")
Back to Journals » Cancer Management and Research » Volume 7
Potential of epigenetic therapies in the management of solid tumors
Authors Valdespino VM, Valdespino PM
Received 16 February 2015
Accepted for publication 20 April 2015
Published 31 July 2015 Volume 2015:7 Pages 241—251
DOI https://doi.org/10.2147/CMAR.S70358
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Kenan Onel
Video abstract presented by Victor Valdespino
Views: 301
Victor Valdespino,1 Patricia M Valdespino2
1Health Attention Department, Universidad Autónoma Metropolitana, Mexico; 2Bacterial Ecology and Epigenetics Laboratory, Universidad Nacional Autónoma de México, Mexico
Abstract: Cancer is a complex disease with both genetic and epigenetic origins. The growing field of epigenetics has contributed to our understanding of oncogenesis and tumor progression, and has allowed the development of novel therapeutic drugs. First-generation epigenetic inhibitor drugs have obtained modest clinical results in two types of hematological malignancy. Second-generation epigenetic inhibitors are in development, and have intrinsically greater selectivity for their molecular targets. Solid tumors are more genetic and epigenetically complex than hematological malignancies, but the transcriptome and epigenome biomarkers have been identified for many of these malignancies. This solid tumor molecular aberration profile may be modified using specific or quasi-specific epidrugs together with conventional and innovative anticancer treatments. In this critical review, we briefly analyze the strategies to select the targeted epigenetic changes, enumerate the second-generation epigenetic inhibitors, and describe the main signs indicating the potential of epigenetic therapies in the management of solid tumors. We also highlight the work of consortia or academic organizations that support the undertaking of human epigenetic therapeutic projects as well as some examples of transcriptome/epigenome profile determination in clinical assessment of cancer patients treated with epidrugs. There is a good chance that epigenetic therapies will be able to be used in patients with solid tumors in the future. This may happen soon through collaboration of diverse scientific groups, making the selection of targeted epigenetic aberration(s) more rapid, the design and probe of drug candidates, accelerating in vitro and in vivo assays, and undertaking new cancer epigenetic-therapy clinical trails.
Keywords: solid tumor, treatment, targeted therapy, epigenetic therapy
Introduction
Epigenetics participate not just in the normal functioning of the cell and its development, but also in diseases like neurological diseases and cancer. Epigenetic therapies can help to resolve different problems of these pathophysiological conditions. Cancer is a complex disease with both genetic and epigenetic origins. The importance of epigenetics in cancer has been recognized, and the field has emerged rapidly in recent years. Epigenetic and genetic alterations contribute to the initiation and progression of cancer. Epigenetic modifications precede genetic changes, and usually occur at an early stage in development of a neoplasm, but may also be involved in its invasion and spread. Recent technological advances in genetics and epigenetics offer a better understanding of the underlying epigenetic alterations during initiation and in the progression process of the human tumors. Techniques like next-generation sequencing allow studies of the DNA methylation status of human cells at nucleotide resolution; RNa-Seq, high-density microarrays for miRNA profiling; and CHIP-Chip and ChIP-Seq for detecting the transcriptome and identifying the binding sites of DNA-associated proteins.1
The main classic and next generation hallmarks of cancer have been recognized and analyzed in recent years,2–4 although changes in the differentiation process have not yet been analyzed. The cell differentiation process is very complex. Towards an integral understanding of this process, it will be necessary to know the heritable and somatic changes in gene expression not related to alterations in DNA sequences, such as key processes of DNA methylation, chromatin modifications, nucleosome positions, and alterations in noncoding RNA profiles. The Encyclopedia of DNA Elements (ENCODE) project has enhanced our understanding of the principles of the genome, epigenome, and chromatin organization by identifying hundreds of thousands of potential regulatory regions and transcription factor-binding sites in normal cells. The ENCODE project has provided some of the strongest and most detailed data regarding organization of the human genome. After understanding the principles of chromatin and epigenome organization in human primary cell types, the next step will be identification of changes (particularly through ongoing epigenome mapping) that have an important role in specific diseases.5
Reprogramming of somatic cells can be used as a simplistic model to understand the cell differentiation process.6 Takahashi and Yamanaka et al devised an accessible model of cell reprogramming from the differentiated to the induced-pluripotent state, in which somatic to pluripotent states are reverted by re-establishing modification of epigenetic markers.7 Similarly, in the last decade, many studies have highlighted the prominent role of altered environments (epigenetic signals) in regulation of gene expression in carcinogenesis. It seems evident that epimutations generally outnumber genetic abnormalities, and often occur much earlier in the development of cancer.1,8 These changes in the epigenetic landscape should be seen as responsive to aberrant signals coming from the environment and/or to abnormal interactions between the mesenchyme/stroma and the parenchyma/cells (“tissue organization field theory”).9
There are substantial parallels between the molecular mechanisms that underpin the reprogramming events leading to generation of induced-pluripotent stem cells and those that lead to oncogenic transformation and phenotype switching, including epithelial–mesenchymal transition (EMT) and mesenchymal–epithelial transition, and regeneration of tumor-initiating or cancer stem cell-like cells.10,11 Oncogenesis is ontogenesis gone awry, and like the small subset of the cancer cell population responsible for tumor initiation and growth, cancer stem cells (CSCs) may also have a similar kind of epigenetic reprogramming phenotype, leading to loss of expression of specific genes that maintain the differentiated state and regain or preserve stem cell-specific characteristics. Reprogramming epigenetic mechanisms in CSCs or in low-grade differentiation tumor cells by inducing them to differentiate from their pluripotent state to a mature differentiated form offers the potential for development of new therapeutic interventions.8,12
Cancer epigenome
Cancer represents a group of over 300 specific diseases that share a number of genetic, epigenetic, and pathological features. Carcinogenesis can be considered a potentially adaptive and a disordered ontogenic/evolutionary process. Cancerous and precancerous cells present different epigenetic abnormalities that modify the transcription of cancer-associated genes through aberrant expression of oncogenes and underexpression of tumor suppressor genes. The main epigenetic alterations in cancer cells are: genome-wide hypomethylation (in the gene body and in repetitive genomic regions), CpG island hypermethylation of gene promoter regions, activation and mobilization of retrotransposons, abnormal non-coding RNA patterns (microRNAs have a prominent role), loss of genomic imprinting, modifications of histone and Polycomb repressive complexes, chromatin remodeling, nucleosomal positioning and chromosomal looping changes, and accessibility of gene transcription regions.
