")
Back to Journals » OncoTargets and Therapy » Volume 14
Potential Biomarkers for the Efficacy of PD-1-PD-L Blockade in Cancer
Authors Grecea M, Soritau O, Dulf D , Ciuleanu TE, Zdrenghea M
Received 12 May 2021
Accepted for publication 24 September 2021
Published 17 November 2021 Volume 2021:14 Pages 5275—5291
DOI https://doi.org/10.2147/OTT.S283892
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Federico Perche
Miruna Grecea,1 Olga Soritau,2 Daniel Dulf,1,2 Tudor Eliade Ciuleanu,1,2 Mihnea Zdrenghea2,3
1Iuliu Hatieganu University of Medicine and Pharmacy, Department of Oncology, Cluj-Napoca, Romania; 2Oncology Institute ‘Prof. Dr. Ion Chiricuta’, Cluj-Napoca, Romania; 3Iuliu Hatieganu University of Medicine and Pharmacy, Department of Hematology, Cluj-Napoca, Romania
Correspondence: Mihnea Zdrenghea
Iuliu Hatieganu University of Medicine and Pharmacy, Department of Hematology, Republicii 34-36, Cluj-Napoca, 400015, Romania
Tel +40741100480
Fax +40264598606
Email [email protected]
Abstract: A decade ago, immune checkpoint blockade emerged as a major breakthrough in oncology, proposing a novel approach by which immune brakes could be released to enhance antitumor responses. Despite apparently modest improvement of the median duration of response, a spectacular doubling of long-term responses as compared to the available standard of care was seen, for instance, in metastatic melanoma. It soon became obvious that the percentage of patients responding to these novel approaches is relatively small, and the importance of an accurate prediction of responders became more and more clear. Strong predictive markers would allow for the administration of immune checkpoint blocker therapy to the patients most likely to benefit from it, and sparing the potential non-responders of a treatment which is far from innocuous, being associated with significant side-effects and, not least, an important price tag. A number of potential response predictors have already been investigated and partly validated, but they do not cover the major unmet need encountered in the current clinical setting. Here, we review biomarkers for immune checkpoint blockade efficacy, either clinically validated and currently in use, or which have been proposed as candidates and are currently under investigation.
Keywords: immune checkpoint blockade, predictive biomarkers, PD-L1 expression, microbiome
Introduction
Lymphocyte interaction with antigen is not enough for the initiation of an adaptive immune response. Half a century ago, the two signal models of lymphocyte activation were proposed, involving, besides relevant antigen presentation, the ligation of co-stimulatory surface molecules like CD28.1 The notion of a third signal is used to describe a pro-activatory cytokine environment. Negative, or co-inhibitory, “second signals” were later identified, credited with a protective role for the host against the over-engagement of immune effector mechanisms. Inhibiting or blocking these “immune brakes” emerged as a paradigm shift in the immunotherapy of cancer, and earned their pioneers, Tasuku Honjo and James Allison, the 2018 Nobel Prize in Physiology or Medicine.2
Immune checkpoint inhibitors (ICIs) have been developed and tested in cancer over the last two decades, the first to be granted clinical approval being ipilimumab, a CTLA-4-blocking monoclonal antibody (mAb), approved by the FDA in 2011 for metastatic melanoma. Later approvals were granted to agents directed against the signaling axis represented by the programmed death (PD)-1 co-inhibitory receptor and its ligands. The advent of ICI revolutionized oncology by the introduction of a new class of agents, active in relapsed/refractory tumors for which extremely limited therapeutic options were available. Immune checkpoint blockade also brought a reconsideration of treatment response criteria,3,4 including the formulation of new criteria for radiologic responses such as iRECIST and imRECIST.5
Despite spectacular results, mostly seen in previously unmanageable tumors, it has quickly become clear that only a minority of patients respond to immune checkpoint blocker therapy.6 Consecutively, the availability of biomarkers capable of predicting a response became of crucial importance, for a treatment approach which is far from innocuous in terms of toxicity, ease of administration, and financial burden.7 While it was somewhat logical to focus on lymphocyte or tumor-related markers like co-inhibitory receptors or ligand expression levels, humoral, systemic markers have also been proposed for predicting outcome.
