")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 15
Potential Effect of DPP-4 Inhibitors Towards Hepatic Diseases and Associated Glucose Intolerance
Authors Sharma A, Virmani T , Sharma A, Chhabra V, Kumar G, Pathak K , Alhalmi A
Received 6 April 2022
Accepted for publication 10 June 2022
Published 16 June 2022 Volume 2022:15 Pages 1845—1864
DOI https://doi.org/10.2147/DMSO.S369712
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Ashwani Sharma,1 Tarun Virmani,1 Anjali Sharma,2 Vaishnavi Chhabra,1 Girish Kumar,1 Kamla Pathak,3 Abdulsalam Alhalmi4
1School of Pharmaceutical Sciences, MVN University, Palwal, Haryana, 121105, India; 2Freelancer, Pharmacovigilance Expert, Uttar Pradesh, India; 3Faculty of Pharmacy, Uttar Pradesh University of Medical Sciences, Uttar Pradesh, 206130, India; 4Department of Pharmaceutical Science, College of Pharmacy, Aden University, Aden, Yemen
Correspondence: Abdulsalam Alhalmi, Department of Pharmaceutical Science, College of Pharmacy, Aden University, Aden, Yemen, Email [email protected]
Abstract: Dipeptidyl-peptidase-4 (DPP-4) is an enzyme having various properties and physiological roles in lipid accumulation, resistance to anticancer agents, and immune stimulation. DPP-4 includes membrane-bound peptidases and is a kind of enzyme that cleaves alanine or proline-containing peptides such as incretins, chemokines, and appetite-suppressing hormones (neuropeptide) at their N-terminal dipeptides. DPP-4 plays a role in the final breakdown of peptides produced by other endo and exo-peptidases from nutritious proteins and their absorption in these tissues. DPP-4 enzyme activity has different modes of action on glucose metabolism, hunger regulation, gastrointestinal motility, immune system function, inflammation, and pain regulation. According to the literature survey, as DPP-4 levels increase in individuals with liver conditions, up-regulation of hepatic DPP-4 expression is likely to be the cause of glucose intolerance or insulin resistance. This review majorly focuses on the cleavage of alanine or proline-containing peptides such as incretins by the DPP-4 and its resulting conditions like glucose intolerance and cause of DPP-4 level elevation due to some liver conditions. Thus, we have discussed the various effects of DPP-4 on the liver diseases like hepatitis C, non-alcoholic fatty liver, hepatic regeneration and stem cell, hepatocellular carcinoma, and the impact of elevated DPP-4 levels in association with liver diseases as a cause of glucose intolerance and their treatment drug of choices. In addition, the effect of DPP-4 inhibitors on obesity and their negative aspects are also discussed in brief.
Keywords: DPP-4, insulin, incretins, glucose intolerance, liver diseases, sitagliptin, DPP-4 inhibitors
Introduction to DPP-4 Enzyme
In 1966, Hopsu-Havu and Glenner found dipeptidyl peptidase-4 (DPP-4) in rat liver during the processing of the cells and commercially enzymatic preparations as an activity that liberates naphthylamine from Gly–Pro-2-naphthylamide, and it was originally called glycylproline naphthylamidase.1 Meanwhile, the protein characteristics and distribution were intensively investigated, and it was rediscovered numerous times as a binding protein and a cellular marker.2 DPP-4 is the enzyme for the immune response which is known as antigen CD26 co-stimulator of T- cell, having a multiuse protein that serves as a binding protein and a ligand for a range of extracellular molecules in addition to its catalytic activity.3 It is a membrane protein that is expressed on cells all over the body, but it is also detached from the membrane and comes into circulation in the plasma as a soluble protein.4,5 Lymphocytes, fibroblasts, endothelial cells, and apical portions of acinar and epithelial cells express DPP-4, which is also found in plasma as in soluble circulating form.6,7
All membrane-bound molecules like proline or alanine-specific exopeptidases have been proposed to have a biological function in the degradation of bioactive peptides,8 but the DPP-4 role has been explored and reported most. In comparison to other peptidase enzymes, like aminopeptidase and carboxypeptidase, which have a limited distribution, DPP-4 is found in almost all vertebrate tissues, but its activity varies greatly.9
The enzyme is found largely in the cortical region and in the brush-border and microvillus portions of the kidney and hepatocytes at the cytoplasmic membrane surrounding bile canaliculi and on epithelial of the bile duct in the liver. It can also be detected on pancreatic duct epithelial cells.10 DPP-4 is thus present in body compartments/fluids engaged in nutrition and excretion (bile, pancreatic fluid, intestinal lumen, urine). As a result, DPP-4 plays a digestive role in the final breakdown of peptides produced by other endo and exo-peptidases from nutritious proteins and their absorption in these tissues.11 In both rats and humans, DPP-4 is a ubiquitous enzyme, including the exocrine pancreas, biliary tract, spleen, small intestine, and brain.12,13 DPP-4 possesses differentially expressed biological functions, as evidenced by its extensive organ distribution. The liver is among the organs with the highest levels of DPP-4 expression.14 DPP-4 marking is high in hepatic acinar zones 2 and 3, but never in zone 1, in a normal healthy liver.15 DPP-4 may be implicated in the control of hepatic metabolism, based on the uneven lobular distribution.16
DPP-4, on the other hand, is in direct touch with hormones flowing in the blood, as it is present on blood vessels’ endothelial cells17 and as a mobile enzyme in plasma. DPP-4 is expressed on excited T-helper lymphocytes18 as well as fractions of macrophages19 among immune system cells.20 DPP-4 is highly expressed in the endocrine organs, but occasionally in parenchymal cells, such as thyroid follicular epithelial cells and luteal cells.21 DPP-4 is expressed in specialized fibroblasts in a variety of tissues, including the skin, mammary gland, and synovia.22 The concentration and activity of DPP-4 in different organs/tissues/cells are shown in Figure 1.
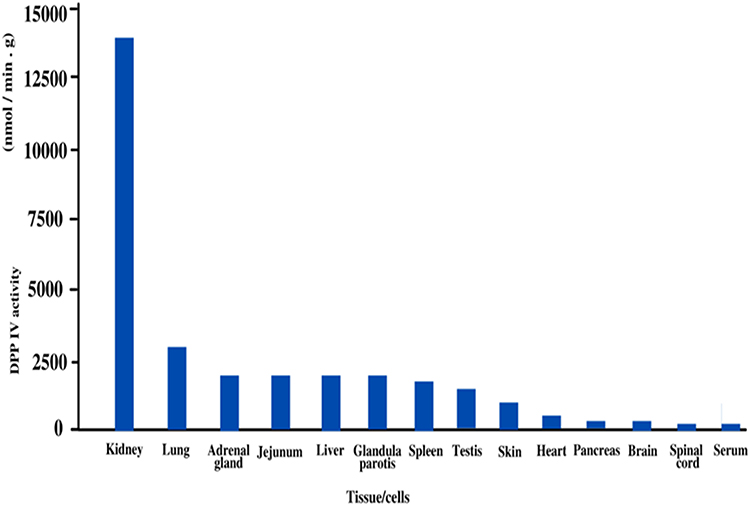 |
Figure 1 Graphical representation of the concentration and activity of DPP-4 in different organs/tissues/cells. |
Molecular Biology of DPP-4
DPP-4 includes membrane-bound peptidases like fibroblast activation protein (FAP)/seprase, resident cytoplasmic enzymes, and nonenzymatic members, which are found in neuronal membranes, as well as prolyl endopeptidase. Despite other major changes in sequence, the position and identity of the residues are crucial for catalytic activity within the C-terminal region of these related enzymes and are highly conserved in prokaryotes and eukaryotes.23 DPP-4 interacts with other membrane proteins and sends signals across cell membranes. The molecular structure of DPP-4 is shown in Figure 2.
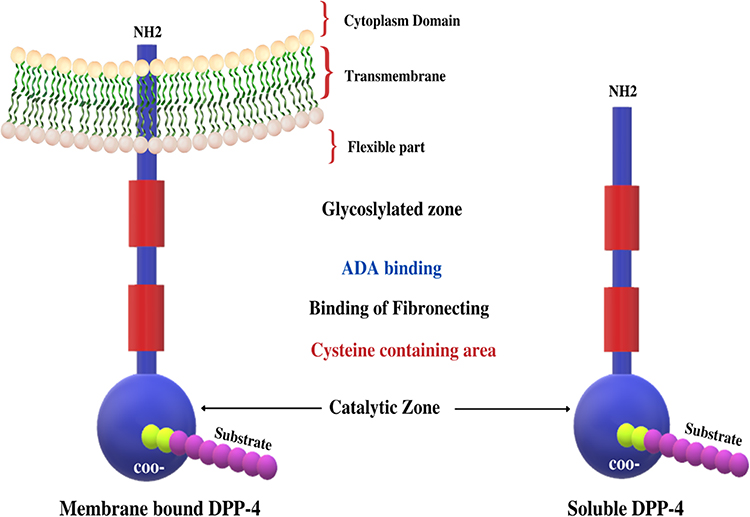 |
Figure 2 Molecular structure of DPP-4. |
Notably, the majority of the protein is extracellular, including the catalytic domain at the C-terminus, a cysteine-rich region, and a large glycosylated region connected to the transmembrane portion by a flexible stalk. Only six amino acids at the N-terminus are expected to reach into the cytoplasm. DPP-4 can form tetramers between two soluble proteins or two membrane-bound proteins, which could alter the efficiency of substrate entrance and cleavage by the catalytic active site or facilitate cell–cell communication, as reported in a study of the protein crystal structure.23
The intracellular signalling of membrane-bound DPP-4 is initiated by the interactions with T-cell antigen CD-45, Adenosine deaminase (ADA), caveolin-1, and the caspase recruitment domain-containing protein 11.24,25 DPP-4 binds to the extracellular matrix proteins, collagen, and fibronectin, as well as ADA, binding to these proteins and ADA, is mediated by amino acid residues that are not part of the substrate-binding site26,27 (Figure 2). DPP-4 which is catalytically active is released from the plasma membrane, resulting in DPP-4 (727 aa), a soluble circulating form that lacks the intracellular tail and transmembrane portions (cytoplasmic domain, flexible stalk)28,29 and accounts for a significant amount of DPP-4 activity in human blood.30 Moreover, both membrane-bound and circulating soluble DPP-4 share some domains such as ADA binding domain, glycosylated region, cytosine-rich domain, catalytic domain, fibronectin domain, and the disulfide bonds.25 Here are some examples of target peptides of DPP-4 as shown in Table 1.
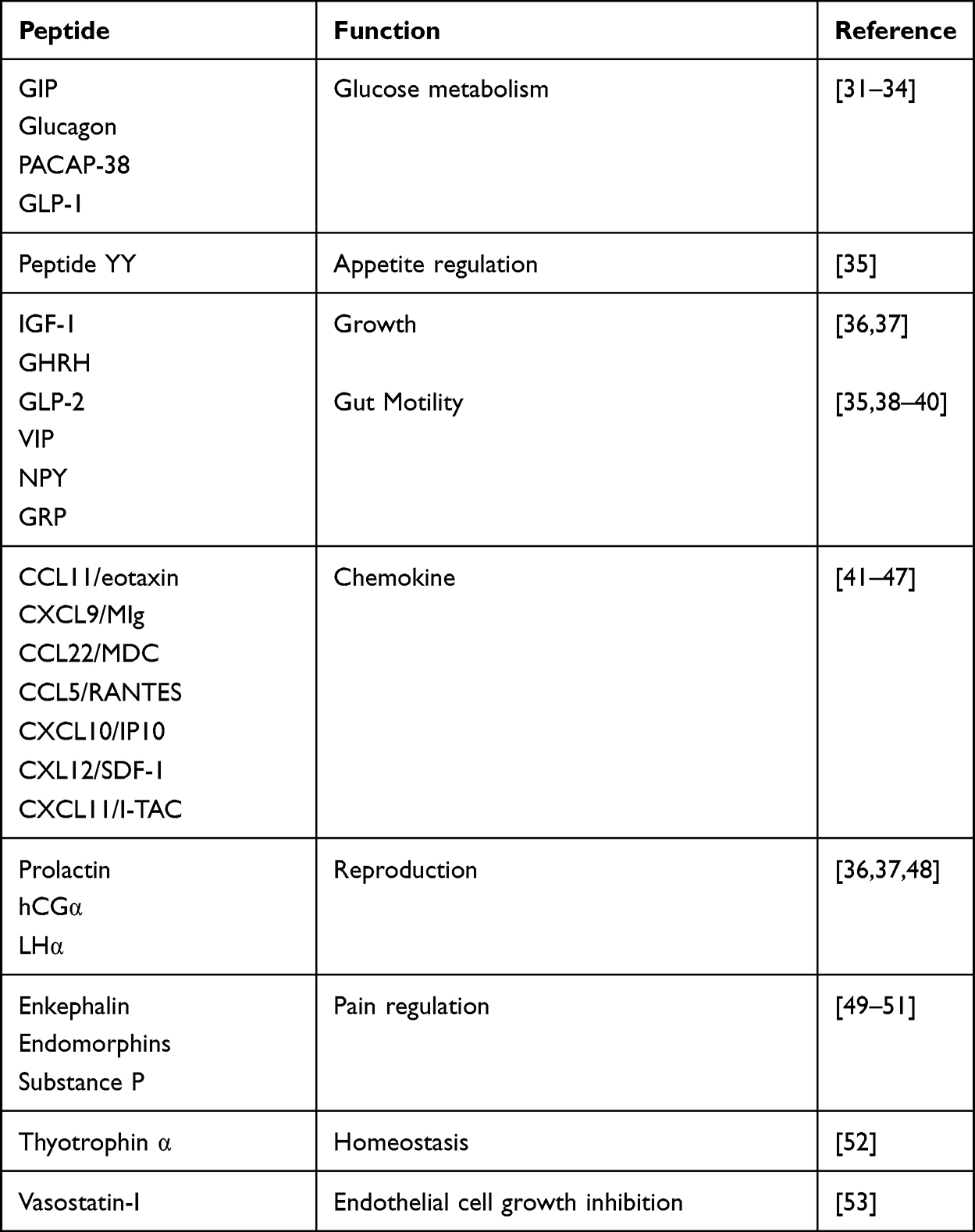 |
Table 1 Various Target Peptide of DPP-4 |
DPP-4 Physiological Properties
DPP-4 is a kind of enzyme that cleaves alanine or proline-containing peptides such as incretin, chemokines, and appetite-suppressing hormones (neuropeptide) at their N-terminal dipeptides. GLP-1, peptide YY, GLP-2, chemokine ligand 12/stromal-derived factor-1 (CXCL12/SDF-1), and substance P are examples of potential targets. Consequently, DPP-4 peptidase activity has different modes of action on glucose metabolism, hunger regulation, gastrointestinal motility, immune system function, inflammation, and pain regulation. Figure 3 shows that DPP-4 has different modes of action on chemokine production and metabolism through its peptidase activity. DPP-4 is also implicated in immunological stimulation, anti-cancer drug resistance, and ECM (Extracellular Matrix) binding and breakdown. DPP-4 also has an impact on lipid build-up.