A wide variety of cancers exhibit the CpG island methylator phenotype (CIMP). This is a hypermethylation pattern of CpG islands at gene promoter regions that is associated with the silencing of numerous cancer-related genes affecting a variety of vital cell processes. CIMP is strongly associated with clinical outcome, suggesting it may be a predictive biomarker of the disease course. In recent years, technological advances have enabled genome-wide DNA methylation analysis and unsupervised clustering of methylome data, which have clearly delineated CIMP-positive groups, including breast, lung, colorectal, endometrial, bladder, oral, and renal cancers.13,14
Modern evidence indicates that epigenetic changes in cancer are present in the normal-appearing tissue surrounding many tumors. Different epigenetic aberrations and mechanisms are associated with premalignant, invasive, and metastatic spreading phenotypes. Some of these epigenetic biomarker profiles may have diagnostic, prognostic, and predictive clinical utility.1,15
Recent studies have shown that the dynamics of repetitive elements or transposable elements may have a positive function by contributing to tissue-specific transcriptional programs as enhancer-like elements. They may also modulate the regulation of higher order chromatin structure.16 These mobile DNAs, also known as “jumping genes”, are widespread in nature and comprise an estimated 45% of the human genome; the long interspersed element-1 (LINE-1, retrotransposon) in particular, is capable of retrotransposing, not just of their own RNA but also of nearby RNAs by encoding reverese transcriptase. Retrotransposition is a common contributor to human genetic variation.17 LINE-1 encodes reverse transcriptase, required for its own mobility, and its expression is inhibited in differentiated tissues, but active in tumors. Experimental evidence indicates that inhibition of LINE-1-derived reverse transcriptase restores differentiation in cancer cells, inhibits tumor progression, and yields globally reprogrammed transcription profiles.18 Epigenetic changes in LINE-1 occur early during carcinogenesis. In cancer cells, the methylation levels of a majority of LINE-1 loci is decreased (44%), with some loci remaining unchanged and a few being increased (56%–61%) when compared with normal cells. Hypomethylation of LINE-1 loci is common in most types of cancer; this increases their mobilization and can drive the mobilization of adjacent coding regions and regulatory elements to new locations. LINE-1 dissemination has been observed to wax and wane during evolution of tumors in humans. LINE-1 hypomethylation levels is a candidate tumor marker for cancer.19,20
Aberrant epigenomics contributes to development of cancer by its involvement in initiation, promotion, invasion, metastasis, and resistance to chemotherapy. More than 300 genes and gene products are epigenetically altered in human cancers. The proteins of these cancer-related genes participate, eg, in the transcriptional inactivation caused by promoter hypermethylation of genes involved in the major cellular pathways (including DNA repair, cell cycle control, cell differentiation, apoptosis, metastasis, detoxification, hormone response, vitamin response, and the p53 network) or in DNA hypomethylation of oncogenes and repetitive regions of the genome (causing genomic instability). These genes also affect histone modification patterns related to gene expression, long noncoding RNAs, and repositioning chromatin involved in complex cellular regulations.
The key processes responsible for epigenetic regulation in eukaryotic cells are DNA methylation and modifications of chromatin (covalent alteration in core histones). Early successes have been achieved with DNA-demethylating drugs and agents supporting histone acetylation. DNA methylation is an epigenetic event that regulates compacting of chromatin and repression of gene expression. In cancer cells, a variety of genes are abnormally silenced by DNA methylation, including tumor suppressor genes and genes controlling the immune response and drug sensitivity. DNA methylation is catalyzed by DNA methyltransferases (DNMTs) and involves the transfer of a methyl group from the methyl donor S-adenosylmethionine to the C-5 of cytosine in DNA. Classical DNMTs are DNMT1, DNMT3A, and DNMT3B. DNMT1 is the most abundant DNMT and maintains methylation during DNA replication, while DNMT3A and DMNT3B are responsible for establishing methylation during embryonic development. Overexpression of the major DNMTs may result in accumulated hypermethylation of DNA for tumor-related genes. Disrupted homeostasis of DNA methylation is a hallmark of cancer, and acts as an alternative and complementary mechanism to gene mutation or deletion, leading to inactivation of specific tumor suppressor genes. DNMT inhibitors can be divided into nucleoside-derived and non-nucleoside inhibitors. The former class includes azacitidine and decitabine, of which low doses have shown clinical benefits.21 Histone modifications include a multitude of covalent variations affecting the histone amino-terminal tails protruding from nucleosomes. The post-translational modifications comprise acetylation, methylation, phosphorylation, sumoylation, and ADP ribosylation, which alter both the electrostatic charge of the histone and its interactions with multiprotein complexes, thus modulating the activity of the associated DNA. Among the post-translational modifications on histone tails, histone acetylation and methylation of specific lysine residues on histones H3 and H4 are the most extensively studied. Histone acetylation is an important determinant of gene expression, acetylation