In this paper, we review established or proposed biomarkers for ICI treatment efficacy and the data that supports their use in the clinical setting.
The Immunotherapy Rationale – from History to Authorization
The immune response consists of two distinct effector arms: the innate and adaptive responses. While the innate immune cells including natural killer (NK) cells, macrophages and neutrophils react promptly, but less specifically, to external antigens, adaptive effectors such as B and T lymphocytes are part of a more accurate and precisely directed immune feedback mechanism. Nonetheless, in order to initiate such a precise action, a certain amount of time is needed. The highly discriminating adaptive immune reactions are offering excellent targets for the triggering of potent and specific immunotherapeutic responses. A model of the immune anticancer response is the cancer immunity cycle, proposed as a fundamental mechanism of immune-mediated cancer elimination.8,9 Briefly, this multistep mechanism is initiated by the release of neoantigens by apoptotic tumor cells within the tumor microenvironment. These neoantigens are perceived as foreign, non-self, by the adaptive immune system. Cell fragments are seized by dendritic cells, which, after drifting through lymphatic vessels to the lymph nodes, present them to T-cells, triggering tumor-specific cytotoxic T-cell responses against the cancer-specific antigens. CD8+ cytotoxic T-cells migrate and infiltrate the tumor microenvironment, specifically binding to cancer antigen targets, killing tumor cells and leading to an additional discharge of tumor-associated antigens.8 The cycle is repetitive and can lead to an optimal antitumor immune response. A disruption of this anticancer immunity course can arise, leading to tumor cells escaping immunosurveillance and consequently promoting tumor growth, progression and metastasis. One of the best known mechanisms of immune evasion to be elucidated and finally targeted is the immune regulatory checkpoints.10,11 Immune regulatory checkpoints are represented by proteins situated on T-cells and antigen presenting cells (APCs),12 some operating in up-regulating the immune response, some in down-regulating it. Tumor cells can escape the immunity cycle by activating immune checkpoint pathways that restrain antitumor immune responses.
A major milestone in oncology was set by the discovery of immune checkpoints such as cytotoxic T lymphocyte associated protein 4 (CTLA-4) and programmed death-1 (PD-1), molecules expressed on effector cells acting as a brake on immune feedbacks including antitumor responses.13,14 CTLA-4 is a co-inhibitory cell surface signaling molecule that competes with CD28, co-stimulatory molecule, for the binding of CD80 and CD86.15,16 PD-1 has two known ligands, programmed death ligands (PD-Ls) 1 and 2, and its engagement counteracts positive signaling through the T-cell receptors (TCR) and CD28.17 By blocking these immune-silencing molecules using ICIs, a restoration of the immune response against tumor cells can be established.
The pivotal shift of immune checkpoint blockade in the clinical setting was started by the recent approval of ICIs such as ipilimumab (anti-CTLA-4) in 2011, followed by inhibitory/blocking antibodies directed at either PD-1 like nivolumab, pembrolizumab and cemiplimab, or at its ligand PD-L1, like atezolizumab, avelumab, and durvalumab18–20 (Table 1). The interest for immune checkpoint blockade surged, with around a thousand clinical stage immune-oncology agents (IO) under investigation in over 3000 ongoing single agent and over 1000 combination therapy trials.21 The very promising clinical results seen with ICIs have, however, some shortcomings: only 20 to 30% of cancer patients show sustainable objective responses6 and some might experience severe immune-related adverse events. The majority of ICIs under investigation are targeted at PD-1 and PD-L1 since the efficacy of this approach has already been proven, thus deflecting attention from other potentially interesting targets. Still, there is no consensus regarding potential biomarkers of the effectiveness of PD-1/PD-L1 blockade. Advancement of this field is mandatory to allow for the best achievable responses with the least immune-related adverse events (irAE).