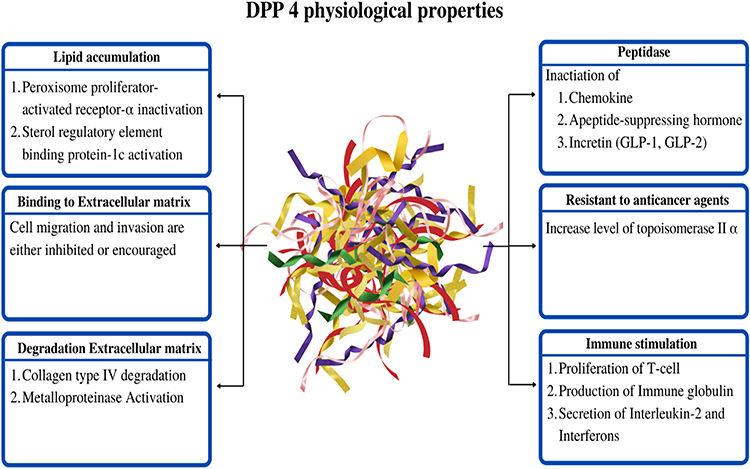 |
Figure 3 Physiological properties of DPP-4 in various regions. |
Role of Incretins and DPP-4 in Glucose Regulation
The functions and abundance of DPP-4 in the body have already been discussed in the above section. But the major focus is on the cleavage of alanine or proline-containing peptides such as incretins by the DPP-4 and its resulting consequences.
Incretins are hormones with an important role in the homeostasis of glucose, type 2 diabetes pathophysiology, and other metabolic disorders.54 These incretin hormones help in lowering the blood glucose level by stimulating the release of insulin and insulin opens the GLUT4 channel so that glucose can enter the cell and is utilized by the cells for energy production.55 There is an interesting fact that oral administration of glucose stimulates more insulin release than the intravenous administration of glucose while the concentration of glucose reaches circulation remains the same.56 This situation is known as the incretin effect and it is credited to specialized cells enteroendocrine present in the gut and coupled with glucose absorption. When glucose is administered orally, it reaches the enteroendocrine cells during absorption, and incretin hormones like glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide (GLP-1) are released from enteroendocrine cells, which stimulate pancreatic β-cells to release insulin.57 On the other hand, in the intravenous administration of glucose, the enteroendocrine cells are bypassed and thus less availability of incretins leads to less stimulation of pancreatic β-cells as compared to oral administration of glucose at the same concentration.56,58 When blood glucose concentrations rise beyond a threshold of roughly 66 mg dL−1, gut hormones including incretins generated in response to dietary absorption of glucose which provides the endocrine signal to the pancreatic β-cells, boosting insulin production and modifying glucagon secretion.59 Incretin hormones stimulate insulin secretion physiologically, whereas physiological degrees of hyperglycemia constitute to provide a stimulus accordingly for the release of insulin.56,60,61 An “isoglycemic” intravenous glucose administration induces an identical increase in arterial blood glucose level just as an oral glucose load leads to a rise in insulin secretion that is around one-third of the stimulation responses induced by oral glucose, which is the combined action of hyperglycemia and incretin hormones.62 The contribution of incretin hormones in the secretion of insulin responses following oral glucose administration is estimated to be in the range of 25% and 75%, depending on the dosage of glucose used. Undoubtedly, this measurable contribution supports incretin hormones’ physiological role in the maintenance of normal glucose homeostasis.56 The endocrine pancreas receives three signals from the gut, which is possible due to three substrates viz. incretin hormones, glucose, and neural signals by the autonomic nervous system.62,63
After the utilization of glucose by the cells throughout the body, insulin release is reduced accordingly and extra available incretins are degraded by the enzyme DPP-4 as a part of homeostasis. However, excess availability of enzyme DPP-4 leads to a condition by unnecessarily inhibiting the activity of incretins, which leads to a reduction in the secretion of insulin, and reduced insulin is not able to open the sufficient amount of glucose channels GLUT4 leads to cause glucose intolerance or hyperglycemia. As the intestinal hormone, glucagon-like peptide-1 (GLP-1) was discovered to be a DPP-4 substrate, the relationship between DPP-4 and glucose homeostasis was discovered.64,65 GLP-1 role in managing glycemia was discovered in 198666 when this unknown peptide was discovered to have dramatic effects on the endocrine pancreas. Denmark and the United States researchers described potent insulinotropic67 and glucagonostatic effects.68 Whenever the level of glucose increases then incretins stimulate the release of insulin which lowers the blood glucose, but when the DPP-4 level increases due to any cause, it metabolizes the GLP-1 and reduces the availability of the incretin hormones. The level of glucose continuously increases but incretin hormones are unable to stimulate insulin release which can result in hyperglycemia or glucose intolerance due to the high availability of DPP-432 (Figure 4). It is observed that the level of DPP-4 is increased in various liver conditions. The pathological role of DPP-4 in liver diseases and associated glucose intolerance with their therapeutic management are discussed below in detail.
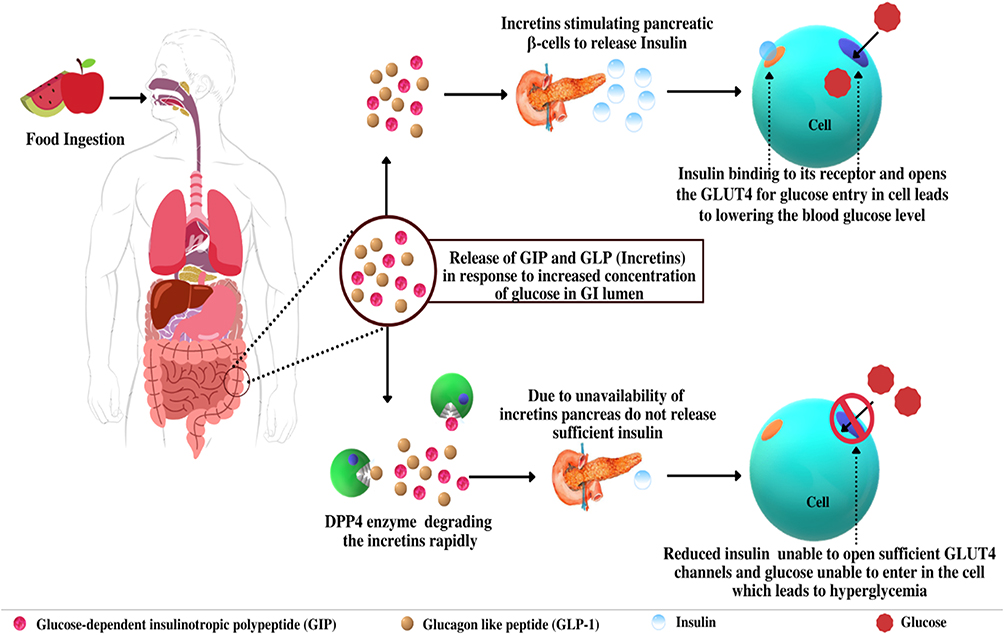 |
Figure 4 Role of Incretins and DPP-4 in glucose regulation. |
DPP-4 in Liver Conditions and the Potential Effect of DPP-4 Inhibitors in Reducing the Risk of Liver Conditions
As per research, as the DPP-4 level increases in individuals with liver conditions69–71 and up-regulation of hepatic DPP-4 expression is likely to be the cause of glucose intolerance or insulin resistance.72,73 The effects of DPP-4 on each liver disease with pathology are described below.
DPP-4 Inhibitors in Hepatitis C Virus (HCV)
HCV is a serious public health concern around the world. Consequently, HCV has a high proclivity for causing severe infection, and chronic hepatitis C affects 58 million people worldwide, with about 1.5 million new infections occurring per year as per reports by WHO. This can progress to severe hepatic fibrosis, cirrhosis, and hepatic cancer in the long run. As a result, in developed countries, HCV is a very common reason for liver transplantation.74 Interferon has always been the cornerstone of HCV treatment for almost two decades. In 1998, ribavirin was added to the medication, and subsequently, in 2001–2002, the interferon (INF) molecule was linked to polyethylene glycol (PEG) to enhance treatment responses.75,76 IP-10 (interferon-inducible protein of 10 kDa), commonly known as chemokine ligand 10 (CXCL10), is a CXC chemokine that binds to chemokine receptor 3 (CXCR3) and plays a vital role in selecting candidates for T lymphocytes and natural killer cells. IP-10 and other chemokines are secreted by hepatocytes infected with the hepatitis C virus to boost the adaptive and innate immune response.20 Surprisingly, elevated blood levels of IP-10, a powerful chemoattractant, have been linked to PEG-IFN and ribavirin therapy failure. IP-10 is usually changed by DPP-4, which produces the antagonist version of IP-10 by cleaving two amino acids from the amino terminal portion of IP-10. Antagonist version of IP-10 has the ability to bind to the IP-10 receptor but does not cause signalling. CD8+ T-cells, which express DPP-4, have also been seen in the portal and periportal areas of patients with HCV infection. In hepatocytes, DPP-4 expression is enhanced in patients with HCV infection.69,77 In patients with HCV infection, a high baseline blood soluble DPP-4 concentration is linked to poor treatment results. The IP-10 and DPP-4 proteins’ expression and binding capabilities are affected by genetic differences in the IP-10 and DPP-4 genes.78,79
According to lymphocyte subset analysis, HCV attacks CD8+ T-cells; hence, HCV-infected T-cells could be blamed for the elevated blood DPP-4 activation in HCV patients. DPP-4 alters the immune response by cleaving two amino acids from the amino-terminal portion of IP-10 which suppress the immune responses toward the HCV which may lead to more severe hepatic infection.80,81 Furthermore, Hepatitis-C is related to hyperglycemia and insulin sensitivity, which is linked to the progression of the disease and prognosis because of elevation in DPP-4 level.82–89 HCV is engaged in the development of insulin resistance by the disruption of signaling pathway substrate,90 in addition to hepatic inflammation and steatosis. Furthermore, Hepatitis-C has been linked to higher DPP-4 expression in the intestinal lumen, hepatic portion, and blood.77,91 Transfection of hepatocyte cell lines with cDNA expressing a portion of the Hepatitis viral non-structural genomic region 4B/5A increases DPP-4 expression.92 HCV infection may directly upregulate DPP-4 activity, resulting in glucose metabolism impairment.16,77 Inhibition of DPP-4 is significant in HCV infection as well as in glucose intolerance as successfully shown in Figure 5.