is generally associated with elevated transcription, and histone deacetylation is often associated with gene repression. Histone deacetylases (HDACs) are critical regulators of gene expression, removing the acetyl group from histones and non-histone proteins. Four classical HDAC classes, ie, I, II, III, and IV, containing eleven HDACs, have been classified according to their homology to yeast proteins, subcellular location, and enzyme activity. The biological outcome of HDAC inhibition is dependent on the HDAC specificity of the compound and intrinsic operation of cell-signaling pathways. HDAC inhibitors (eg, vorinostat and romidepsin) exert their anticancer activity by promoting acetylation of histones, and are the best characterized anticancer drugs. These first-generation epigenetic inhibitors have a broad target specificity for the mechanisms of action of the eleven HDACs.22
Over the last few years, many mutations have been identified in genes that modify the epigenome. Epigenetic changes in tumors are generally of a clonal nature, and occur in the early generation of cancer cells. It is established that 5-methylcytosine residues are “hot spots” for mutations, because 5-methylcytosine is highly mutable by deamination. Environmental and micro-environmental changes such as oxidative stress can contribute to tumor development not only through genetic mechanisms but also through epigenetic mechanisms.1 Recently, new sequencing technologies have identified driver mutations in epigenetic regulators, providing a mechanistic link between the cancer epigenome and genetic alterations. Many genetic alterations and post-translational modifications in cancer-targeted epigenetic regulators cause cancer-associated phenotypes via epigenetic dysfunction. Most of the structural data of the epigenetic enzyme classes is well known and is used in the application of targetable molecules. (Table 1). Mutations in epigenetic modifiers comprise a large proportion of all genetic events in many cancers. Oncogenic activating mutations occur in a number of epigenetic regulators (eg, IDH1/2, EZH2, DNMT3A) or inactivation mutations of tumor suppressors/chromatin modifiers (eg, KDM6A, CREBBP/EP300, SMARCB1). Modifier proteins with frequent driver mutations in cancer can be identified by specific function and target site: DNA methylation and hydroxymethylation modifiers (DNMT3A, TET2, IDH1/2); three covalent histone modifier types, ie, reader (recognizes and recruits), writer (adds modifications, eg, EZH2, SETD2, MLLs, NSD1/2; CREBBP, EP300), and eraser (removes modifications, eg, KDM6A, other KDMs; HDACs); and chromatin remodeling modifiers, such as the SWI/SNF complex (ARIDs, SMARCs, PBRM1). Low recurrent mutations in genes encoding the histones have also been identified (HIST1H1B, HIST1HIC); most striking is the location of these mutations at amino acid residues near critical sites for post-translational modifications on the histone tail (eg, H3K27, H3K36).22
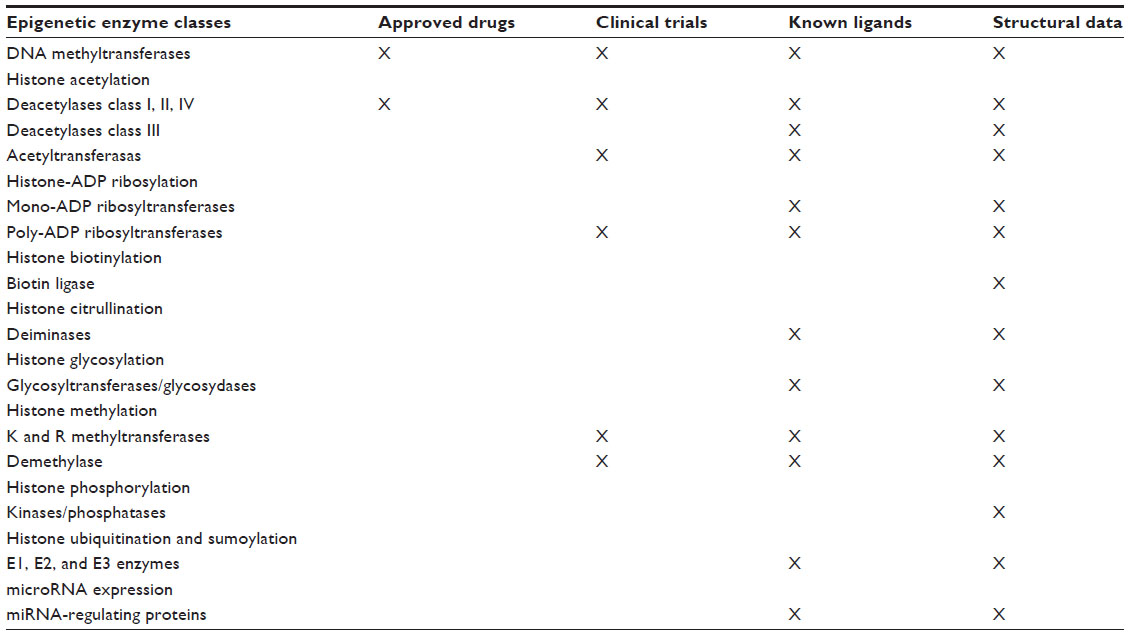 | Table 1 Targetable epigenetic enzyme classes and connections with drug discovery information for the treatment of cancer |
Several studies have shown that the mutations in epigenetic modifiers are both incredibly diverse and ubiquitous in different types of cancer. Several neoplasms rely on few or many mutations in selected genes that drive tumorigenesis through an altered epigenome, at least in part. Mutations in regulators of DNA methylation have a strong cancer-specific prevalence, whereas genetic alteration in histone modifiers is widespread across many cancers. Understanding how genetic alterations give rise to the cancer epigenome will offer new possibilities for developing better prognostic and therapeutic strategies. Epigenomic alterations associated with these driver gene mutations could lead to the discovery of novel pathways involved in tumorigenesis, which may themselves possess unique vulnerabilities for therapeutic intervention.23
Epigenotyping collaboration in the molecular pathological epidemiology of cancer
To better understand the interplay between etiological factors, cellular-molecular characteristics, and disease evolution, the field of “molecular pathological epidemiology” (MPE) has emerged as an interdisciplinary integration of molecular pathology and epidemiology. MPE is founded on the unique disease principle that each disease process results from a unique profile of exposomes, epigenomes, transcriptomes, proteomes, metabolomes, microbiomes, and interactomes in relation to the macro-environment and the tissue micro-environment. The MPE paradigm (in cancer) represents an integrative interdisciplinary science of molecular pathology (genetic patterns of germlines and genomic patterns of somatic cells) and epidemiology (data on risk factors). The epigenetic patterns of tumor (somatic) cells have recently been added to this paradigm.24 The epigenetic epidemiology of cancer includes the study of variation in epigenetic traits and the risk and progression of disease using large-scale, population-based data. Epigenotyping can classify cancers affecting different organs into distinct groups, with different clinical, pathological, and molecular characteristics. Further, identification of diagnostic epimarkers that enable early detection of disease and its prognosis can be used to apply targetable treatments. Risk factors associated with epigenetic diagnostic and prognostic biomarkers have been studied in lung, breast, colorectal, and bladder cancer.25 Nowadays, the holistic MPE approach in cancer patients analyses the genome, methylome, epigenome, metabolome, transcriptome, proteome, microbiome, immunity, and the interactome.
