cells. J Gen Virol. 2009;90(Pt 3):693–701. | ||
Liu C, Suksanpaisan L, Chen YW, Russell SJ, Peng KW. Enhancing cytokine-induced killer cell therapy of multiple myeloma. Exp Hematol. 2013;41(6):508–517. | ||
Ong HT, Hasegawa K, Dietz AB, Russell SJ, Peng KW. Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther. 2007;14(4):324–333. | ||
Haralambieva I, Iankov I, Hasegawa K, Harvey M, Russell SJ, Peng KW. Engineering oncolytic measles virus to circumvent the intracellular innate immune response. Mol Ther. 2007;15(3):588–597. | ||
Liu C, Russell SJ, Peng KW. Systemic therapy of disseminated myeloma in passively immunized mice using measles virus-infected cell carriers. Mol Ther. 2010;18(6):1155–1164. | ||
Liu YP, Tong C, Dispenzieri A, Federspiel MJ, Russell SJ, Peng KW. Polyinosinic acid decreases sequestration and improves systemic therapy of measles virus. Cancer Gene Ther. 2012;19(3):202–211. | ||
Peng KW, Dogan A, Vrana J, et al. Tumor-associated macrophages infiltrate plasmacytomas and can serve as cell carriers for oncolytic measles virotherapy of disseminated myeloma. Am J Hematol. 2009;84(7):401–407. | ||
Dingli D, Peng KW, Harvey ME, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103(5):1641–1646. | ||
Hallak LK, Merchan JR, Storgard CM, Loftus JC, Russell SJ. Targeted measles virus vector displaying echistatin infects endothelial cells via αvβ3 and leads to tumor regression. Cancer Res. 2005;65(12):5292–5300. | ||
Ohno S, Ono N, Seki F, et al. Measles virus infection of SLAM (CD150) knockin mice reproduces tropism and immunosuppression in human infection. J Virol. 2007;81(4):1650–1659. | ||
Dingli D, Peng KW, Harvey ME, et al. Interaction of measles virus vectors with Auger electron emitting radioisotopes. Biochem Biophys Res Commun. 2005;337(1):22–29. | ||
Dispenzieri A, Tong C, LaPlant B, et al. Phase I trial of systemic administration of Edmonston strain of measles virus genetically engineered to express the sodium iodide symporter in patients with recurrent or refractory multiple myeloma. Leukemia. 2017;31(12):2791–2798. | ||
Russell SJ, Federspiel MJ, Peng KW, et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc. 2014;89(7):926–933. | ||
Myers RM, Greiner SM, Harvey ME, et al. Preclinical pharmacology and toxicology of intravenous MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clin Pharmacol Ther. 2007;82(6):700–710. | ||
Lichty BD, Stojdl DF, Taylor RA, et al. Vesicular stomatitis virus: a potential therapeutic virus for the treatment of hematologic malignancy. Hum Gene Ther. 2004;15(9):821–831. | ||
Goel A, Carlson SK, Classic KL, et al. Radioiodide imaging and radiovirotherapy of multiple myeloma using VSV(Δ51)-NIS, an attenuated vesicular stomatitis virus encoding the sodium iodide symporter gene. Blood. 2007;110(7):2342–2350. | ||
McGowan JJ, Wagner RR. Inhibition of cellular DNA synthesis by vesicular stomatitis virus. J Virol. 1981;38(1):356–367. | ||
Zhang L, Steele MB, Jenks N, et al. Safety studies in tumor and non-tumor-bearing mice in support of clinical trials using oncolytic VSV-IFNβ-NIS. Hum Gene Ther Clin Dev. 2016;27(3):111–122. | ||
Yarde DN, Naik S, Nace RA, Peng KW, Federspiel MJ, Russell SJ. Meningeal myeloma deposits adversely impact the therapeutic index of an oncolytic VSV. Cancer Gene Ther. 2013;20(11):616–621. | ||
Naik S, Nace R, Barber GN, Russell SJ. Potent systemic therapy of multiple myeloma utilizing oncolytic vesicular stomatitis virus coding for interferon-β. Cancer Gene Ther. 2012;19(7):443–450. | ||
Naik S, Nace R, Federspiel MJ, Barber GN, Peng KW, Russell SJ. Curative one-shot systemic virotherapy in murine myeloma. Leukemia. 2012;26(8):1870–1878. | ||
Yarde DN, Nace RA, Russell SJ. Oncolytic vesicular stomatitis virus and bortezomib are antagonistic against myeloma cells in vitro but have additive anti-myeloma activity in vivo. Exp Hematol. 2013;41(12):1038–1049. | ||