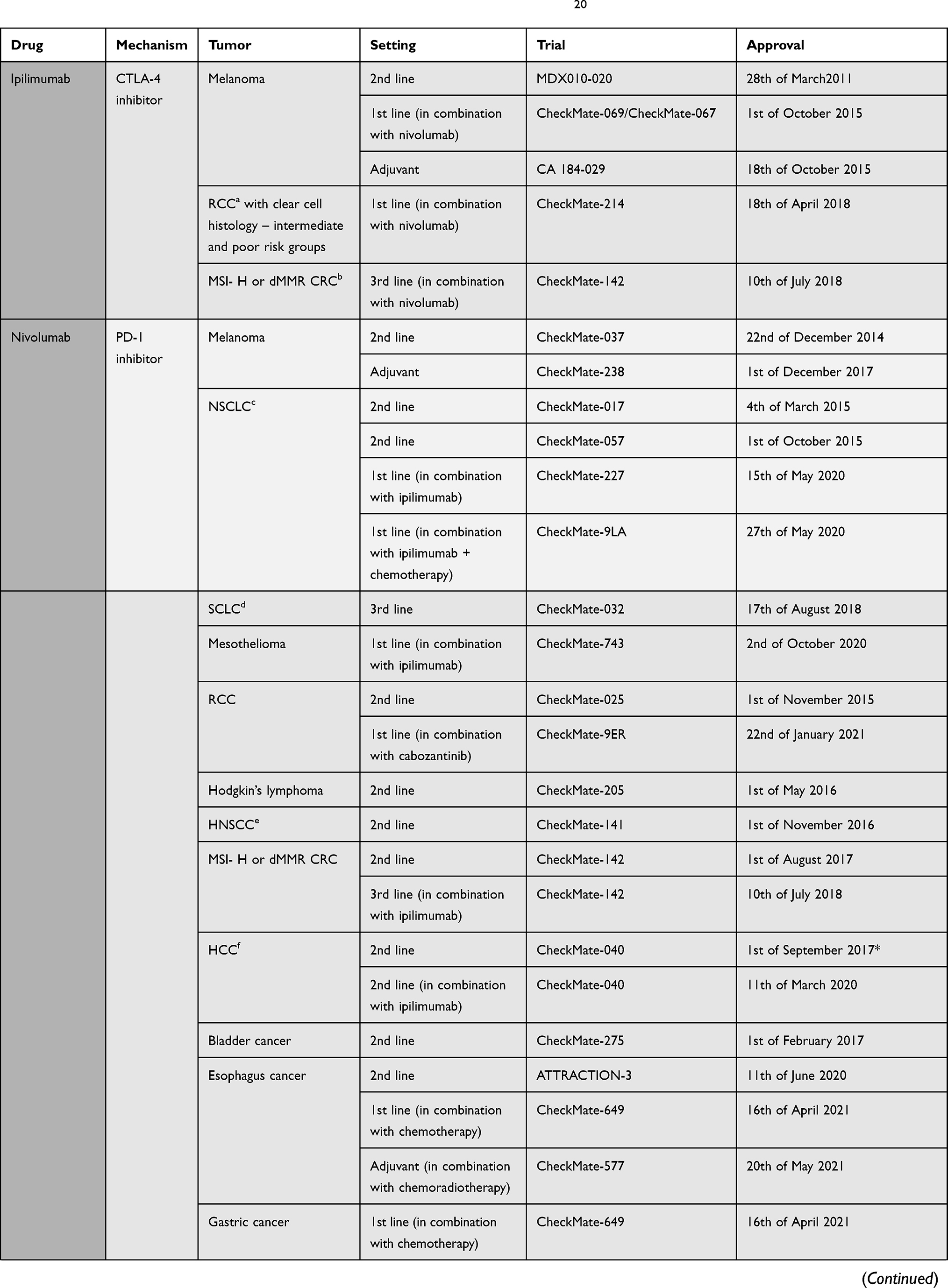 | 