The holistic MPE approach enables us to link potential etiological factors to specific molecular pathologies, and gain novel pathogenetic insights on causality. The main epigenome analyses encompass methylome patterns, such as the promoter CIMP, global methylation, and LINE-1 methylation epigenotypes as well as the histone post-translational profile, including acetylation/methylation of specific lysines of H3 and H4. Biomarkers detected using molecular biology tools allow molecular characterization of cancer signatures and provide information relevant for personalized treatment. Molecular biomarkers based on the molecular profile of the tumor can be used as prognostic biomarkers or molecularly targeted therapy, and for evaluation of the toxic effects of anticancer drugs.26,27
Molecular diagnostics have become an essential component in clinical cancer decision-making. However, tumor evolution and behavior cannot be accurately predicted, despite numerous research studies reporting promising tumor biomarkers. To advance in molecular diagnostics, it is essential to have a better understanding of intratumor and intertumor heterogeneity, and the extracellular matrix–non-neoplastic host cell interactions influenced by genomic variation, hormones, dietary, lifestyle, and environmental exposure. We can better classify tumors by molecular methods, and move closer toward precision medicine for cancer.28 Recently, President Obama announced a research initiative that aims to accelerate progress toward a new era of precision medicine and bring us closer to being able to cure diseases like cancer and diabetes. This initiative will encourage and support scientists’ efforts to develop creative new approaches for detecting, measuring, and analyzing a vast amount of biomedical information. These research activities will propel our understanding of diseases and their origins and mechanisms, and open up opportunities for prevention and treatment.29
Therapeutically targeting the cancer epigenome
Conceptually, epigenetic changes are reversible, so they are rational targets to approach. To date, five epigenetic agents have been approved by the US Food and Drug Administration, and comprise the first generation of epigenetic inhibitors (Table 2), ie, two DNMT inhibitors and three HDAC inhibitors. The DNMT inhibitors, 5-azacytidine (Vidaza®) and 5-aza-2′deoxycytidine (decitabine, Dacogen®), are analogs of cytidine and have demonstrated clinical utility in myelodysplastic syndrome and leukemia, and the HDAC inhibitors, suberoylanilide hydroxamic acid (vorinostat, Zolinza®), the natural product of the Chromobacterium violaceum (romidepsin, Istodax®), and PXD-101 (belinostat, Beleodaq®) have shown clinical utility in the treatment of rare cutaneous T-cell lymphoma.30 DNMT inhibitors act as irreversible covalent inhibitors of all catalytically active DNMT isoforms following incorporation into DNA.31 HDAC inhibitors are a diverse group of compounds that vary in structure, biological activity, and specificity, and novel therapeutics are now targeting zinc-containing HDAC enzymes in the treatment of various types of cancer.32,33
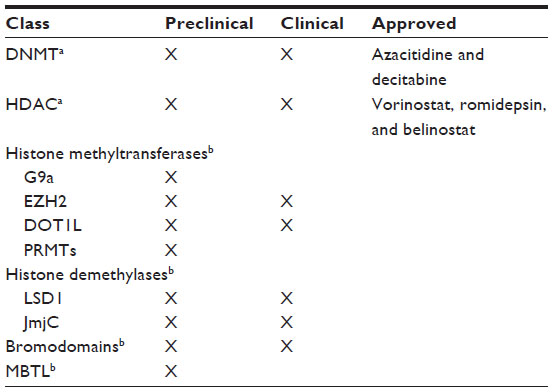 | Table 2 First-generation and second-generation epigenetic inhibitors for the treatment of cancer |
Many other DNMT inhibitors and HDAC inhibitors are being used in clinical trials. Because cross talk can occur between DNA methylation and histone deacetylation, a combination of these two epigenetic modifications has been used too. The first generation of epigenetic inhibitors, being drug-like molecules, has shown significant but limited utility in hematological malignancies due to toxicity and off target effects (Table 2).
Developing therapeutic strategies targeting epigenetic aberrations (in much the same way as the successful development of the targeted protein kinase inhibitors currently used in the treatment of cancer) keeps expanding and is guiding research efforts for many investigators. Efforts to derive more selective and/or less pleiotropic inhibitor scaffolds are continuing, and may provide efficacy in a broader set of tumors. Similarly, the development of a second generation of epigenetic inhibitors (Table 2), which have a sufficient preclinical therapeutic index to allow clinical evaluation, is a growing area. These second-generation compounds hold more promise, because they have greater intrinsic selectivity for their molecular targets and will be developed in indications where the target is known to be a driver or a key mediator of the malignancy. Second-generation epigenetic inhibitors are exciting new drugs that target other reader, writer, and eraser histones which represent epigenetic aberrations in tumor cells (alterations in epigenetic proteins of tumors).34 Most second-generation epigenetic inhibitors are: histone methyltransferase inhibitors such as G9a, responsible for the dimethylation of lysine 9 on histone H3 (H3K9me2); EZH2 inhibitors that catalyze methylation of H3K27 and carry out its function as part of the Polycomb repressive complex 2; DOT1L inhibitors that catalyze the transfer of one, two, or three methyl groups to H3K79; LSD1 inhibitors that catalyze the demethylation of monomethylated and dimethylated H3K4 and H3K9; and Jumonji inhibitors, which represent the largest group of lysine demethylases. Other second-generation epigenetic inhibitors (in the study phase) are the epigenetic protein inhibitors, as the acetyl lysine readers (BET subfamily members), and the methyl lysine readers.31 Recently, a rapidly expanding list of reported somatic gene alterations (recurrent point mutations and chromosomal translocations) have been identified in both hematological and solid tumors.