Tesfay MZ, Kirk AC, Hadac EM, et al. PEGylation of vesicular stomatitis virus extends virus persistence in blood circulation of passively immunized mice. J Virol. 2013;87(7):3752–3759. | ||
Bennette JG, Bush PV, Steele RD. Characteristics of a newborn runt disease induced by neonatal infection with an oncolytic strain of reovirus type 3 (REO3MH) – II: immunological aspects of the disease in mice. Br J Exp Pathol. 1967;48(3):267–284. | ||
Mettler NE, Clarke DH, Casals J. Virus inoculation in mice bearing Ehrlich ascitic tumors: antigen production and tumor regression. Infect Immun. 1982;37(1):23–27. | ||
Mahalingam D, Fountzilas C, Moseley J, et al. A phase II study of Reolysin (pelareorep) in combination with carboplatin and paclitaxel for patients with advanced malignant melanoma. Cancer Chemother Pharmacol. 2017;79(4):697–703. | ||
Cohn DE, Sill MW, Walker JL, et al. Randomized phase IIB evaluation of weekly paclitaxel versus weekly paclitaxel with oncolytic reovirus (Reolysin) in recurrent ovarian, tubal, or peritoneal cancer: an NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. 2017;146(3):477–483. | ||
Noonan AM, Farren MR, Geyer SM, et al. Randomized phase 2 trial of the oncolytic virus pelareorep (Reolysin) in upfront treatment of metastatic pancreatic adenocarcinoma. Mol Ther. 2016;24(6):1150–1158. | ||
Sborov DW, Nuovo GJ, Stiff A, et al. A phase I trial of single-agent Reolysin in patients with relapsed multiple myeloma. Clin Cancer Res. 2014;20(23):5946–5955. | ||
Thirukkumaran CM, Luider JM, Stewart DA, et al. Reovirus oncolysis as a novel purging strategy for autologous stem cell transplantation. Blood. 2003;102(1):377–387. | ||
Thirukkumaran CM, Shi ZQ, Luider J, et al. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clin Cancer Res. 2012;18(18):4962–4972. | ||
Thirukkumaran CM, Shi ZQ, Luider J, et al. Reovirus as a successful ex vivo purging modality for multiple myeloma. Bone Marrow Transplant. 2014;49(1):80–86. | ||
Kelly KR, Espitia CM, Zhao W, et al. Junctional adhesion molecule-A is overexpressed in advanced multiple myeloma and determines response to oncolytic reovirus. Oncotarget. 2015;6(38):41275–41289. | ||
Stiff A, Caserta E, Sborov DW, et al. Histone deacetylase inhibitors enhance the therapeutic potential of reovirus in multiple myeloma. Mol Cancer Ther. 2016;15(5):830–841. | ||
Kelly KR, Espitia CM, Mahalingam D, et al. Reovirus therapy stimulates endoplasmic reticular stress, NOXA induction, and augments bortezomib-mediated apoptosis in multiple myeloma. Oncogene. 2012;31(25):3023–3038. | ||
Kelly KR, Espitia CM, Zhao W, et al. Oncolytic reovirus sensitizes multiple myeloma cells to anti-PD-L1 therapy. Leukemia. Epub 2017 Aug 23. | ||
Senac JS, Doronin K, Russell SJ, Jelinek DF, Greipp PR, Barry MA. Infection and killing of multiple myeloma by adenoviruses. Hum Gene Ther. 2010;21(2):179–190. | ||
Chen CY, Senac JS, Weaver EA, et al. Species D adenoviruses as oncolytics against B-cell cancers. Clin Cancer Res. 2011;17(21):6712–6722. | ||
Fernandes MS, Gomes EM, Butcher LD, et al. Growth inhibition of human multiple myeloma cells by an oncolytic adenovirus carrying the CD40 ligand transgene. Clin Cancer Res. 2009;15(15):4847–4856. | ||
Tong Y, Zhu W, Huang X, et al. PI3K inhibitor LY294002 inhibits activation of the Akt/mTOR pathway induced by an oncolytic adenovirus expressing TRAIL and sensitizes multiple myeloma cells to the oncolytic virus. Oncol Rep. 2014;31(4):1581–1588. | ||
Haddad D. Genetically engineered vaccinia viruses as agents for cancer treatment, imaging, and transgene delivery. Front Oncol. 2017;7:96. | ||
Thorne SH. Immunotherapeutic potential of oncolytic vaccinia virus. Front Oncol. 2014;4:155. | ||
Mastrangelo MJ, Maguire HC Jr, Eisenlohr LC, et al. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999;6(5):409–422. | ||
Marshall JL, Hoyer RJ, Toomey MA, et al. Phase I study in advanced cancer patients of a diversified prime-and-boost vaccination protocol using recombinant vaccinia virus and recombinant nonreplicating avipox virus to elicit anti-carcinoembryonic antigen immune responses. J Clin Oncol. 2000;18(23):3964–3973. | ||