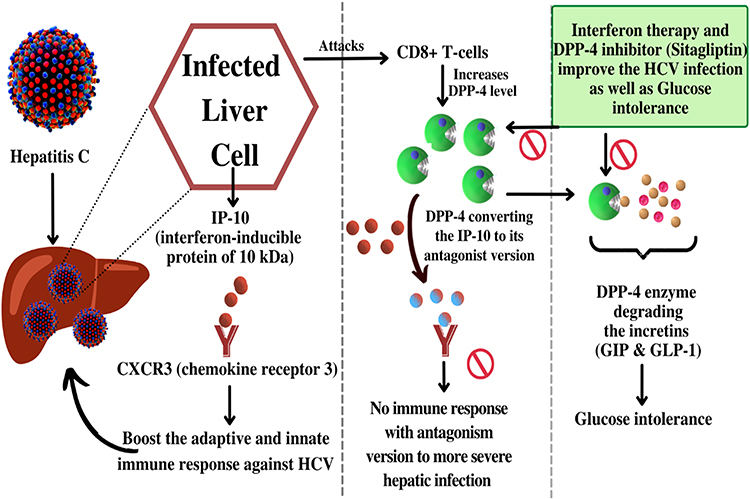 |
Figure 5 Schematic representation of HCV infected hepatocytes releases IP-10 responsible for an immune response towards HCV infection but DPP-4 level elevated due to CD8+ cells attacked by HCV. Increased DPP-4 converted the IP-10 into an inactive form which suppresses the immune response and on the other hand DPP-4 results in glucose intolerance by degrading incretins. Interferon and DPP-4 inhibitors are found to be significant in both HCV resulting conditions. |
Hence, interferon therapy for HCV eradication lowers serum DPP-4 levels and helps in treating the HCV,90,93–96 and Sitagliptin treatment dramatically improves HCV-related glucose intolerance.97,98
DPP-4 Inhibitors in Non-Alcoholic Fatty Liver Disease (NAFLD)/Nonalcoholic Steatohepatitis (NASH)
NAFLD is the most prevalent cause of chronic liver disease.99–102 It is a hepatic expression of metabolic syndrome. Whereas many factors contribute to the formation of NAFLD, elevated blood glucose has been observed, stimulated by DPP-4 expression in hepatoma cells (HepG2), and the amount of liver DPP-4 mRNA activity in the liver is much higher in NAFLD patients than in healthy subjects.103 Cui et al 2016 conducted a randomized controlled trial for NAFLD by DPP-4 inhibitor (sitagliptin) versus placebo. Researchers randomized, double-blind, placebo-controlled clinical study to compare the effectiveness of sitagliptin (100 mg/day orally) versus an identical placebo for 24 weeks to improve hepatic steatosis as measured by MRI-PDFF (Magnetic Resonance Imaging Proton Density Fat Fraction), which is a proven, precise, and quantifiable biomarker for hepatic steatosis. Fifty patients of NAFLD were randomised to receive sitagliptin and placebo from January 2014 to March 2015. The research included 84 patients in total. The primary outcomes of their study towards the liver fat which is measured by MRI-PDFF, when compared to the placebo group, was not substantially lowered in the sitagliptin group. Sitagliptin was not really substantially superior than placebo for lowering liver fat as evaluated by MRI-PDFF in this randomised, double-blind, placebo-controlled clinical study. Sitagliptin did not outperform placebo in terms of improving supplementary targets such as LDL, AST, ALT, and HOMA IR. Sitagliptin did not markedly reduce fibrosis as determined by MRE, despite the fact that participants in the placebo group had more fibrosis. In the conclusion, it is reported that sitagliptin was shown to be safe but ineffective in lowering liver fat in persons with NAFLD who were pre-diabetic or diabetic, and this trial was observed for 24 weeks only.104 On the other hand, Alam et al105 conducted a randomized controlled trial for the impact of sitagliptin on nonalcoholic steatohepatitis patient’s hepatic histological activity and fibrosis which was observed for 12 months in a randomized control study. That randomized controlled research found that using sitagliptin (100 mg daily) for one year, a DPP-4 inhibitor reduces steatosis and swelling in NASH patients. The NAS (score for NASH) in coupled biopsy samples was considerably reduced as a result of these two adjustments. This intervention did not affect fibrosis. The control group’s NAS was likewise reduced by steatosis reduction, although hepatocyte ballooning remained the same. The sitagliptin group was shown to have a much larger reduction in steatosis and NAS than the control group. Regardless of diabetes condition, sitagliptin (100 mg once daily) for a year reduces NAS through alleviating steatosis and hepatocyte enlargement. Sitagliptin has a more powerful effect than weight loss. Sitagliptin has identical safety profile to the control. To validate and solidify these findings, future major, double-blind, randomised control clinical studies are recommended. In a study of fructose-fed rats with metabolic syndrome, sitagliptin shown to be reduced liver steatosis, β-cell apoptosis, and insulin sensitivity.106 Another animal research in Japan found that sitagliptin helps to reduce hepatic steatosis in mice fed a high-fructose diet and prevents the growth of NAFLD by suppressing inflammatory cytokines and the expression levels of genes involved in lipid production in the liver.107 The study’s most important conclusion was that sitagliptin reduced the severity of hepatocyte ballooning hepatic histopathology. Ballooning degradation, which was identified as a characteristic of steatohepatitis, is connected to cytoskeletal damage in NASH and is associated with cell swelling.108,109 As a result, it is tempting to say that DPP-4 inhibitors may improve histology activity by lowering steatosis and swelling. Another uncontrolled experimental trial from Turkey found a similar histologically verified advantage.110
Apart from DPP-4 inhibitors, Glucagon-like peptide-1 receptor agonists (GLP-1 RAs) are a kind of glucose-lowering medication that has been authorized to treat Type 2 diabetes.111 Large randomized controlled trials on GLP-1 RAs have also consistently shown that these medicines reduce the risk of adverse cardiovascular events, all-cause morbidity, and nephropathy worsening in T2DM patients112,113 GLP-1 RAs reduce body weight and insulin sensitivity while improving glycemic management.111 A number of RCTs have recently investigated the putative positive hepatic effects of liraglutide and other long-acting injectable GLP-1 RAs among individuals with NAFLD, regardless of diabetes status. GLP-1 RAs were studied for their effectiveness and safety in treating NAFLD or NASH in people either with or without pre-existing T2DM. Mantovani et al114 compared and conducted the largest and most up-to-date systematic review and meta-analysis of RCTs that used different GLP-1 RAs (including two new long-acting injectable GLP-1 RAs, such as dulaglutide and semaglutide) for the treatment of NAFLD or NASH, regardless of T2DM status. Treatment given with GLP-1RAs was observed to be related to a substantial improvement in the absolute percentage of liver fat content, as measured by magnetic resonance-based methods, as well as blood liver enzymes (particularly serum ALT and GGT levels), as compared to control or standard therapy. The current meta analysis does not include a detailed examination of the hypothesized molecular pathways via which GLP-1 RAs may help people with NAFLD. However, it is plausible to infer that liraglutide’s and other GLP-1 RAs’ good effects on individual NASH histologic scores are multidimensional and a result of their combined effects on hyperglycemia or insulin resistance, weight loss, and a direct positive impact on the liver (beyond the reduction in body weight and hyperglycemia). In reality, GLP-1 RAs are effective in the treatment of T2DM and can also help people lose weight (on mean 4–5 kg).115 GLP-1 RAs are also able to alleviate hepatic steatosis through lowering de novo lipogenesis, boosting fatty acid oxidation, and improving several aspects of the insulin signaling pathways, according to experimental findings based on both human hepatocytes and animal models.116–120 Furthermore, preclinical NASH investigations have revealed that GLP-1 RAs may lower hepatic inflammation via independent pathways, at least in part, of body weight loss.121 Obesity could be a reason for NAFLD and for that cause GLP-1 RAs could be a choice, as recent clinical studies have been shown to successfully promote weight loss in diabetic individuals. The existing evidence suggests that weight loss caused by GLP-1R agonism in humans is mostly due to reduced food consumption. GLP-1 (glucagon-like peptide-1) is known as an endogenous peptide produced in the gastrointestinal tract by enteroendocrine specifically by L cells. GLP-1RAs can help with glucoregulation by promoting satiety, delaying stomach emptying, and lowering calorie intake. The only GLP-1RA licensed for the treatment of obesity is liraglutide. Semaglutide’s first Phase III clinical trial has finished, and the results indicated a considerable weight loss benefit. GLP-1RAs have been shown in clinical studies to be effective and safe, and they are regarded as potential anti-obesity medications.122 On the other side, according to Velija-Asimi et al 2013, it is found that DP-4 inhibitors DPP-4 inhibitors in combination with metformin were related to improved glycaemic control and a decrease in body weight in obese adults with type 2 diabetes.123
The increase of intrahepatic triglycerides (TGs) is the major symptom of NAFLD, which affects 75–90% of people with type 2 diabetes.124,125 NAFLD can proceed to NASH, which is marked by extensive histologic transformation, such as hepatocellular ballooning, lobular inflammation, fibrosis, and an increased risk of hepatocellular carcinoma. Various pharmacotherapies are being explored since insulin resistance, oxidative stress, lipotoxicity, immunology, mitochondrial damage, the cytokine system, and apoptosis are all implicated in the pathophysiology of NASH. Although no medicine is available for the evidence-based therapy of NASH, antidiabetic therapies may be beneficial in individuals who also have diabetes mellitus. Several investigations have found a relationship between DPP-4 and hepatic insulin sensitivity. Upregulation of DPP-4 in hepatocytes is linked to hepatic insulin resistance and liver steatosis as observed in rats,73 whereas knocking down DPP-4 optimizes insulin sensitivity and lowers lipid buildup in cultured hepatocytes.126 DPP-4 has also been linked to the occurrence of insulin sensitivity and glucose intolerance in the liver and adipose tissue, according to other research. Obesity and accompanying visceral adipose tissue inflammation cause insulin sensitivity in mice, a process that appears to be driven by increased hepatic DPP-4 production and release, since abolishing hepatocyte DPP-4 expression reduces inflammation and improves insulin sensitivity. DPP-4 is thought to be a new adipokine that affects insulin sensitivity in both autocrine and paracrine ways. DPP-4 release is closely correlated with adipocyte size, suggesting that adipocytes may be a major source of DPP-4.127 The more fat in the liver, the higher the activation of hepatokine DPP-4, which might lead to NAFLD and subsequently, NASH in a paracrine and autocrine manner. Thus, omarigliptin may inhibit the activity of DPP-4, which is abundantly released from the liver in NAFLD/NASH, preventing the stimulation of adipose inflammation and insulin resistance in the liver.128 According to Wang et al 2021, study findings show that the major cause of hepatic inflammation like NFκB pathway activation, oxidative stress, and cell apoptosis inhibition reduces hepatic inflammation. In the study, sitagliptin was found to be restricting the DPP-4 activity in hepatocytes reducing NFκB pathway activation and oxidative stress, as well as cell apoptosis, in diabetic conditions, and sitagliptin’s ROS cleaning function promotes NFκB pathway deactivation; additionally, sitagliptin can reduce Streptozotocin chronic hepatotoxicity and oxidative stress. Under diabetes circumstances, sitagliptin inhibits DPP4 activity in hepatocytes, resulting in reduced NFκB pathway activation, oxidative stress, and cell death.122 The inactivation of the NFκB pathway is promoted by sitagliptin’s ROS cleansing action and DPP-4 inhibitors are also known for the reduction in body weight in obese adults with type 2 diabetes.122 But there is vildagliptin, which is also a strong and selective DPP-4 inhibitor that is weight neutral in type 2 diabetic patients in several solotherapy and combined studies. Because of its glucose-dependent mode of action, vildagliptin has a reduced risk of hypoglycemia, which eliminates the “defensive eating” that can emerge with insulin injections or independent glucose-insulin secretagogues. More data show that vildagliptin may affect postprandial lipid and lipoprotein metabolism by decreasing the absorption of triglyceride from the gut and boosting sympathetically triggered lipid mobilization and catabolism in the postabsorptive phase. Additional research into these pathways might offer a molecular foundation for understanding the weight-loss benefits of vildagliptin medication.129 Vildagliptin is an important DPP-4 inhibitor that may be used for lowering the risk or decreasing hepatic inflammation without body weight reduction.
In reality, hepatic DPP-4 expression and serum DPP-4 activity are linked to hepatic steatosis and fatty liver grading.130,131 Furthermore, as compared to wild-type rats, DPP-4 deficient animals have lower levels of liver pro-inflammatory and pro-fibrotic cytokines, as well as less hepatic steatosis. These beneficial alterations in lipid metabolism are not caused by changes in glucose metabolism.132 In individuals with NAFLD, DPP-4 activity in serum and liver specimens correlates with indicators of hepatic injury like blood gamma-glutamyl transferase (GGT) and alanine aminotransferase amounts, but not with fasting blood glucose levels or glycosylated hemoglobin (HbA1c) values, similar to the findings in animal studies. As a result, hepatic DPP-4 expression in NAFLD could be linked to hepatic lipogenesis and liver damage.133,134 In humans and rodents, a DPP-4 inhibitor has been shown to ameliorate hepatic steatosis.135 The activity of DPP-4 inhibitors is successfully shown in Figure 6.
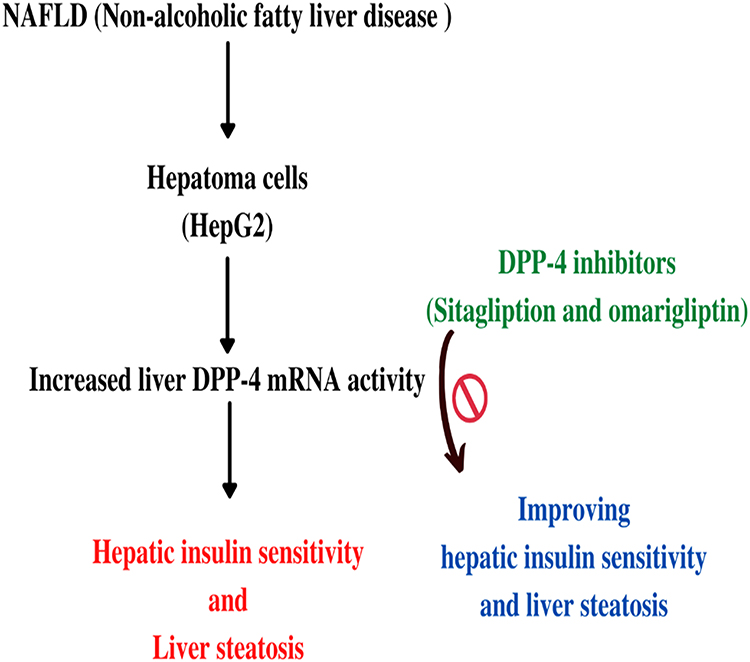 |
Figure 6 Non-alcoholic fatty liver disease results in an increased level of DPP-4 expression leads to hepatic insulin sensitivity and liver steatosis but sitagliptin and omarigliptin improve the conditions. |
A case of refractory fatty liver that was successfully treated with sitagliptin, a DPP-4 inhibitor.136 In addition, omarigliptin and sitagliptin have been shown to reduce liver enzymes and hepatocyte ballooning in patients with NASH.110,128 These data suggest that DPP-4 inhibitors may help patients with NAFLD with hepatic damage and glucose intolerance.