According to the model of cellular differentiation deregulation in carcinogenesis process, normal adult somatic stem cells and particularly CSCs can be modified using molecular epigenetic interventionism targeting oncogenic signaling cascades. CSCs represent into the tumor, a small subset of the cancerous population responsible for tumor initiation and growth. They possess the characteristic properties of quiescence, indefinite self-renewal, and intrinsic resistance to chemotherapy and radiotherapy. Somatic stem cells and CSCs share common signaling pathways for the retention of their stem cell properties. The most important self-renewal signaling pathways include WNT/b-catenin, NOTCH, and Hedgehog, each of which has multiple downstream target proteins and genes.12 The generation of CSCs in the process of carcinogenesis involves epigenetic reprogramming mechanisms, which lead to loss of expression of genes (specific to the differentiated states) and regaining of stem cell-specific characteristics. In the reprogramming of CSCs, epigenetic changes regulate the genesis and progression of cancer growth. Many epigenetic alterations in CSCs modify structural patterns of DNA methylation/chromatin remodeling and post-transcriptional regulation of non-coding RNAs. Most of the epigenetic alterations in CSCs are caused by epigenetic enzyme alterations, which are frequently used as targets for anticancer therapy. Epigenetic enzymes are often dysregulated in human tumors through mutation, altered expression, or inappropriate recruitment to certain loci. Identification of these enzymes and their partner proteins has driven the rapid development of small-molecule inhibitors that target the cancer epigenome (Table 1). Regulation of histone post-translational modifications and DNA methylation is controlled and catalyzed by many different classes of enzymes, the existence and functions of which have been elucidated with a rapid progression in the last decade. In the years to come, identification of somatic gene alterations highlighting epigenetic deregulation may be possible for many cancers, and in the same way as the successful development of the targeted protein kinase inhibitors currently in use, the possibility of developing “personalized epigenetic medicine” will be achieved.
Epigenetic modifications are reversible nuclear chemical reactions that occur due to the ability of enzymes to exercise opposing catalytic effects (eg, using specific inhibitors). For now, the Structural Genomics Consortium (SGC; http://www.thesgc.org), a non-profit organization, formed in 2004 to determine the three-dimensional structures of proteins of medical relevance, and in particular its epigenetics program, is working to generate pharmacological inhibitors of human proteins that regulate epigenetic signaling. The SGC develops chemical probes and antibodies to study the role that different protein domains play in histone modifications and DNA methylation. SGC accelerates the development of new or not very well understood areas of human genome research, creating collaboration networks between nine global pharmaceutical companies, scientists, and universities.31
Cancer pathologies have been associated to many different epigenetic changes. Identification of specific epigenetic alterations is necessary for designing specific anticancer drugs. Each type of cancer may have an epigenetic alteration profile, and patients with the same type of neoplasm may have a particular epigenetic profile. Epigenetic aberrations arise early in the carcinogenesis process, preceding classical gene mutations identified in the specific pathophysiology of each of the 300 types of human cancer. Epigenetic-related aberrations associated with changes in genomic, transcriptomic or signaling pathways (particularly of the oncogenes and tumor-suppressor genes), should be used for epigenetic analysis and as epigenetic targets for anticancer therapy. For example, the estrogen receptor and human epidermal growth factor 2 are epidrug targets in the treatment of breast cancer; chromosomal instability, miR-34b, miR-148-a, and CIMP are targets in colorectal cancer; and the epidermal growth factor receptor in non-small-cell lung carcinoma.35 Nevertheless, different tumor types express CSC-like markers, such as CD133, CD44, CD34, and CD24, and pluripotency genes such as OCT3/4, Nanog, SOX2, and MYC. Also, other expressed markers of invasiveness such as vimentin, N-cadherin, snail, Twist, and Zeb1 as well as markers of drug resistance such as aldehyde dehydrogenase and ABC transporters.12,36 Different combinations of these markers are characteristic for identification of the different types of tumor tissues and are used to design epigenetic targets for anticancer therapy.
On the other hand, the selection of specific anticancer epidrugs needs to transit through long analytic studies to to obtain clinical therapeutic utility. This developing research on epidrugs requires: target selection (altered epigenetic proteins); epigenetic chemical probes; analyses of drug candidates or small-molecule inhibitors; in vitro assays (development of cells); in vivo biological/pharmacological/toxicological assays (novel animal models with specific epigenetic aberrations); and studies progressing through Phase I, II, III, and IV clinical trials.34,37
As with other cancer therapies, the deployment of epigenetic therapies will likely require development of a rational combination of chemotherapy agents, target kinase inhibitors, inmmunotherapies, or different classes of epigenetic drugs. Epigenetic therapy should also consider the biological evolution of the specific cancer and the cytotoxic agents previously used in patients. This strategy has been used to reverse resistance to cisplatin in patients with ovarian cancer cells, and in other tumor types.31
First-generation epigenetic inhibitors in hematopoietic tumors
Epigenetic therapies may play an important role in the future management of solid tumors. This observation is based on the limited clinical utility of epidrugs in treating defined hematopoietic neoplasms, our better understanding of epigenetic modifications in normal and cancer cells, the development of promising second-generation epigenetic inhibitor, and data from preclinical and clinical studies. Many regulatory proteins involved in abnormal DNA methylation and chromatin coiling in cancer epigenome, including DNMTs, HDACs, methyl-CpG-binding domains, Polycomb-group, and other proteins, are targetable and represent active opportunities in cancer therapy.38 Pathophysiological (genomic and epigenomic) and phenotypic changes are more numerous and complex in solid tumors than in hematopoietic tumors, so carcinomas and sarcomas have more targetable molecules that can be used as therapeutic targets. Differing patterns of DNA methylation abnormalities are emerging from deep sequencing analyses of different tumor types and subtypes; and similar conditions may be identified in chromatin coiling. These aberrant epigenetic patterns will guide clinical approaches to the treatment of specific solid tumors. A rational design of chemical epidrugs, in addition to rigorous preclinical and clinical analyses, will pave the way for clinical success. Drugs targeting the epigenome are promising new cancer treatment modalities, but not all patients receive the same benefit from these drugs. In contrast with conventional chemotherapy, responses may not be apparent until several months after initiation of treatment. Changes in specific epigenetic biomarkers in tumor cells (eg, LINE-1 methylation pattern elements, histone acetylation patterns) may be used as predictor clinical biomarkers in the assessment of anticancer responses. Assessment of these epigenetic markers, together with specific subclinical cancer biomarkers, such as circulating tumor cells, or minimal residual disease (gene expression signature of a specific solid tumor) in treated cancer patients improve our ability to predict the clinical response.39,40 Ideally, identification of good predictive biomarkers would allow selection of a personalized therapy, and thereby maximize the benefit of treatment.41 At present, despite our comprehensive knowledge of the biology and function of epigenetic therapies, the search for specific biomarkers for response and survival is still key. Novel high-throughput epigenetic screening methodologies should progress to achieve these objectives.