Mukherjee S, Haenel T, Himbeck R, et al. Replication-restricted vaccinia as a cytokine gene therapy vector in cancer: persistent transgene expression despite antibody generation. Cancer Gene Ther. 2000;7(5):663–670. | ||
Eder JP, Kantoff PW, Roper K, et al. A phase I trial of a recombinant vaccinia virus expressing prostate-specific antigen in advanced prostate cancer. Clin Cancer Res. 2000;6(5):1632–1638. | ||
Sanda MG, Smith DC, Charles LG, et al. Recombinant vaccinia-PSA (Prostvac) can induce a prostate-specific immune response in androgen-modulated human prostate cancer. Urology. 1999;53(2):260–266. | ||
Conry RM, Khazaeli MB, Saleh MN, et al. Phase I trial of a recombinant vaccinia virus encoding carcinoembryonic antigen in metastatic adenocarcinoma: comparison of intradermal versus subcutaneous administration. Clin Cancer Res. 1999;5(9):2330–2337. | ||
Futami M, Sato K, Miyazaki K, Suzuki K, Nakamura T, Tojo A. Efficacy and safety of doubly-regulated vaccinia virus in a mouse xenograft model of multiple myeloma. Mol Ther Oncolytics. 2017;6:57–68. | ||
Deng H, Tang N, Stief AE, et al. Oncolytic virotherapy for multiple myeloma using a tumour-specific double-deleted vaccinia virus. Leukemia. 2008;22(12):2261–2264. | ||
Paul S, Geist M, Dott K, et al. Specific tumor cell targeting by a recombinant MVA expressing a functional single chain antibody on the surface of intracellular mature virus (IMV) particles. Viral Immunol. 2007;20(4):664–671. | ||
Chan WM, Bartee EC, Moreb JS, Dower K, Connor JH, McFadden G. Myxoma and vaccinia viruses bind differentially to human leukocytes. J Virol. 2013;87(8):4445–4460. | ||
Lei W, Wang S, Yang C, et al. Combined expression of miR-34a and Smac mediated by oncolytic vaccinia virus synergistically promote anti-tumor effects in multiple myeloma. Sci Rep. 2016;6:32174. | ||
Kawa A, Arakawa S. The effect of attenuated vaccinia virus AS strain on multiple myeloma: a case report. Jpn J Exp Med. 1987;57(1):79–81. | ||
Ito T, Wang DQ, Maru M, et al. Antitumor efficacy of vaccinia virus-modified tumor cell vaccine. Cancer Res. 1990;50(21):6915–6918. | ||
Ito T, Okabayashi M, Osada Y, Wakamiya N, Kato S. Antitumor efficacy of two kinds of tumor vaccine modified with vaccinia virus. In Vivo. 1988;2(5):325–329. | ||
Fujiwara H, Shimizu Y, Takai Y, et al. The augmentation of tumor-specific immunity by virus help – I: demonstration of vaccinia virus-reactive helper T cell activity involved in enhanced induction of cytotoxic T lymphocyte and antibody responses. Eur J Immunol. 1984;14(2):171–175. | ||
Shimizu Y, Fujiwara H, Ueda S, Wakamiya N, Kato S, Hamaoka T. The augmentation of tumor-specific immunity by virus help – II: enhanced induction of cytotoxic T lymphocyte and antibody responses to tumor antigens by vaccinia virus-reactive helper T cells. Eur J Immunol. 1984;14(9):839–843. | ||
Ueda S, Wakamiya N, Wu KS, Kato S, Fujiwara H, Hamaoka T. Additional evidence for the augmented induction of tumor-specific resistance in vaccinia virus-primed mice by immunization with vaccinia virus-modulated syngeneic tumor cells. Biken J. 1984;27(1):1–7. | ||
Shimizu Y, Hasumi K, Masubuchi K, Okudaira Y. Establishment of tumor-specific immunotherapy model utilizing vaccinia virus-reactive helper T cell activity. Eur J Immunol. 1988;18(11):1773–1778. | ||
Fenner F, Ratcliffe FN. Myxomatosis. Cambridge: Cambridge University Press; 1965. | ||
Kerr PJ, Best SM. Myxoma virus in rabbits. Rev Sci Tech. 1998;17(1):256–268. | ||
Bartee E, Chan WM, Moreb JS, Cogle CR, McFadden G. Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol Blood Marrow Transplant. 2012;18(10):1540–1551. | ||
Kim M, Madlambayan GJ, Rahman MM, et al. Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia. 2009;23(12):2313–2317. | ||
Stanford MM, Shaban M, Barrett JW, et al. Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol Ther. 2008;16(1):52–59. | ||