DPP-4 Inhibitors in Hepatic Regeneration and Stem Cell
The cirrhotic liver has been shown to have increased hepatic DPP-4 expression.128,137 Although the consequence of increased DPP-4 expression is unknown, recently showed that human liver stem cells express DPP-4 but not CD34 or CD45, which are markers of hematopoietic stem and endothelial progenitor cells.138 If we understand the concept of Cell-released chemokines, cytokines, and other growth-modulating substances that elicit their effects through particular receptor-mediated intracellular signaling modulate hematopoietic progenitor cell (HPC) and hematopoietic stem cell (HSC) functions in a paracrine manner.139 Other progenitor and stem cell types are regulated by these proteins, and also impact the more mature cell’s function. On HPCs expressing CD26, inhibiting DPP4 enzymatic activity with short peptides such diprotin A (ILE-PRO-ILE) or VAL-PYR improves chemotaxis to the chemokine stromal cell-derived factor-1 (SDF-1/CXCL12)140 as well as homing and engraftment of HSCs.141–143 CXCL12 with a DPP4 truncation lacked chemotactic efficacy but prevented chemotaxis triggered by full-length SDF-1.140 A pilot clinical trial evaluated the effects of sitagliptin (inhibitor of DPP4 used to treat type 2 diabetes)144 administration to patients with high-risk hematologic malignancies receiving single-unit cord blood transplants. With the findings that DPP4 has a detrimental effect on CSFs6, which nourish immature cell types in the bone marrow, attempts are being made to change the dosing schedule of sitagliptin to improve the time to engraftment of cord blood.145 Chemokines are important for degranulation, angiogenesis, and leukocyte trafficking in the immune system,146 and DPP4 may have a major impact on the activity of chemokine. DPP4 induces negative feedback by lowering CCL22/MDC activity, similar to its actions on CXCL.140,147,148 CCL22 purportedly possesses anti–HIV-1 action and attracts activated lymphocytes, dendritic cells, natural killer cells, and monocytes. In CCR4-transfected cells, DPP4-truncated CCL22 fails to desensitize calcium mobilization by full-length CCL22 or thymus and activation-regulated chemokine.149 HUT-78 T-cell chemotactic activity is reduced by truncated CCL22, which is 100 times less effective than full-length CCL22. As a result, DPP4ʹs N-terminal truncation of CCL22 has various effects on its multiple immunologic roles. Eosinophils are drawn to allergic inflammation and parasite infections by the CCL11 (eotaxin) and, CC chemokine. When DPP4 truncates it, its chemotactic potency for signaling capability and blood eosinophils through CCR3 are lowered 30-fold.44 These examples show the importance of DPP4 in infectious processes and inflammatory, as well as in steady-state hematopoiesis. It has been documented that the DPP4-truncated versions of the chemokines studied (CCL2, CCL3, CXCL8/IL-8, and CXCL9) lost their suppressive effect and blocked myelosuppression in vitro and in vivo when compared to their full-length counterparts. The shortened molecule functions as a dominant-negative or competitive inhibitor form of the full-length molecule in both circumstances. This could lead to feedback regulation of their full-length molecules’ actions. It’s also possible that DPP4 truncation enhances a molecule’s stimulatory or inhibitory activity beyond that of the full-length version.145 It’s critical to double-check protein sequences in databases containing potential DPP4 truncation domains on a regular basis to make sure they have not been altered. TGF-, for example, once had a DPP4 truncation site; however, the sequence has since been changed and no longer possesses a DPP4 site. Finally, biochemical and biological (in vitro and in vivo) studies are needed to confirm whether the putative DPP4 truncation sites are true truncation sites for each protein, especially when different alanine, proline, serine, or other potential DPP4 truncation sites are present at the N-terminus of every molecule. If that is the case, it is crucial to figure out whether the abbreviated form’s activity differs from that of its full-length counterpart, and if so, how. Overall understanding of the in vitro and in vivo control of various stem, progenitor, and more mature hematopoietic and other kinds of cells might result from such studies. This data might have therapeutic implications.145
Through activation of insulin resistance (IR), obesity-related inflammation raises the risk of type 2 diabetes mellitus (T2DM), obstructive sleep apnea syndrome (OSAS), and polycystic ovary syndrome (PCOS).150 In obesity-related NAFLD, IR is nearly universally found, leading to the development of the metabolic syndrome and hepatocarcinoma.151 Stem cell growth factor-beta (SCGF-β) has been shown to have activity on macrophage/granulocyte progenitor cells.152,153 C-reactive protein (CRP) levels were found to be elevated only in one-third of obese patients in the investigation, indicating a link with SCGF. The study characterizes itself by the prediction of homeostatic metabolic assessment (HOMA) values by SCGF levels, possibly mediated by indicators of inflammation, offering some insight on processes inducing/worsening IR in male patients with obesity-related NAFLD. M-CSF, TNF-, IL-12p40, and IL-6, among other pro-inflammatory cytokines, were not linked with HOMA values, with the exception of IL-6, which predicted a reduced chronic inflammation state. The small rise in CRP levels supports this notion. According to the study of Tarantino et al 2020, suggest that barely raised CRP levels might make IL-10 more accessible in an attempt to partially decrease inflammation, the major cause of IR, in line with data that CRP affects the anti-inflammatory or pro-inflammatory balance, exacerbating inflammation. In this regard, we would like to call attention to our results, which include the presence of IR in almost half of the obese individuals, increased levels of IL-10, and IL-12p40ʹs defensive response. SCGF- serum concentrations might also be due to hematopoietic stem or progenitor cells’ limited autocrine/paracrine activity. It is thought that by switching M1 to M2, inflammation could be reversed and IR reduced. Even though our median HOMA values overlapped according to gender, individuals with a more prominent HOMA had a greater frequency of moderate-to-severe steatosis than those with a HOMA below the median. The finding that SCGF levels solely predicted the severity of hepatic steatosis in men might indicate that these patients’ obesity influences their inflammatory state and/or immune system. As a result, only males’ CRP and IL-6 levels predicted SCGF-concentrations. These findings support the observation that SCGF levels solely predict IR, as measured by HOMA, in males. CRP’s mediating involvement is conceivable when we consider its functional role in inflammation. In summary, this study is characterized by the estimation of HOMA values by SCGF levels, which is likely mediated by inflammation, providing insights on processes worsening IR in male patients having obesity-related NAFLD.154 As a result, DPP-4 is a particular marker of adult hepatic stem and progenitor cells, suggesting that it may play a role in liver regeneration in chronically inflamed patients. CXCL12/SDF-1 is a chemokine that promotes the homing of hematopoietic stem cells (HSCs) and is critical for hepatic regeneration.155,156 CXCL12/SDF-1 is a DPP-4 target peptide, and inhibiting cell-surface DPP-4 activity promotes CXCL12/SDF-1 directed chemotaxis, homing, and engraftment in HSC/hematopoietic progenitor cell populations. As a result, inhibiting DPP-4 might be a good way to improve the efficacy and success of HSC/hematopoietic progenitor cell transplantation.157 DPP-4 suppression also increases the number of progenitor cells, and DPP-4 inhibition can stabilize endogenous CXCL12/SDF-1, which could be a promising technique for increasing the sequestration of regenerative stem cells.158
DPP-4 Inhibitors in Hepatocellular Carcinoma
Breast cancer,159,160 malignant mesothelioma,161 lung cancer,162 and squamous cell laryngeal carcinoma163 are all known to have increased DPP-4 expression. Increased DPP-4 expression is also found in liver tissues and serum from rats164 and humans with hepatocellular carcinoma (HCC).165
Higurashi et al (2016) conducted a multicentre double-blind, placebo-controlled, randomized Phase 3 trial for the chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes and it is observed that non-diabetic patients were given a small dose of metformin for a year with no side effects. After polypectomy, a small dose of metformin decreased the prevalence and quantity of metachronous adenomas or polyps. Metformin shows the potential to prevent colorectal cancer through chemoprevention. However, further large-scale, long-term studies are required to draw definitive results.166
Kawakita et al (2021) observed the potential influence of DPP-4 inhibitors and DPP-4 on cancer with diabetes and states that there is currently no obvious link between DPP-4 inhibitors and cancer incidence or prognosis in diabetic individuals, according to available clinical evidence. However, the safety profile of a DPP-4 inhibitor (which is the same as different anti-diabetic medications) on cancer development or recurrence has yet to be shown. The results suggested for further mechanistic studies into the relationship between DPP-4 inhibitors and cancer biology, particularly in diabetic situations, are an important study subject in both diabetes and oncology.167 Zhao et al 2017 worked on a meta-analysis of randomized clinical trials on DPP-4 inhibitors and cancer risk in patients with type 2 diabetes and there were 72 studies in all, with 35,768 and 33,319 patients recruited in the DPP-4 inhibitors and comparator medicine trials, respectively. In comparison to the usage of other active medicines or placebo, no significant connections between DPP-4 inhibitor use and cancer development were found. The findings were similar in pre-defined subgroups stratified by DPP-4 inhibitor type, cancer kind, comparative medication, trial duration, or baseline characteristics. The findings of this meta-analysis reveal that people with type 2 diabetes who take DPP-4 inhibitors have no increased risk of cancer than people who take a placebo or other medicines. Wilson et al 2021 provide clear evidence data that the currently authorized medication sitagliptin (DPP-4 inhibitors) can boost antitumor immunity in a syngeneic ovarian cancer mouse model, lowering metastatic burden and lengthening longevity. Our findings suggest a method for improving immune responses in ovarian cancer patients, as well as a justification for using DPP4 inhibitors as a fast translatable 2nd line therapy for this illness.168
According to Hsu et al 2021, DPP-4 inhibitors can lower the incidence of hepatocellular carcinoma in individuals with chronic hepatitis C infection with type 2 diabetes. In this study, individuals with type 2 diabetes and persistent HCV infection who used DPP-4 inhibitors had a decreased risk of HCC. DPP-4 inhibitors were associated with a greater incidence of HCC-free patients. This suggests that DPP-4 inhibitors may help people with type 2 diabetes and persistent HCV infection avoid developing HCC. DPP-4 inhibitors may be used as a second-line treatment after metformin for individuals with type 2 diabetes with persistent HCV infection.69
DPP-4 inhibition suppresses tyrosine kinase in human hepatoma cells, resulting in anti-apoptotic effects.165 Recently, a case has been discussed in which a patient with HCV-related chronic hepatitis experienced remarkable HCC reduction following four weeks of treatment with a DPP-4 inhibitor (Figure 7). Although it is unclear whether the DPP-4 inhibitor is directly involved in the regression of HCC, a significant invasion of CD8+ T-cells around the HCC tissue was observed, suggesting that the DPP-4 inhibitor may have improved the immune response, which has been compromised by chronic HCV infection.169 Whereas treatment with exogenous insulin or sulfonylureas raises the risk of HCC,85 treatment with a DPP-4 inhibitor had no tumor-promoting effects in mice.170 As a result, a DPP-4 inhibitor may have a safe effect on HCV-related HCC through modulating immunity.
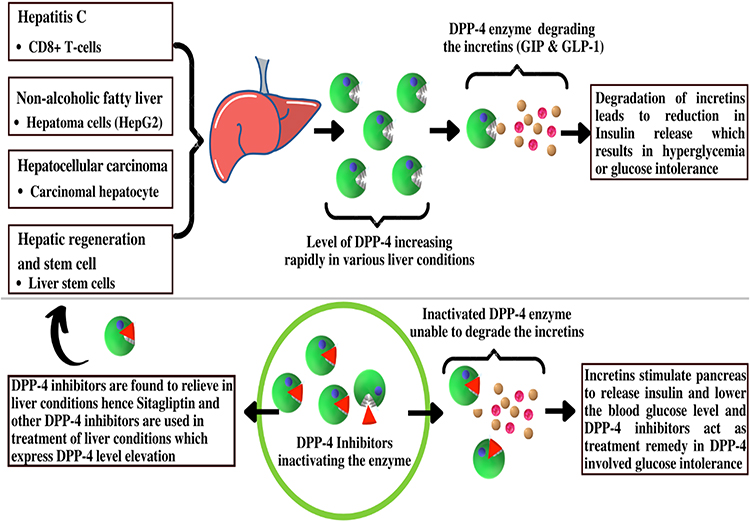 |
Figure 7 Liver diseases cause an increase in DPP-4, which causes glucose intolerance and DPP-4 inhibitors lead to relief in glucose intolerance as well as in liver conditions. |
This review discussed the various liver conditions and glucose intolerance management with DPP-4 inhibitors. The summarizing table with the mechanism of action and treatment of liver conditions associated with DPP-4 is given in Table 2.