First and second generations of epigenetic inhibitors in treatment of solid tumors
Human gene expression patterns are controlled and coordinated by the activity of a diverse array of epigenetic regulators, including histone methyltransferases, acetyltransferases, and chromatin remodelers. Deregulation of these epigenetic pathways can lead to genome-wide changes in gene expression. In recent years, research has suggested that cross talk between genomic and epigenomic factors may drive the etiology of both hematological malignancies and solid tumors. Epigenetic alterations are associated with different stages of tumor formation and progression in many types of malignancy. Epigenetic alterations are frequently observed in cancers associated with chronic inflammation and/or persistent infection with infection with viral or other pathogenic micro-organisms, and with cigarette smoking. To obtain a comprehensive picture of the standard epigenome profiles of normal tissues, the International Human Epigenome Consortium (http://www.ihec-epigenomes.org) has set the ambitious goal of decoding at least 1,000 epigenomes in the years to come.42 The International Human Epigenome Consortium coordinates the production of reference maps of human epigenomes for key cellular states that are relevant to health and disease. New epigenetic regulators, whose contribution to cancer initiation and progression has been identified, and many second generation epigenetic inhibitors are currently in preclinical and clinical studies.34,43
The experience with epigenetic therapy in hematopoietic and solid tumors to date has been restricted to DNA-demethylating drugs (decitabine, 5-azacitidine) and HDAC inhibitors (romidepsin, vorinostat, belinostat). These first-generation epidrugs have had very modest anti-tumor efficacy as monotherapy in Phase I and II clinical trials in patients with solid tumors, and the combination of epidrugs with other therapies would enhance or restore sensitivity to such therapies. These drugs have shown very modest antitumor efficacy, with some responses seen in isolated cases (3% of patients responded to epidrugs alone, 20% of patients responded to epidrugs in combination with other chemotherapies). The disadvantage of epidrugs for the management of cancer is genome-wide effects, which may cause unwanted upregulation of, eg, prometastatic genes. Development of gene-targeted epigenetic modifications in dysregulation of tumor-associated genes of specific tumors can provide a novel approach to prevent such unwanted events.38,45,46 At the present time, 72 clinical trials using first-generation and second-generation epidrugs, alone or in combination, as epigenetic cancer therapy are registered at ClinicalTrials.gov (http://www.clinicaltrials.gov). A review by Nie et al of decitabine-based epigenetic therapy in patients with 12 different types of solid tumors showed that cervical carcinoma, pleural tumors, ovarian carcinoma, and colorectal cancer registered improved response rates (8%–35%).47 In another interesting study, Li et al showed that the strong immunomodulatory intracellular role of 5-azacitidine in cancer cell lines and in biopsies from carcinoma patients, this condition can restore functional expression of the components involved in the biosynthesis and assembly of the peptide/Beta-2-microglobulin/human leukocyte antigen class I heavy chain complex.48,49 These changes in expression may be screened for in the future to identify and handle epigenetic priming for immune therapy.
Future directions
Epigenetic therapy is emerging as a potentially effective therapy for solid tumors. In solid tumors, simultaneous multilayer/integrative omics analyses (including genome, epigenome, and transcriptome abnormalities) will be useful to elucidate the molecular background of pathogenesis and to explore possible therapeutic targets. This organization model has been implemented by the Program for Promotion of Fundamental Studies in Health Sciences at the National Institute of Biomedical Innovation.42 As mentioned earlier, in the coming years, the International Human Epigenome Consortium together with information from Gene Expression Human Maps will provide reference maps of human epigenomes and transcriptomes for key cellular types and states. Also, specific gene sets participating in oncogenic signaling pathways, oncogenic phenotype cell characteristics, and/or cancer reprogramming cells should be analyzed to obtain a wide and specific landscape of cancer cells, particularly with regard to their epigenomic profile.