Tosic V, Thomas DL, Kranz DM, et al. Myxoma virus expressing a fusion protein of interleukin-15 (IL15) and IL15 receptor alpha has enhanced antitumor activity. PLoS One. 2014;9(10):e109801. | ||
Wu Y, Lun X, Zhou H, et al. Oncolytic efficacy of recombinant vesicular stomatitis virus and myxoma virus in experimental models of rhabdoid tumors. Clin Cancer Res. 2008;14(4):1218–1227. | ||
Kinn VG, Hilgenberg VA, MacNeill AL. Myxoma virus therapy for human embryonal rhabdomyosarcoma in a nude mouse model. Oncolytic Virother. 2016;5:59–71. | ||
Woo Y, Kelly KJ, Stanford MM, et al. Myxoma virus is oncolytic for human pancreatic adenocarcinoma cells. Ann Surg Oncol. 2008;15(8):2329–2335. | ||
Wennier ST, Liu J, Li S, Rahman MM, Mona M, McFadden G. Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Mol Ther. 2012;20(4):759–768. | ||
Lun X, Alain T, Zemp FJ, et al. Myxoma virus virotherapy for glioma in immunocompetent animal models: optimizing administration routes and synergy with rapamycin. Cancer Res. 2010;70(2):598–608. | ||
Lun X, Yang W, Alain T, et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005;65(21):9982–9990. | ||
Weng M, Gong W, Ma M, et al. Targeting gallbladder cancer: oncolytic virotherapy with myxoma virus is enhanced by rapamycin in vitro and further improved by hyaluronan in vivo. Mol Cancer. 2014;13:82. | ||
Villa NY, Wasserfall CH, Meacham AM, et al. Myxoma virus suppresses proliferation of activated T lymphocytes yet permits oncolytic virus transfer to cancer cells. Blood. 2015;125(24):3778–3788. | ||
Lilly CL, Villa NY, de Matos AL, et al. Ex vivo oncolytic virotherapy with myxoma virus arms multiple allogeneic bone marrow transplant leukocytes to enhance graft versus tumor. Mol Ther Oncolytics. 2017;4:31–40. | ||
Dunlap KM, Bartee MY, Bartee E. Myxoma virus attenuates expression of activating transcription factor 4 (ATF4) which has implications for the treatment of proteasome inhibitor-resistant multiple myeloma. Oncolytic Virother. 2015;4:1–11. | ||
Bartee MY, Dunlap KM, Bartee E. Myxoma virus induces ligand independent extrinsic apoptosis in human myeloma cells. Clin Lymphoma Myeloma Leuk. 2016;16(4):203–212. | ||
Au GG, Lincz LF, Enno A, Shafren DR. Oncolytic Coxsackievirus A21 as a novel therapy for multiple myeloma. Br J Haematol. 2007;137(2):133–141. | ||
Hadac EM, Kelly EJ, Russell SJ. Myeloma xenograft destruction by a nonviral vector delivering oncolytic infectious nucleic acid. Mol Ther. 2011;19(6):1041–1047. | ||
Ruiz AJ, Hadac EM, Nace RA, Russell SJ. MicroRNA-detargeted mengovirus for oncolytic virotherapy. J Virol. 2016;90(8):4078–4092. | ||
Russell SJ, Peng KW. Measles virus for cancer therapy. Curr Top Microbiol Immunol. 2009;330:213–241. | ||
Evgin L, Acuna SA, de Souza CT, et al. Complement inhibition prevents oncolytic vaccinia virus neutralization in immune humans and cynomolgus macaques. Mol Ther. 2015;23(6):1066–1076. | ||
Magge D, Guo ZS, O’Malley ME, Francis L, Ravindranathan R, Bartlett DL. Inhibitors of C5 complement enhance vaccinia virus oncolysis. Cancer Gene Ther. 2013;20(6):342–350. | ||
Jelinek T, Mihalyova J, Kascak M, Duras J, Hajek R. PD-1/PD-L1 inhibitors in haematological malignancies: update 2017. Immunology. 2017;152(3):357–371. | ||
Badros A, Hyjek E, Ma N, et al. Pembrolizumab, pomalidomide, and low-dose dexamethasone for relapsed/refractory multiple myeloma. Blood. 2017;130(10):1189–1197. | ||
Lesokhin AM, Ansell SM, Armand P, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase IB study. J Clin Oncol. 2016;34(23):2698–2704. | ||
Hofgaard PO, Jodal HC, Bommert K, et al. A novel mouse model for multiple myeloma (MOPC315.BM) that allows noninvasive spatiotemporal detection of osteolytic disease. PLoS One. 2012;7(12):e51892. | ||
Asosingh K, Radl J, Van Riet I, Van Camp B, Vanderkerken K. The 5TMM series: a useful in vivo mouse model of human multiple myeloma. Hematol J. 2000;1(5):351–356. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.