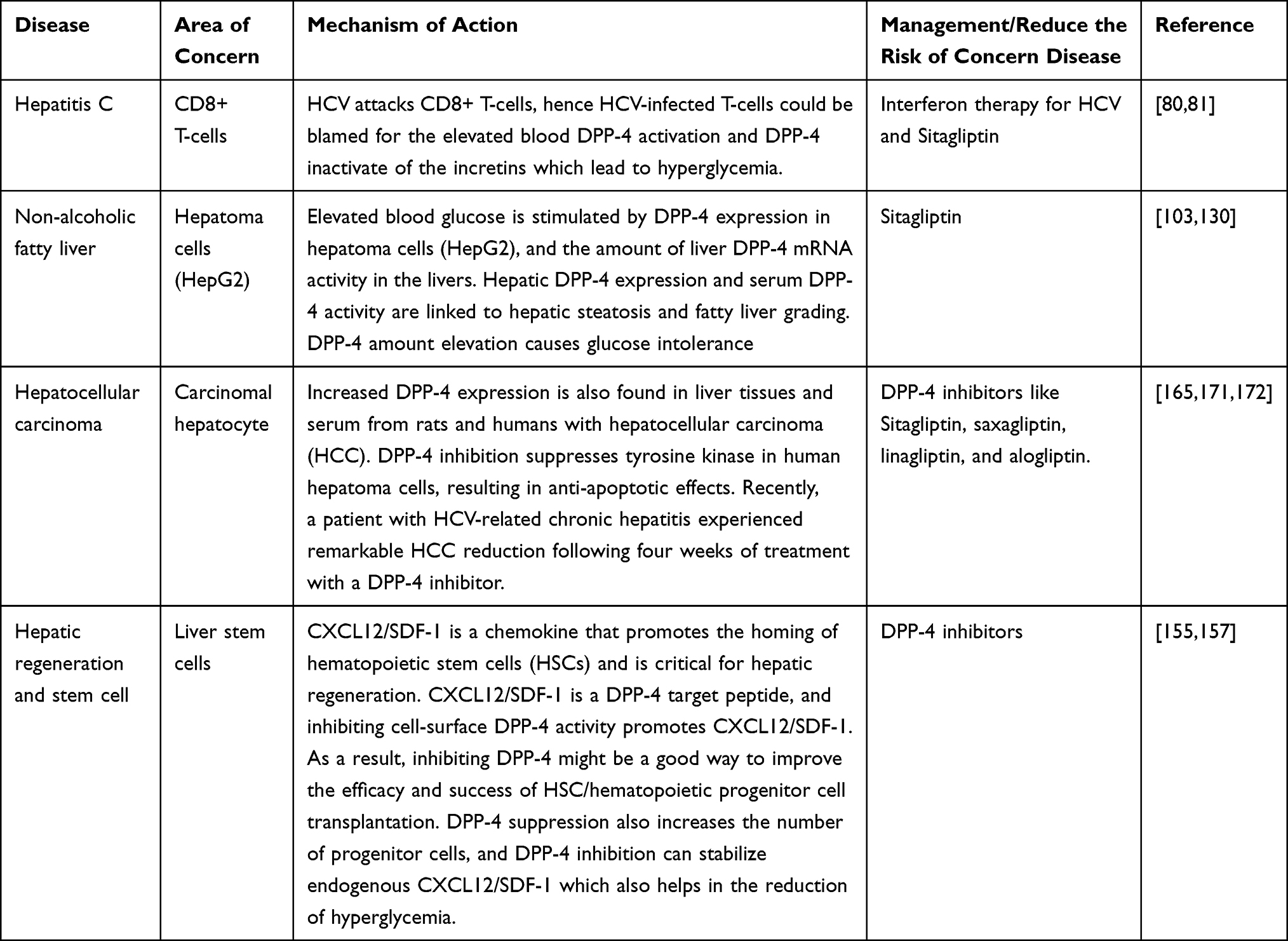 |
Table 2 Various Mechanisms of Action and Management of Some DPP-4-Associated Liver Diseases |
DPP-4 elevation could be considered a biomarker for diabetes and is a very interesting molecule in understanding the relationship between diabetes and liver or other organs, and inhibition of DPP-4 could help to reduce the risk of its associated diseases but, on the other hand, DPP-4 inhibitors have some negative aspects. DPP-4 inhibitors have been linked to an increase in gastrointestinal side effects in 24-week research, 1091 T2DM patients were randomly assigned to different combinations of sitagliptin and metformin.173 There have been a number of instances of allergic responses occurring spontaneously in people using sitagliptin and angioedema has also been documented with DPP-4 inhibitors, usually commonly within the first three months of therapy, with some responses occurring even before the first dosage.174–176 As per the study design of saxagliptin (2.5mg/day v/s 5mg/day v/s 10mg/day) with placebo on metformin for 24 weeks revealed that skin disorders, nasopharyngitis, headache, sinusitis, urinary tract infection, and arthralgia are the adverse effects produced by saxagliptin which are in high proportion than the placebo.176 Alogliptin versus placebo (Population 5380 and duration is 18 months) study showed the adverse effects of alogliptin at more proportion than placebo such as acute and chronic pancreatitis, angioedema, malignancy, renal dialysis, and hypoglycemia but without a comparison of proportions of alogliptin and placebo showed non-fatal myocardial infarction or non-fatal stroke.177 Similarly, other DPP-4 inhibitors also showed some side effects such as musculoskeletal disorders, infections (immune-related disorders such as irritable bowel syndrome, arthritis, and multiple sclerosis because of their potential influence on immunological function), nervous system (Headache and dizziness), Fertility (A 39-year-old physician started on sitagliptin, he had issues with spermatogenesis, according to a case study), and Blood effects (increase in white blood cell count).178
Conclusion
In glucose regulation, the role of incretins (GIP & GLP-1) is very important. They are released from the GIT lumen in response to the increased level of glucose during absorption and then stimulate pancreatic beta-cells to release insulin which lowers the blood glucose level by enhancing the entry of glucose in the cell through the GLUT4 channel and the cell utilizes the glucose to form energy. But there is an enzyme that inhibits this process by degrading the incretins and creating low availability of incretins which leads to reduced signaling towards pancreatic β-cells to release insulin resulting in an increased level of blood glucose as glucose remains in the blood, unable to enter in the cell through GLUT4. Apart from that, it is commonly observed that in various liver disorders such as hepatitis C, Non-alcoholic fatty liver, hepatocellular carcinoma, hepatic regeneration, and stem cell the serum level of DPP-4 is increased and leads to glucose intolerance. It is observed and reported that DPP-4 inhibitors are commonly used as a reliever in glucose intolerance and diabetes and have potential activities to improve liver conditions also. Hence, DPP-4 inhibitors like Sitagliptin could be a choice of drug in DPP-4-associated glucose intolerance because of various liver conditions and also in the therapy of liver conditions.
Abbreviations
GIP, Glucose-dependent insulinotropic peptide; GLP, Glucagon-like peptide; VIP, Vasoactive intestinal peptide; PACAP-38, Pituitary adenylate cyclase-activating polypeptide-38; GRP, Gastrin-releasing peptide; NPY, Neuropeptide Y; RANTES, Regulated upon activation; CCL, Chemokine (C-C motif) ligand; CXCL, Chemokine (C-X-C motif) ligand; SDF-1, Stromal-derived factor-1; MDC, Macrophage-derived chemokine; MIg, Monokine induced by gamma interferon; IP-10, Protein 10 from interferon (γ)-induced cell line; GHRH, Growth hormone-releasing hormone; I-TAC, Interferon-inducible T-cell α chemoattractant; LHα, Leutinizing hormone α chain; IGF-1, Insulin-like growth factor-1; CGRP, Calcitonin-related peptide; hCGα, Human chorionic gonadotropin α subunit.
Disclosure
The authors declare no conflicts of interest in relation to this work.
References
1. Hopsu-Havu VK, Glenner GG. A new dipeptide naphthylamidase hydrolyzing glycyl–prolyl-b-naphthylamide. Histochemie. 1966;201:197–201.
2. Heymann E, Mentlein R. Liver dipeptidyl aminopeptidase IV hydrolyzes substance P. FEBS Lett. 1978;91(2):360–364. doi:10.1016/0014-5793(78)81210-1
3. Nicotera R, Casarella A, Longhitano E, et al. Antiproteinuric effect of DPP-IV inhibitors in diabetic and non-diabetic kidney diseases. Pharmacol Res. 2020;159:105019. doi:10.1016/j.phrs.2020.105019
4. Anderluh M, Kocic G, Tomovic K, Kocic H, Smelcerovic A. DPP-4 inhibition: a novel therapeutic approach to the treatment of pulmonary hypertension? Pharmacol Ther. 2019;201:1–7. doi:10.1016/j.pharmthera.2019.05.007
5. Yu DMT, Yao T, Chowdhury S, et al. The dipeptidyl peptidase IV family in cancer and cell biology. FEBS J. 2010;277:1126–1144. doi:10.1111/j.1742-4658.2009.07526.x
6. Holst JJ. Glucagon-like peptide-1: from extract to agent. The Claude Bernard Lecture, 2005. Diabetologia. 2006;49:253–260. doi:10.1007/s00125-005-0107-1
7. Iwanaga T, Nio-Kobayashi J. Cellular expression of CD26/dipeptidyl peptidase IV. Biomed Res. 2021;42(6):229–237. doi:10.2220/BIOMEDRES.42.229
8. Vanhoof G, Goossens F, Meester D, Hendriks D, Scharpé S. Proline motifs in peptides and their biological processing. FEBS J. 1995;9:736–744.
9. Singh AK, Yadav D, Sharma N, Jin JO. Dipeptidyl peptidase (Dpp)‐iv inhibitors with antioxidant potential isolated from natural sources: a novel approach for the management of diabetes. Pharmaceuticals. 2021;14(6):586. doi:10.3390/ph14060586
10. Gupta S, Sen U. More than just an enzyme: dipeptidyl peptidase-4 (DPP-4) and its association with diabetic kidney remodelling. Methods Mol Biol. 2019;176(5):139–148. doi:10.1016/j.phrs.2019.104391.More
11. Koh JA, Ong JH, Manan FA, Ee KY, Wong FC, Chai TT. Discovery of bifunctional anti-dpp-iv and anti-ace peptides from housefly larval proteins after in silico gastrointestinal digestion. Biointerface Res Appl Chem. 2022;12(4):4929–4944. doi:10.33263/BRIAC124.49294944
12. Heike M, Mobius U, Knuth A, Meuer S, Zum KM, Medizinische BI. Tissue distribution of the T cell activation antigen Tal. Serological, immunohistochemical and biochemical investigations. Clin Exp Immunol. 1988;1640:431–434.