Each type of solid tumor has particular oncogenic phenotype characteristics based on a specific genomic/epigenomic program for initiation of its transformation. Each patient with this type of malignancy may show a specific phenotypic or personalized subtype, which includes particular genomic/epigenomic/transcriptomic aberrations (epigenome mechanisms show heterogeneity among tissues and cell lineages). This specific profile should be identified to determine the specific pathophysiology of the neoplastic process, which could be potentially applicable in disease prevention, diagnosis, and therapy. Once this profile is identified, it is possible to explore rational therapeutic targets. Pathophysiological (genomic/epigenomic/transcriptomic) aberrations in solid tumor types may be suspected considering the biological behavior of the clinical history and the histopathological subtype image.38 For example, in most solid cancers, metastatic disease is the main cause of mortality and morbidity. Metastasis is a complex process requiring tumor cells to invade the surrounding tissue, gain access to the vasculature, survive transport, exit the vasculature, and then grow in a foreign tissue environment. Our understanding of metastasis indicates that cancer cells acquire EMT. This is a cellular program orchestrated by a set of pleiotropic transcriptional factors such as Twist, Snail, Slug, and Zeb1/2 that together form an intricate transcriptional circuit which acts as a transcriptional repressor of E-cadherin and zona occludens-1, leading to dissolution of adherens and tight junctions. Induction of EMT is accompanied by dynamic reprogramming of the epigenome, involving changes in DNA methylation and several post-translational histone modifications. Different epigenetic regulators involved in EMT could be used in individualized epigenetic tumor therapy, eg, specific small-molecule inhibitors.50 As clinical response indicators, patients treated with epidrugs may show results several months after starting the treatment. In these patients, it is relevant to identify molecular biomarkers associated with positive or negative clinical response in a post-treatment stage. Quantitative changes in signaling molecules in the oncogenic pathway can be used as predictive biomarkers, particularly tumor-associated gene expression and molecular epigenetic biomarkers, such as DNA methylation patterns in tumor-associated genes, or histone post-translational changes like CIMP in specific genes; these have also been used as criteria for prognostication in patients.51 An example of assessment of tumor-associated gene expression was shown by Li et al in response to use of the epidrug 5-azacitidine, surveying the differential determination of 15 immunomodulatory genes expression.48
A promising new second-generation of epigenetic inhibitors is being developed, and the support and collaboration of the Structural Genomics Consortium or Consortium-like organizations in the design, generation, and analysis of pharmacological inhibitors of epigenetic-targeted molecules, which will enhance the progress of testing preclinical and clinical assays.
Other scientific advances in genome engineering and disease modeling are becoming available. The application of genome engineering techniques in human gene-therapy will be critical to obtain improved therapeutic results (eg, bacterial RNA-guided CRISPR-Cas system).52 Induced-pluripotent stem cell technology can be employed in various genetic/degenerative diseases and types of cancer for disease modeling and gene therapy. Use of induced-pluripotent stem cells is a better alternative to conventional tests of toxicology and drug research, since they provide an environment that is more similar to human physiological conditions.53
More clinical trials on epigenetic therapy for cancer are necessary for design and contribution to further progress of this therapeutic strategy. The proven clinical utility of DNMT inhibitors and pan-HDAC inhibitors, as well as the rapid preclinical advancement of second-generation epigenetic modulators, offer an optimistic outlook for future epigenetic drug discovery and development. More clinical experience with these agents will serve to guide strategies for therapeutic application in targeted patient populations.34 CSC-targeted therapeutic approaches might improve the chances of patient survival by reducing the frequency of tumor relapse. With the president’s budget for 2016, the National Cancer Institute will have increased funding to support clinical trials in order to link their genomic findings to clinical data, and to connect them with lifestyle risks and environmental conditions. The translational and therapeutic programs at the National Cancer Institute, such as the Molecular Analysis for Therapy Choice (NCI-MATCH) Program, and others with similar purposes, will become stronger in their use of molecular methods to identify drugs that will deliver optimum results for treating tumors.
The use of epigenetic inhibitors in the treatment of cancer has multiple therapeutic challenges, particularly with regard to identification of genome/epigenome signaling pathways in each tumor type and subtype and in personalized patient profiles. Biomarkers and clinical criteria should be used, such as identification of class- and subclass-specific targets, pharmacological dose and dosage schedule, and use of clinical protocols. Epigenetic therapy for each cancer patient should consider the tumor-profile specific type or subtype. The selection of the appropriate epidrug (with minimal toxic effects) will lead to an improved therapeutic response.
Disclosure
The authors report no conflicts of interest in this work.
References
Kanwal R, Gupta K, Gupta S. Cancer epigenetics: an introduction. Methods Mol Biol. 2015;1238:3–25. | |
Hanahan D, Weinberg R. The hallmarks of cancer. Cell. 2000;100:57–70. | |
Valdespino VM, Valdespino VE. Alteraciones celulares y moleculares no clasicas en el desarrollo del cáncer [Non classical cellular and molecular deficits in cancer development]. Gac Med Mex. 2010;146:185–198. Spanish. | |
Hanahan D, Weinberg R. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. | |
Sieggens L, Ekwall K. Epigenetics, chromatin and genome organization: recent advances from the ENCODE project. J Intern Med. 2014;276:201–214. | |
Valdespino V, Valdespino PM, Valdespino V Jr. Main phenotype subphases in reprogramming somatic cells as a model of cellular differentiation process. Am J Biomed Res. 2013;1:48–56. | |
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblast by defined factors. Cell. 2007;131:861–872. | |