13. Mentzel S, Dijkman HB, Van Son JP, Koene RA, Assmann KJ. Organ distribution of aminopeptidase A and dipeptidyl peptidase IV in normal mice. Journal Histochem Cytochem. 1996;44(5):445–461. doi:10.1177/44.5.8627002
14. Deacon CF. Physiology and pharmacology of DPP-4 in glucose homeostasis and the treatment of type 2 diabetes. Front Endocrinol. 2019;10:1–14. doi:10.3389/fendo.2019.00080
15. Röhrborn D, Wronkowitz N, Eckel J. DPP4 in diabetes. Front Immunol. 2015;6:1–20. doi:10.3389/fimmu.2015.00386
16. Itou M, Kawaguchi T, Taniguchi E, Sata M. Dipeptidyl peptidase-4: a key player in chronic liver disease. World J Gastroenterol. 2013;19(15):2298–2306. doi:10.3748/wjg.v19.i15.2298
17. Avogaro A, Kreutzenberg S, Fadini G. Dipeptidyl-peptidase 4 inhibition: linking metabolic control to cardiovascular protection. Curr Pharm Des. 2014;20(14):2387–2394. doi:10.2174/13816128113199990474
18. Casrouge A, Sauer AV, Barreira da Silva R, et al. Lymphocytes are a major source of circulating soluble dipeptidyl peptidase 4. Clin Exp Immunol. 2018;194(2):166–179. doi:10.1111/cei.13163
19. Jackman HL, Tan F, Schraufnagel D, et al. Plasma membrane-bound and lysosomal peptidases in human alveolar macrophages. Am J Respir Cell Mol Biol. 1995;13(2):196–204. doi:10.1165/ajrcmb.13.2.7626287
20. Shao S, Xu Q, Yu X, Pan R, Chen Y. Dipeptidyl peptidase 4 inhibitors and their potential immune modulatory functions. Pharmacol Ther. 2020;209:107503. doi:10.1016/j.pharmthera.2020.107503
21. Trzaskalski NA, Fadzeyeva E, Mulvihill EE. Dipeptidyl peptidase-4 at the interface between inflammation and metabolism. Clin Med Insights Endocrinol Diabetes. 2020;13:1–10. doi:10.1177/1179551420912972
22. Lee SY, Wu ST, Liang YJ, et al. Soluble dipeptidyl peptidase-4 induces fibroblast activation through proteinase-activated receptor-2. Front Pharmacol. 2020;11:1–13. doi:10.3389/fphar.2020.552818
23. Engel M, Hoffmann T, Wagner L, et al. The crystal structure of dipeptidyl peptidase IV (CD26) reveals its functional regulation and enzymatic mechanism. Proc Natl Acad Sci U S A. 2003;100(9):5063–5068. doi:10.1073/pnas.0230620100
24. Ohnuma K, Uchiyama M, Yamochi T, et al. Caveolin-1 triggers T-cell activation via CD26 in association with CARMA1. J Biol Chem. 2007;282(13):10117–10131. doi:10.1074/jbc.M609157200
25. Mulvihill EE, Drucker DJ. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev. 2014;35(6):992–1019. doi:10.1210/er.2014-1035
26. Stulc T, Sedo A. Inhibition of multifunctional dipeptidyl peptidase-IV: is there a risk of oncological and immunological adverse effects? Diabetes Res Clin Pract. 2010;88(2):125–131. doi:10.1016/j.diabres.2010.02.017
27. Zhong J, Kankanala S, Rajagopalan S. DPP4 inhibition: insights from the bench and recent clinical studies. Curr Opin Lipidol. 2017;176(5):139–148. doi:10.1097/MOL.0000000000000340.DPP4
28. Shi S, Koya D, Kanasaki K. Dipeptidyl peptidase-4 and kidney fibrosis in diabetes. Fibrogenes Tissue Repair. 2016;9(1):1–10. doi:10.1186/s13069-016-0038-0
29. Durinx C, Lambeir AM, Bosmans E, et al. Molecular characterization of dipeptidyl peptidase activity in serum soluble CD26/dipeptidyl peptidase IV is responsible for the release of X-pro dipeptides. Eur J Biochem. 2000;267(17):5608–5613. doi:10.1046/j.1432-1327.2000.01634.x
30. Cordero OJ, Salgado FJ, Nogueira M. On the origin of serum CD26 and its altered concentration in cancer patients. Cancer Immunol Immunother. 2009;58(11):1725–1749. doi:10.1007/s00262-009-0728-1
31. Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl‐peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon‐like peptide‐ 1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem. 1993;214(3):829–835. doi:10.1111/j.1432-1033.1993.tb17986.x
32. Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes. 1995;44(9):1126–1131. doi:10.2337/diab.44.9.1126
33. Brubaker PL, Drucker DJ. Structure-function of the glucagon receptor family of G protein-coupled receptors: the glucagon, GIP, GLP-1, and GLP-2 receptors. Recept Channels. 2002;8(3–4):179–188. doi:10.3109/10606820213687
34. Ballantyne GH. Peptide YY(1-36) and peptide YY(3-36): part II. Changes after gastrointestinal surgery and bariatric surgery. Obes Surg. 2006;16(6):795–803. doi:10.1381/096089206777346619
35. Byrd JB, Touzin K, Sile S, et al. Dipeptidyl peptidase IV in angiotensin-converting enzyme inhibitor-associated angioedema. Hypertension. 2008;51(1):141–147. doi:10.1161/HYPERTENSIONAHA.107.096552
36. Mentlein R. Dipeptidyl-peptidase IV (CD26) -role in the inactivation of regulatory peptides. Regul Pept. 1999;85:9–24. doi:10.1016/S0167-0115(99)00089-0
37. Liu X, Murali SG, Holst JJ, Ney DM. Enteral nutrients potentiate the intestinotrophic action of glucagon-like peptide-2 in association with increased insulin-like growth factor-I responses in rats. Am J Physiol. 2008;295(6):1794–1802. doi:10.1152/ajpregu.90616.2008
38. Walsh NA, Yusta B, Dacambra MP, Anini Y, Drucker DJ, Brubaker PL. Glucagon-like peptide-2 receptor activation in the rat intestinal mucosa. Endocrinology. 2003;144(10):4385–4392. doi:10.1210/en.2003-0309
39. Mentlein R, Roos T. Proteases involved in the metabolism of angiotensin II, bradykinin, calcitonin gene-related peptide (CGRP), and neuropeptide Y by vascular smooth muscle cells. Peptides. 1996;17(4):709–720. doi:10.1016/0196-9781(96)00066-6
40. Ahrén B, Hughes TE. Inhibition of dipeptidyl peptidase-4 augments insulin secretion in response to exogenously administered glucagon-like peptide-1, glucose-dependent insulinotropic polypeptide, pituitary adenylate cyclase-activating polypeptide, and gastrin-releasing peptide in mice. Endocrinology. 2005;146(4):2055–2059. doi:10.1210/en.2004-1174
41. Lun SWM, Wong CK, Ko FWS, Hui DSC, Lam CWK. Increased expression of plasma and CD4+ T lymphocyte costimulatory molecule CD26 in adult patients with allergic asthma. J Clin Immunol. 2007;27(4):430–437. doi:10.1007/s10875-007-9093-z
42. Liu Z, Christensson M, Forslöw A, De Meester I, Sundqvist K-G. A CD26-controlled cell surface cascade for regulation of T cell motility and chemokine signals. J Immunol. 2009;183(6):3616–3624. doi:10.4049/jimmunol.0804336
43. Rai AK, Thakur CP, Kumar P, Mitra DK. Impaired expression of CD26 compromises T-cell recruitment in human visceral leishmaniasis. Eur J Immunol. 2012;42(10):2782–2791. doi:10.1002/eji.201141912
44. Struyf S, Proost P, Schols D, et al. CD26/dipeptidyl-peptidase IV down-regulates the eosinophil chemotactic potency, but not the anti-HIV activity of human eotaxin by affecting its interaction with CC chemokine receptor 3. J Immunol. 1999;162:4903–4909.
45. Lambeir AM, Proost P, Durinx C, et al. Kinetic investigation of chemokine truncation by CD26/dipeptidyl peptidase iv reveals a striking selectivity within the chemokine family. J Biol Chem. 2001;276(32):29839–29845. doi:10.1074/jbc.M103106200
46. Wong PTY, Wong CK, Tam LS, Li EK, Chen DP, Lam CWK. Decreased expression of T lymphocyte co-stimulatory molecule cd26 on invariant natural killer t cells in systemic lupus erythematosus. Immunol Invest. 2009;38(5):350–364. doi:10.1080/08820130902770003
47. Crane M, Oliver B, Matthews G, et al. Immunopathogenesis of hepatic flare in HIV/hepatitis B virus (HBV)-coinfected individuals after the initiation of HBV-active antiretroviral therapy. J Infect Dis. 2009;199(7):974–981. doi:10.1086/597276
48. Faidley TD, Leiting B, Pryor KD, Lyons K, Hickey GJ, Thompson DR. Inhibition of dipeptidyl-peptidase IV does not increase circulating IGF-1 concentrations in growing pigs. Exp Biol Med. 2006;231:1373–1378. doi:10.1177/153537020623100811
49. Sakurada C, Sakurada S, Hayashi T, Katsuyama S, Tan-No K, Sakurada T. Degradation of endomorphin-2 at the supraspinal level in mice is initiated by dipeptidyl peptidase IV: an in vitro and in vivo study. Biochem Pharmacol. 2003;66(4):653–661. doi:10.1016/S0006-2952(03)00391-5
50. Király K, Szalay B, Szalai J, et al. Intrathecally injected Ile-Pro-Ile, an inhibitor of membrane ectoenzyme dipeptidyl peptidase IV, is antihyperalgesic in rats by switching the enzyme from hydrolase to synthase functional mode to generate endomorphin 2. Eur J Pharmacol. 2009;620(1–3):21–26. doi:10.1016/j.ejphar.2009.08.018
51. Guieu R, Fenouillet E, Devaux C, et al. CD26 modulates nociception in mice via its dipeptidyl-peptidase IV activity. Behav Brain Res. 2006;166(2):230–235. doi:10.1016/j.bbr.2005.08.003
52. Tian L, Gao J, Hao J, et al. Reversal of new-onset diabetes through modulating inflammation and stimulating β-cell replication in nonobese diabetic mice by a dipeptidyl peptidase IV inhibitor. Endocrinology. 2010;151(7):3049–3060. doi:10.1210/en.2010-0068
53. Zhang XY, De Meester I, Lambeir AM, et al. Study of the enzymatic degradation of vasostatin I and II and their precursor chromogranin A by dipeptidyl peptidase IV using high-performance liquid chromatography/electrospray mass spectrometry. J Mass Spectrom. 1999;34(4):255–263. doi:10.1002/(SICI)1096-9888(199904)34:4<255::AID-JMS752>3.0.CO;2-7
54. Radbakhsh S, Atkin SL, Simental-Mendia LE, Sahebkar A. The role of incretins and incretin-based drugs in autoimmune diseases. Int Immunopharmacol. 2021;98:107845. doi:10.1016/j.intimp.2021.107845
55. Gallwitz B. Emerging DPP-4 inhibitors: focus on linagliptin for type 2 diabetes. Diabetes Metab Syndr Obes Targets Ther. 2013;6:1–9. doi:10.2147/dmso.s23166
56. Nauck MA, Meier JJ. The incretin effect in healthy individuals and those with type 2 diabetes: physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol. 2016;4(6):525–536. doi:10.1016/S2213-8587(15)00482-9
57. Andukuri R, Drincic A, Rendell M. Alogliptin: a new addition to the class of DPP-4 inhibitors. Diabetes Metab Syndr Obes Targets Ther. 2009;2:117–126. doi:10.2147/dmsott.s4312
58. Holst JJ. The incretin system in healthy humans: the role of GIP and GLP-1. Metabolism. 2019;96:46–55. doi:10.1016/j.metabol.2019.04.014
59. Salvatore T, Nevola R, Pafundi PC, et al. Incretin hormones: the link between glycemic index and cardiometabolic diseases. Nutrients. 2019;11(8):1–12. doi:10.3390/nu11081878
60. Kishimoto M. Teneligliptin: a DPP-4 inhibitor for the treatment of type 2 diabetes. Diabetes Metab Syndr Obes Targets Ther. 2013;6:187–195. doi:10.2147/DMSO.S35682
61. Ahrén B. Use of DPP-4 inhibitors in type 2 diabetes: focus on sitagliptin. Diabetes Metab Syndr Obes Targets Ther. 2010;3:31–41. doi:10.2147/dmso.s7327
62. Rehfeld JF. The origin and understanding of the incretin concept. Front Endocrinol. 2018;9:1–7. doi:10.3389/fendo.2018.00387
63. Kim W, Egan JM. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev. 2008;60(4):470–512. doi:10.1124/pr.108.000604
64. Bell GI, Santerre RF, Mullenbach GT. Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature. 1983;302(5910):716–718. doi:10.1038/302716a0
65. Ørskov C, Holst JJ, Knuhtsen S, Baldissera FGA, Poulsen SS, Nielsen OV. Glucagon-like peptides GLP-1 and GLP-2, predicted products of the glucagon gene, are secreted separately from pig small intestine but not pancreas. Endocrinology. 1986;119(4):1467–1475. doi:10.1210/endo-119-4-1467
66. Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest. 1987;79(2):616–619. doi:10.1172/JCI112855
67. Holst JJ, Ørskov C, Vagn Nielsen O, Schwartz TW. Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett. 1987;211(2):169–174. doi:10.1016/0014-5793(87)81430-8
68. Ørskov C, Holst JJ, Nielsen OV. Effect of truncated glucagon-like peptide-1 [proglucagon-(78-107) amide] on endocrine secretion from pig pancreas, antrum, and nonantral stomach. Endocrinology. 1988;123(4):2009–2013. doi:10.1210/endo-123-4-2009
69. Hsu WH, Sue SP, Liang HL, et al. Dipeptidyl peptidase 4 inhibitors decrease the risk of hepatocellular carcinoma in patients with chronic hepatitis C infection and type 2 diabetes mellitus: a Nationwide Study in Taiwan. Front Public Heal. 2021;9:1–6. doi:10.3389/fpubh.2021.711723
70. Eggstein S, Kreisel W, Gerok W, Eggstein M. Dipeptidyl aminopeptidase IV in hospitalized patients and in galactosamine hepatitis of the rat: activity and lectin affinity chromatography in serum and hepatic plasma membranes. J Clin Chem Clin Biochem. 1989;27:547–554.
71. Nilius R, Stuhec K, Dietrich R. Changes of dipeptidylpeptidase IV as a membrane marker of lymphocytes in acute and chronic liver diseases–biochemical and cytochemical investigations. Physiol Res. 1991;40:95–102.
72. Matsumoto Y, Bishop GA, McCaughan GW. Altered zonal expression of the CD26 antigen (dipeptidyl peptidase IV) in human cirrhotic liver. Hepatology. 1992;15(6):1048–1053. doi:10.1002/hep.1840150613
73. Baumeier C, Schlüter L, Saussenthaler S, et al. Elevated hepatic DPP4 activity promotes insulin resistance and non-alcoholic fatty liver disease. Mol Metab. 2017;6(10):1254–1263. doi:10.1016/j.molmet.2017.07.016
74. Thomas DL. Global control of hepatitis C: where challenge meets opportunity. Nat Med. 2013;19(7):850–858. doi:10.1038/nm.3184
75. Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358(9286):958–965. doi:10.1016/S0140-6736(01)06102-5
76. Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2013;347(13):1673–1680.
77. Itou M, Kawaguchi T, Taniguchi E, et al. Altered expression of glucagon-like peptide-1 and dipeptidyl peptidase IV in patients with HCV-related glucose intolerance. J Gastroenterol Hepatol. 2008;23(2):244–251. doi:10.1111/j.1440-1746.2007.05183.x
78. Casrouge A, Decalf J, Ahloulay M, et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J Clin Invest. 2011;121(1):308–317. doi:10.1172/JCI40594
79. Charles ED, Dustin LB. Chemokine antagonism in chronic hepatitis C virus infection. J Clin Invest. 2011;121(1):25–27. doi:10.1172/JCI45610
80. Dimitropoulou D, Karakantza M, Tsamandas AC, Mouzaki A, Theodorou G, Gogos CA. T-lymphocyte subsets in peripheral blood and liver tissue of patients with chronic hepatitis B and C. In vivo. 2011;25(5):833–840.
81. Rahman W, Huang P, Belov L, et al. Analysis of human liver disease using a cluster of differentiation (CD) antibody microarray. Liver Int. 2012;32(10):1527–1534. doi:10.1111/j.1478-3231.2012.02854.x
82. Sakata M, Kawahara A, Kawaguchi T, et al. Decreased expression of insulin and increased expression of pancreatic transcription factor PDX-1 in islets in patients with liver cirrhosis: a comparative investigation using human autopsy specimens. J Gastroenterol. 2013;48(2):277–285. doi:10.1007/s00535-012-0633-9
83. Miyajima I, Kawaguchi T, Fukami A, et al. Chronic HCV infection was associated with severe insulin resistance and mild atherosclerosis: a population-based study in an HCV hyperendemic area. J Gastroenterol. 2013;48(1):93–100. doi:10.1007/s00535-012-0610-3
84. Eslam M, Kawaguchi T, Del Campo JA, Sata M, Abo-Elneen Khattab M, Romero-Gomez M. Use of HOMA-IR in hepatitis C. J Viral Hepat. 2011;18(10):675–684. doi:10.1111/j.1365-2893.2011.01474.x
85. Fukushima N, Kuromatsu R, Arinaga-Hino T, et al. Adipocytokine involvement in hepatocellular carcinoma after sustained response to interferon for chronic hepatitis C. Hepatol Res. 2010;40(9):911–922. doi:10.1111/j.1872-034X.2010.00699.x
86. Sumie S, Kawaguchi T, Komuta M, et al. Significance of glucose intolerance and SHIP2 expression in hepatocellular carcinoma patients with HCV infection. Oncol Rep. 2007;18(3):545–552. doi:10.3892/or.18.3.545
87. Kawaguchi T, Ide T, Taniguchi E, et al. Clearance of HCV improves insulin resistance, beta-cell function, and hepatic expression of insulin receptor substrate 1 and 2. Am J Gastroenterol. 2007;102(3):570–576. doi:10.1111/j.1572-0241.2006.01038.x
88. Kawaguchi T, Nagao Y, Tanaka K, et al. Causal relationship between hepatitis C virus core and the development of type 2 diabetes mellitus in a hepatitis C virus hyperendemic area: a pilot study. Int J Mol Med. 2005;16(1):109–114. doi:10.3892/ijmm.16.1.109
89. Kawaguchi T, Yoshida T, Harada M, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am J Pathol. 2004;165(5):1499–1508. doi:10.1016/S0002-9440(10)63408-6
90. Pazienza V, Clément S, Pugnale P, et al. The hepatitis C virus core protein of genotypes 3a and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology. 2007;45(5):1164–1171. doi:10.1002/hep.21634
91. Nishina S, Hino K. CD26/DPP4 as a therapeutic target in nonalcoholic steatohepatitis associated hepatocellular carcinoma. Cancers. 2022;14(2):454. doi:10.3390/cancers14020454
92. Shintani Y, Fujie H, Miyoshi H, et al. Hepatitis C virus infection and diabetes: direct involvement of the virus in the development of insulin resistance. Gastroenterology. 2004;126:840–848. doi:10.1053/j.gastro.2003.11.056
93. Stone SF, Lee S, Keane NM, Price P, French MA. Association of increased hepatitis C virus (HCV)-specific IgG and soluble CD26 dipeptidyl peptidase IV enzyme activity with hepatotoxicity after highly active antiretroviral therapy in human immunodeficiency virus-HCV-coinfected patients. J Infect Dis. 2002;186(10):1498–1502. doi:10.1086/344892
94. Andrieu T, Thibault V, Malet I, et al. Similar increased serum dipeptidyl peptidase IV activity in chronic hepatitis C and other viral infections. J Clin Virol. 2003;27(1):59–68. doi:10.1016/S1386-6532(02)00128-2
95. Maes M, Bonaccorso S. Lower activities of serum peptidases predict higher depressive and anxiety levels following interferon-alpha-based immunotherapy in patients with hepatitis C. Acta Psychiatr Scand. 2004;109(2):126–131. doi:10.1046/j.0001-690X.2003.00230.x
96. Yang SS, Fu LS, Sen CC, Yeh HZ, Chen GH, Kao JH. Changes of soluble CD26 and CD30 levels correlate with response to interferon plus ribavirin therapy in patients with chronic hepatitis C. J Gastroenterol Hepatol. 2006;21(12):1789–1793. doi:10.1111/j.1440-1746.2006.04677.x
97. Arase Y, Suzuki F, Kobayashi M, et al. Efficacy and safety in sitagliptin therapy for diabetes complicated by chronic liver disease caused by hepatitis C virus. Hepatol Res. 2011;41(6):524–529. doi:10.1111/j.1872-034X.2011.00798.x
98. Kaku K, Kadowaki T, Terauchi Y, et al. Sitagliptin improves glycaemic excursion after a meal or after an oral glucose load in Japanese subjects with impaired glucose tolerance. Diabetes Obes Metab. 2015;17(11):1033–1041. doi:10.1111/dom.12507
99. Sumida Y, Yoneda M, Hyogo H, et al. A simple clinical scoring system using ferritin, fasting insulin, and type IV collagen 7S for predicting steatohepatitis in nonalcoholic fatty liver disease. J Gastroenterol. 2011;46(2):257–268. doi:10.1007/s00535-010-0305-6
100. Sumida Y, Yoneda M, Hyogo H, et al. Validation of the FIB4 index in a Japanese nonalcoholic fatty liver disease population. BMC Gastroenterol. 2012;12(1):2. doi:10.1186/1471-230X-12-2
101. Eguchi Y, Hyogo H, Ono M, et al. Prevalence and associated metabolic factors of nonalcoholic fatty liver disease in the general population from 2009 to 2010 in Japan: a multicenter large retrospective study. J Gastroenterol. 2012;47(5):586–595. doi:10.1007/s00535-012-0533-z
102. Powell EE, Wong VWS, Rinella M. Non-alcoholic fatty liver disease. Lancet. 2021;397(10290):2212–2224. doi:10.1016/S0140-6736(20)32511-3
103. Miyazaki M, Kato M, Tanaka K, et al. Increased hepatic expression of dipeptidyl peptidase-4 in non-alcoholic fatty liver disease and its association with insulin resistance and glucose metabolism. Mol Med Rep. 2012;5(3):729–733. doi:10.3892/mmr.2011.707
104. Cui J, Philo L, Nguyen P, et al. Sitagliptin vs. placebo for non-alcoholic fatty liver disease: a randomized controlled trial. J Hepatol. 2016;65(2):369–376. doi:10.1016/j.jhep.2016.04.021
105. Alam S, Kabir J, Mustafa G, Gupta U, Hasan S, Alam A. Effect of sitagliptin on hepatic histological activity and fibrosis of nonalcoholic steatohepatitis patients: a 1-year randomized control trial. Hepatic Med Evid Res. 2018;10(1):23–31. doi:10.4103/1319-3767.173762
106. Maiztegui B, Borelli MI, Madrid VG, et al. Sitagliptin prevents the development of metabolic and hormonal disturbances, increased β-cell apoptosis and liver steatosis induced by a fructose-rich diet in normal rats. Clin Sci. 2011;120(2):73–80. doi:10.1042/CS20100372
107. Sujishi T, Fukunishi S, Ii M, et al. Sitagliptin can inhibit the development of hepatic steatosis in high-fructose diet-fed ob/ob mice. J Clin Biochem Nutr. 2015;57(3):244–253. doi:10.3164/jcbn.15-84
108. Lackner C. Hepatocellular ballooning in nonalcoholic steatohepatitis: the pathologist’s perspective. Expert Rev Gastroenterol Hepatol. 2011;5(2):223–231. doi:10.1586/egh.11.8
109. Caldwell S, Ikura Y, Dias D, et al. Hepatocellular ballooning in NASH. J Hepatol. 2010;53(4):719–723. doi:10.1016/j.jhep.2010.04.031
110. Yilmaz Y, Yonal O, Deyneli O, Celikel CA, Kalayci C, Duman DG. Effects of sitagliptin in diabetic patients with nonalcoholic steatohepatitis. Acta Gastroenterol Belg. 2012;75(2):240–244.
111. Nauck MA, Quast DR, Wefers J, Meier JJ. GLP-1 receptor agonists in the treatment of type 2 diabetes e state-of-The-art. Mol Metab. 2021;46:101102. doi:10.1016/j.molmet.2020.101102
112. Sachinidis A, Nikolic D, Pantea A, et al. Cardiovascular outcomes trials with incretin-based medications: a critical review of data available on GLP-1 receptor agonists and DPP-4 inhibitors. Metabolism. 2020;111:154343. doi:10.1016/j.metabol.2020.154343
113. Li J, Albajrami O, Zhuo M, Hawley CE, Paik JM. Decision algorithm for prescribing SGLT2 inhibitors and GLP-1 receptor agonists for diabetic kidney disease. Clin J Am Soc Nephrol. 2020;15(11):1678–1688. doi:10.2215/CJN.02690320
114. Mantovani A, Petracca G, Beatrice G, Csermely A, Lonardo A, Targher G. Glucagon-like peptide-1 receptor agonists for treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: an updated meta-analysis of randomized controlled trials. Metabolites. 2021;11(2):73. doi:10.3390/metabo11020073
115. American Diabetes Association. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes—2021. Diabetes Care. 2021;44:S111–S124. doi:10.2337/dc21-S009
116. Ding X, Saxena NK, Lin S, Gupta N, Anania FA. Exendin-4, a Glucagon-Like Protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Gerontology. 2006;43(1):173–181. doi:10.1002/hep.21006.Exendin-4
117. Gupta NA, Mells J, Dunham RM, et al. Glucagon-like Peptide-1 Receptor (GLP-1R) is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology. 2010;51(5):1584–1592. doi:10.1163/9789087905132_002
118. Dhir G, Cusi K. Glucagon like peptide-1 receptor agonists for the management of obesity and non-alcoholic fatty liver disease: a novel therapeutic option. J Investig Med. 2018;66(1):7–10. doi:10.1136/jim-2017-000554
119. Svegliati-Baroni G, Saccomanno S, Rychlicki C, et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011;31(9):1285–1297. doi:10.1111/j.1478-3231.2011.02462.x
120. Trevaskis JL, Griffin PS, Wittmer C, et al. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am J Physiol. 2012;302(8):762–772. doi:10.1152/ajpgi.00476.2011
121. Rakipovski G, Rolin B, Nøhr J, et al. The GLP-1 analogs liraglutide and semaglutide reduce atherosclerosis in ApoE −/− and LDLr −/− mice by a mechanism that includes inflammatory pathways. JACC Basic to Transl Sci. 2018;3(6):844–857. doi:10.1016/j.jacbts.2018.09.004
122. Wang X, Ke J, Zhu YJ, et al. Dipeptidyl peptidase-4 (DPP4) inhibitor sitagliptin alleviates liver inflammation of diabetic mice by acting as a ROS scavenger and inhibiting the NFκB pathway. Cell Death Discov. 2021;7(1):1–10. doi:10.1038/s41420-021-00625-7
123. Velija-Asimi Z, Izetbegovic S, Karamehic J, et al. The effects of dipeptidyl peptidase-4 inhibitors in treatment of obese patients with type 2 diabetes. Med Arh. 2013;67(5):365–367. doi:10.5455/medarh.2013.67.365-367
124. Klöting N, Fasshauer M, Dietrich A, et al. Insulin-sensitive obesity. Am J Physiol. 2010;299(3):506–515. doi:10.1152/ajpendo.00586.2009
125. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. doi:10.1002/hep.28431
126. Rufinatscha K, Radlinger B, Dobner J, et al. Dipeptidyl peptidase-4 impairs insulin signaling and promotes lipid accumulation in hepatocytes. Biochem Biophys Res Commun. 2017;485(2):366–371. doi:10.1016/j.bbrc.2017.02.071
127. Lamers D, Famulla S, Wronkowitz N, et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes. 2011;60(7):1917–1925. doi:10.2337/db10-1707
128. Hattori S, Nomoto K, Suzuki T, Hayashi S. Beneficial effect of omarigliptin on diabetic patients with non-alcoholic fatty liver disease/non-alcoholic steatohepatitis. Diabetol Metab Syndr. 2021;13(1):1–6. doi:10.1186/s13098-021-00644-5
129. Foley JE, Jordan J. Weight neutrality with the DPP-4 inhibitor, vildagliptin: mechanistic basis and clinical experience. Vasc Health Risk Manag. 2010;6(1):541–548. doi:10.2147/vhrm.s10952
130. Balaban YH, Korkusuz P, Simsek H, et al. Dipeptidyl peptidase IV (DDP IV) in NASH patients. Ann Hepatol. 2007;6(4):242–250. doi:10.1016/s1665-2681(19)31905-2
131. Barchetta I, Ceccarelli V, Cimini FA, et al. Circulating dipeptidyl peptidase-4 is independently associated with the presence and severity of NAFLD/NASH in individuals with and without obesity and metabolic disease. J Endocrinol Invest. 2021;44(5):979–988. doi:10.1007/s40618-020-01392-5
132. Ben-Shlomo S, Zvibel I, Shnell M, et al. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J Hepatol. 2011;54(6):1214–1223. doi:10.1016/j.jhep.2010.09.032
133. Firneisz G, Varga T, Lengyel G, et al. Serum dipeptidyl peptidase-4 activity in insulin resistant patients with non-alcoholic fatty liver disease: a novel liver disease biomarker. PLoS One. 2010;5(8):e12226. doi:10.1371/journal.pone.0012226
134. Lee M, Shin E, Bae J, et al. Dipeptidyl peptidase-4 inhibitor protects against non-alcoholic steatohepatitis in mice by targeting TRAIL receptor-mediated lipoapoptosis via modulating hepatic dipeptidyl peptidase-4 expression. Sci Rep. 2020;10(1):1–13. doi:10.1038/s41598-020-75288-y
135. Shirakawa J, Fujii H, Ohnuma K, et al. Diet-induced adipose tissue inflammation and liver steatosis are prevented by DPP-4 inhibition in diabetic mice. Diabetes. 2011;60(4):1246–1257. doi:10.2337/db10-1338
136. Itou M, Kawaguchi T, Taniguchi E, Oriishi T, Sata M. Dipeptidyl peptidase IV inhibitor improves insulin resistance and steatosis in a refractory nonalcoholic fatty liver disease patient: a case report. Case Rep Gastroenterol. 2012;6(2):538–544. doi:10.1159/000341510
137. Reynard MP, Turner D, Navarrete CV. Allele frequencies of polymorphisms of the tumour necrosis factor-α, interleukin-10, interferon-γ and interleukin-2 genes in a North European Caucasoid group from the UK. Eur J Immunogenet. 2000;27(4):241–249. doi:10.1046/j.1365-2370.2000.00227.x
138. Lee JH, Park HJ, Kim YA, et al. The phenotypic characteristic of liver-derived stem cells from adult human deceased donor liver. Transplant Proc. 2012;44(4):1110–1112. doi:10.1016/j.transproceed.2012.02.020
139. Shaheen MBH. Hematopoietic cytokines and growth factors. In: Cord Blood Biology, Transplantation, Banking, and Regulation. AABB Press; 2011:35–74.