Campos-Sanchez E, Cobaleda C. Tumoral reprogramming: plasticity takes a walk on the wild side. Biochim Biophys Acta. 2015;1849:436–447. | |
Burgio E, Migliore L. Towards a systemic paradigm in carcinogenesis: linking epigenetics and genetics. Mol Biol Rep. 2015;42:777–790. | |
Goding CR, Pei D, Lu X. Cancer: pathological nuclear reprogramming? Nat Rev Cancer. 2014;14:568–573. | |
Kanherkar RR, Bhatia-Dey N, Makarev E, Csoka AB. Cellular reprogramming for understanding and treating human disease. Front Cell Dev Biol. 2014;2:67. | |
Shukla S, Meeran SM. Epigenetics of cancer stem cells: pathways and therapeutics. Biochim Biophys Acta. 2014;1840:3494–3502. | |
Suzuki H, Yamamoto E, Maruyama R, Niinuma T, Kai M. Biological significance of the CpG island methylator phenotype. Biochim Biophys Res Commun. 2014;455:35–42. | |
Colussi D, Brandi G, Bazzoli F, Ricciardiello L. Molecular pathways involved in colorectal cancer: implications for disease behavior and prevention. Int J Mol Sci. 2013;14:16365–16385. | |
Jones P. The cancer epigenome. Genome. 2013;56:540–541. | |
Bodega B, Orlando V. Repetitive elements dynamics in cell identity programming, maintenance and disease. Curr Opin Cell Biol. 2014;31C: 67–73. | |
Hancks DC, Kazazian HH Jr. Active human retrotransposons: variation and disease. Curr Opin Genet Dev. 2012;22:191–203. | |
Sciamanna I, Gualtieri A, Piazza PV, Spadafora C. Regulatory roles of LINE-1-encoded reverse transcriptase in cancer onset and progression. Oncotarget. 2014;5:8039–8051. | |
Kitkumthorn N, Mutirangura A. Long interspersed nuclear element-1 hypomethylation in cancer: biology and clinical applications. Clin Epigenet. 2011;2:315–330. | |
Turbio JM, Li Y, Ju YS, et al. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 2014;345:1251343. | |
Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13:679–692. | |
West A, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–39. | |
Roy DM, Walsh LA, Chan TA. Driver mutations of cancer epigenomes. Protein Cell. 2014;5:265–296. | |
Ogino S, Lochhead P, Chan AT, et al. Molecular pathological epidemiology of epigenetics: emerging integrative science to analyze environment, host, and disease. Mod Pathol. 2013;26:465–484. | |
Barrow TM, Michels KB. Epigenetic epidemiology of cancer. Biochem Biophys Res Commun. 2014;455:70–83. | |
Kalia M. Biomarkers for personalized oncology: recent advances and future challenges. Metabolism. 2015;64(3 Suppl 1):S16–S21. | |
Nalejska E, Maczynska E, Lewandowska MA. Prognostic and predictive biomarkers: tools in personalized oncology. Mol Diagn Ther. 2014;18:273–284. | |
Ogino S, Fuchs CS, Giovannucci E. How many molecular subtypes? Implications of the unique tumor principle in personalized medicine. Expert Rev Mol Diagn. 2012;12:621–628. | |
Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–795. | |
Lee HZ, Kwitkowski VE, Del Valle PL, et al. FDA approval: belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin Cancer Res. March 23, 2015. [Epub ahead of print.] | |
Dhanak D, Jackson P. Development and classes of epigenetic drugs for cancer. Biochem Biophys Res Commun. 2014;455:58–69. | |
Lakshmaiah KC, Jacob LA, Aparna S, Lokanatha D, Saldanha SC. Epigenetic therapy of cancer with histone deacetylase inhibitors. J Cancer Res Ther. 2014;10:469–478. | |
De Souza C, Chatterji BP. HDAC inhibitors as novel anti-cancer therapeutics. Recent Pat Anticancer Drug Discov. March 17, 2015. [Epub ahead of print.] | |
Campbell RM, Tummino P. Cancer epigenetics drug discovery and development: the challenge of hitting the mark. J Clin Invest. 2014;124:64–70. | |
Andreoli F, Barbosa AJ, Parenti MD, Del Rio A. Modulation of epigenetic targets for anticancer therapy: clinicopathological relevance, structural data and drug discovery perspectives. Curr Pharm Des. 2013;19:578–613. | |
Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716–727. | |
Verma SK. Recent progress in the discovery of epigenetic inhibitors for the treatment of cancer. Methods Mol Biol. 2015;1238:677–688. | |
Azad N, Zahnow CA, Rudin C, Baylin SB. The future of epigenetic therapy in solid tumours – lessons from the past. Nat Rev Clin Oncol. 2013;10:256–266. | |
Alix-Panabieres C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer. 2014;14:623–631. | |
Ben Lassoued A, Nivaggioni V, Gabert J. Minimal residual disease testing in hematologic malignancies and solid cancer. Expert Rev Mol Diagn. 2014;14:699–712. | |
Treppendahl MB, Kristen LS, Gronbaek. Predicting response to epigenetic therapy. J Clin Invest. 2014;124:47–55. | |
Kanai Y, Arai E. Multilayer-omics analyses of human cancers: exploration of biomarkers and drug targets based on the activities of the International Human Epigenome Consortium. Front Genet. 2014;5:24. | |
Wee S, Dhanak D, Li H, et al. Targeting epigenetic regulators for cancer therapy. Ann N Y Acad Sci. 2014;1309:30–36. | |
Ahuja N, Easwaran H, Baylin SB. Harnessing the potential of epigenetic therapy to target solid tumors. J Clin Invest. 2014;124:56–63. | |
Falahi F, van Kruchten M, Martinet N, Hospers G, Rots MG. Current and upcoming approaches to exploit the reversibility of epigenetic mutations in breast cancer. Breast Cancer Res. 2014;16:412. | |
Neureiter D, Jager T, Ocker M, Kiesslich T. Epigenetics and pancreatic cancer: pathophysiology and novel treatment aspects. World J Gastroenterol. 2014;20:7830–7848. | |
Nie J, Liu L, Li Xiang, Han W. Decitabine, a new star in epigenetic therapy: the clinical application and biological mechanism in solid tumors. Cancer Lett. 2014;354:12–20. | |
Li H, Chiappinelli KB, Guzzetta AA, et al. Immune regulation by low doses of DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587–598. | |
Sigalotti L, Fratta E, Coral S, Maio M. Epigenetic drugs as immunomodulators for combination therapies in solid tumors. Pharmacol Ther. 2014;142:339–350. | |
Bedi U, Mishra VK, Wasilewski D, Schell C, Johnsen S. Epigenetic plasticity: a central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget. 2014;5:2016–2029. | |
Tian Y, Arai E, Gotoh M, Komiyama M, Fujimoto H, Kanai Y. Prognostication of patients with clear cell renal cell carcinomas based on quantification of DNA methylation levels of CpG island methylator phenotype marker genes. BMC Cancer. 2014;14:772. | |
Doudna J, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. | |
Singh VK, Kalsan M, Kumar N, Saini A, Chandra R. Induced pluripotent stem cells applications in regenerative medicine, disease modeling, and drug discovery. Front Cell Dev Biol. 2015;3:2. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.