140. Christopherson KW, Hangoc G, Broxmeyer HE. Cell surface peptidase CD26/dipeptidylpeptidase IV regulates CXCL12/stromal cell-derived factor-1α-mediated chemotaxis of human cord blood CD34 + progenitor cells. J Immunol. 2002;169(12):7000–7008. doi:10.4049/jimmunol.169.12.7000
141. Christopherson KW, Hangoc G, Mantel CR, Broxmeyer HE. Modulation of hematopoietic stem cell homing and engraftment by CD26. Science (80-). 2004;305(5686):1000–1003. doi:10.1126/science.1097071
142. Kawai T, Choi U, Liu PC, Whiting-Theobald NL, Linton GF, Malech HL. Diprotin A infusion into nonobese diabetic/severe combined immunodeficiency mice markedly enhances engraftment of human mobilized CD34+ peripheral blood cells. Stem Cells Dev. 2007;16(3):361–370. doi:10.1089/scd.2007.9997
143. Wyss BK, Donnelly AFW, Zhou D, Sinn AL, Pollok KE, Goebel WS. Enhanced homing and engraftment of fresh but not ex vivo cultured murine marrow cells in submyeloablated hosts following CD26 inhibition by Diprotin A. Exp Hematol. 2009;37(7):814–823. doi:10.1016/j.exphem.2009.03.005
144. Herman GA, Bergman A, Stevens C, et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91(11):4612–4619. doi:10.1210/jc.2006-1009
145. Ou X, O’Leary HA, Broxmeyer HE. Implications of DPP4 modification of proteins that regulate stem/progenitor and more mature cell types. Blood. 2013;122(2):161–169. doi:10.1182/blood-2013-02-487470
146. Blanchet X, Langer M, Weber C, Koenen R, von Hundelshausen P. Touch of chemokines. Front Immunol. 2012;3:1–18. doi:10.3389/fimmu.2012.00175
147. Lambeir AM, Díaz Pereira JF, Chacón P, et al. A prediction of DDP IV/CD26 domain structure from a physico-chemical investigation of dipeptidyl peptidase IV (CD26) from human seminal plasma. Biochim Biophys Acta. 1997;1340(2):215–226. doi:10.1016/S0167-4838(97)00045-9
148. Broxmeyer HE, Hoggatt J, Leary HAO, et al. Dipeptidylpeptidase 4 negatively regulates colony- stimulating factor activity and stress hematopoiesis. Nat Med. 2012;18(12):1786–1796. doi:10.1038/nm.2991
149. Proost P, Struyf S, Schols D, et al. Truncation of macrophage-derived chemokine by CD26/dipeptidyl-peptidase IV beyond its predicted cleavage site affects chemotactic activity and CC chemokine receptor 4 interaction. J Biol Chem. 1999;274(7):3988–3993. doi:10.1074/jbc.274.7.3988
150. Ye J. Mechanisms of insulin resistance in obesity. Front med. 2013;7(1):14–24. doi:10.1007/s11684-013-0262-6
151. Utzschneider KM, Kahn SE. The role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2006;91(12):4753–4761. doi:10.1210/jc.2006-0587
152. Zuk PA, Zhu M, Ashjian P, et al. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002;13:4279–4295. doi:10.1091/mbc.E02
153. Sciences M, Sugimura A, Seki T. Cloning, expression, and characterization of a cDNA encoding a novel human growth factor for primitive hematopoietic progenitor cells. Proc Natl Acad Sci USA. 1997;94:7577–7582. doi:10.1073/pnas.94.14.7577
154. Tarantino G, Citro V, Balsano C, Capone D. Could SCGF-beta levels be associated with inflammation markers and insulin resistance in male patients suffering from obesity-related NAFLD? Diagnostics. 2020;10(6):395. doi:10.3390/diagnostics10060395
155. Mavier P, Martin N, Couchie D, Préaux AM, Laperche Y, Zafrani ES. Expression of stromal cell-derived factor-1 and of its receptor CXCR4 in liver regeneration from oval cells in rat. Am J Pathol. 2004;165(6):1969–1977. doi:10.1016/S0002-9440(10)63248-8
156. Duarte N, Coelho I, Holovanchuk D, Inês Almeida J, Penha-Gonçalves C, Paula Macedo M. Dipeptidyl peptidase-4 is a pro-recovery mediator during acute hepatotoxic damage and mirrors severe shifts in Kupffer cells. Hepatol Commun. 2018;2(9):1080–1094. doi:10.1002/hep4.1225
157. Campbell TB, Broxmeyer HE. CD26 inhibition and hematopoiesis: a novel approach to enhance transplantation. Front Biosci. 2008;13:1795–1805. doi:10.2741/2800
158. Jungraithmayr W, De Meester I, Matheeussen V, Baerts L, Arni S, Weder W. CD26/DPP-4 inhibition recruits regenerative stem cells via stromal cell-derived factor-1 and beneficially influences ischaemia-reperfusion injury in mouse lung transplantation. Eur J Cardio-Thoracic Surg. 2012;41(5):1166–1173. doi:10.1093/ejcts/ezr180
159. Kawakita E, Yang F, Kumagai A, et al. Metformin mitigates DPP-4 inhibitor-induced breast cancer metastasis via suppression of mTOR signaling. Mol Cancer Res. 2021;19(1):61–73. doi:10.1158/1541-7786.MCR-20-0115
160. Wilson CH, Abbott CA. Expression profiling of dipeptidyl peptidase 8 and 9 in breast and ovarian carcinoma cell lines. Int J Oncol. 2012;41(3):919–932. doi:10.3892/ijo.2012.1522
161. Aoe K, Amatya VJ, Fujimoto N, et al. CD26 overexpression is associated with prolonged survival and enhanced chemosensitivity in malignant pleural mesothelioma. Clin Cancer Res. 2012;18(5):1447–1456. doi:10.1158/1078-0432.CCR-11-1990
162. Zhang T, Tong X, Zhang S, et al. The roles of dipeptidyl peptidase 4 (DPP4) and DPP4 inhibitors in different lung diseases: new evidence. Front Pharmacol. 2021;12:1–9. doi:10.3389/fphar.2021.731453
163. Starska K, Głowacka E, Kulig A, Lewy-Trenda I, Bryś M, Lewkowicz P. The role of tumor cells in the modification of T lymphocytes activity - The expression of the early CD69+, CD71+ and the late CD25+, CD26+, HLA/DR+ activation markers on TCD4+ and CD8+ cells in squamous cell laryngeal carcinoma. Part I. Folia Histochem Cytobiol. 2011;49(4):579–592. doi:10.5603/FHC.2011.0081
164. McCaughan GW, Siah CL, Abbott C, Wickson J, Ballesteros M, Bishop G. Dipeptidyl peptidase IV is down‐regulated in rat hepatoma cells at the mRNA level. J Gastroenterol Hepatol. 1993;8(2):142–145. doi:10.1111/j.1440-1746.1993.tb01505.x
165. Nishina S, Yamauchi A, Kawaguchi T, et al. Dipeptidyl peptidase 4 inhibitors reduce hepatocellular carcinoma by activating lymphocyte chemotaxis in mice. Cell Mol Gastroenterol Hepatol. 2019;7(1):115–134. doi:10.1016/j.jcmgh.2018.08.008
166. Higurashi T, Hosono K, Takahashi H, et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. 2016;17(4):475–483. doi:10.1016/S1470-2045(15)00565-3
167. Kawakita E, Koya D, Kanasaki K. CD26/DPP-4: type 2 diabetes drug target with potential influence on cancer biology. Cancers. 2021;13:2191. doi:10.3390/cancers13092191
168. Ming Z, Jiayi C, Yanyan Y, Zuquan Z, Lai X, Ra DM. Dipeptidyl peptidase-4 inhibitors and cancer risk in patients with type 2 diabetes: a meta-analysis of randomized clinical trials. Sci Rep. 2017;7:8273. doi:10.1038/s41598-017-07921-2
169. Penaforte-Saboia JG, Couri CEB, Albuquerque NV, et al. Emerging roles of dipeptidyl peptidase-4 inhibitors in delaying the progression of type 1 diabetes mellitus. Diabetes Metab Syndr Obes Targets Ther. 2021;14:565–573. doi:10.2147/DMSO.S294742
170. Kissow H, Hartmann B, Holst JJ, et al. Glucagon-like peptide-1 (GLP-1) receptor agonism or DPP-4 inhibition does not accelerate neoplasia in carcinogen treated mice. Regul Pept. 2012;179(1–3):91–100. doi:10.1016/j.regpep.2012.08.016
171. Stecca BA, Nardo B, Chieco P, Mazziotti A, Bolondi L, Cavallari A. Aberrant dipeptidyl peptidase IV (DPP IV/CD26) expression in human hepatocellular carcinoma. J Hepatol. 1997;27(2):337–345. doi:10.1016/S0168-8278(97)80180-8
172. Kawakubo M, Tanaka M, Ochi K, et al. Dipeptidyl peptidase-4 inhibition prevents nonalcoholic steatohepatitis–associated liver fibrosis and tumor development in mice independently of its anti-diabetic effects. Sci Rep. 2020;10(1):1–11. doi:10.1038/s41598-020-57935-6
173. Iwamoto Y, Tajima N, Kadowaki T, et al. Efficacy and safety of sitagliptin monotherapy compared with voglibose in Japanese patients with type 2 diabetes: a randomized, double-blind trial. Diabetes Obes Metabol. 2010;12:613–622. doi:10.1111/j.1463-1326.2010.01197.x
174. Desai S, Brinker A, Joslyn Swann IS. Sitagliptin-associated drug allergy: review of spontaneous adverse event reports. Arch Intern Med. 2010;170(13):1169–1171. doi:10.1001/archinternmed.2010.188
175. Hamasaki H, Yanai H. The development of angioedema in a patient with type 2 diabetes due to a novel dipeptidyl peptidase-IV inhibitor, anagliptin. Int J Cardiol. 2013;168(3):e106. doi:10.1016/j.ijcard.2013.07.257
176. Gosmanov AR, Fontenot EC. Sitagliptin-associated angioedema. Diabetes Care. 2012;35:2012. doi:10.2337/dc12-0574
177. White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Al E. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327–1335. doi:10.1056/NEJMoa1305889
178. Filippatos TD, Athyros VG, Elisaf MS. The pharmacokinetic considerations and adverse effects of DDP-4 inhibitors. Expert Opin Drug Metab Toxicol. 2014;10(6):787–812. doi:10.1517/17425255.2014.907274
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.