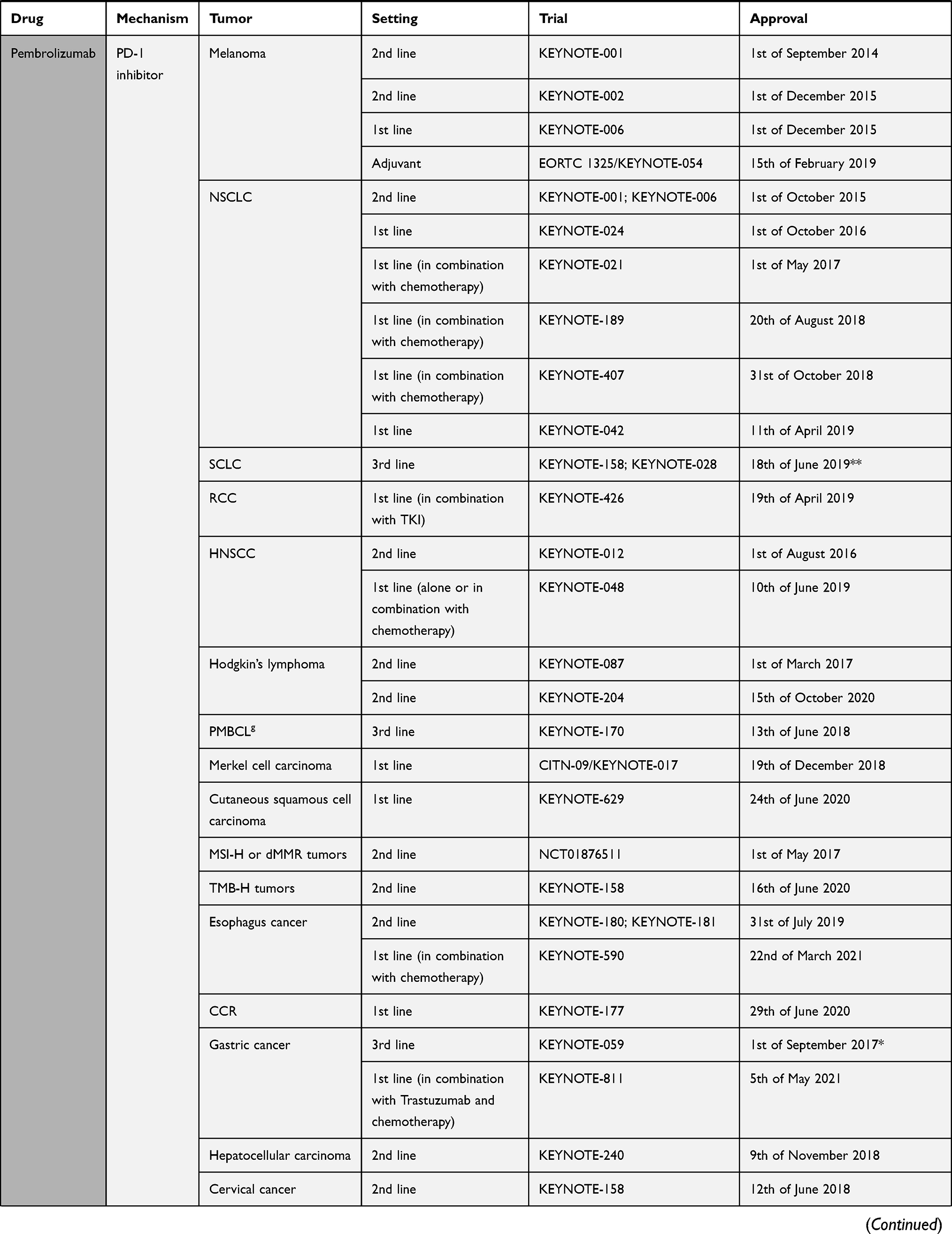 | 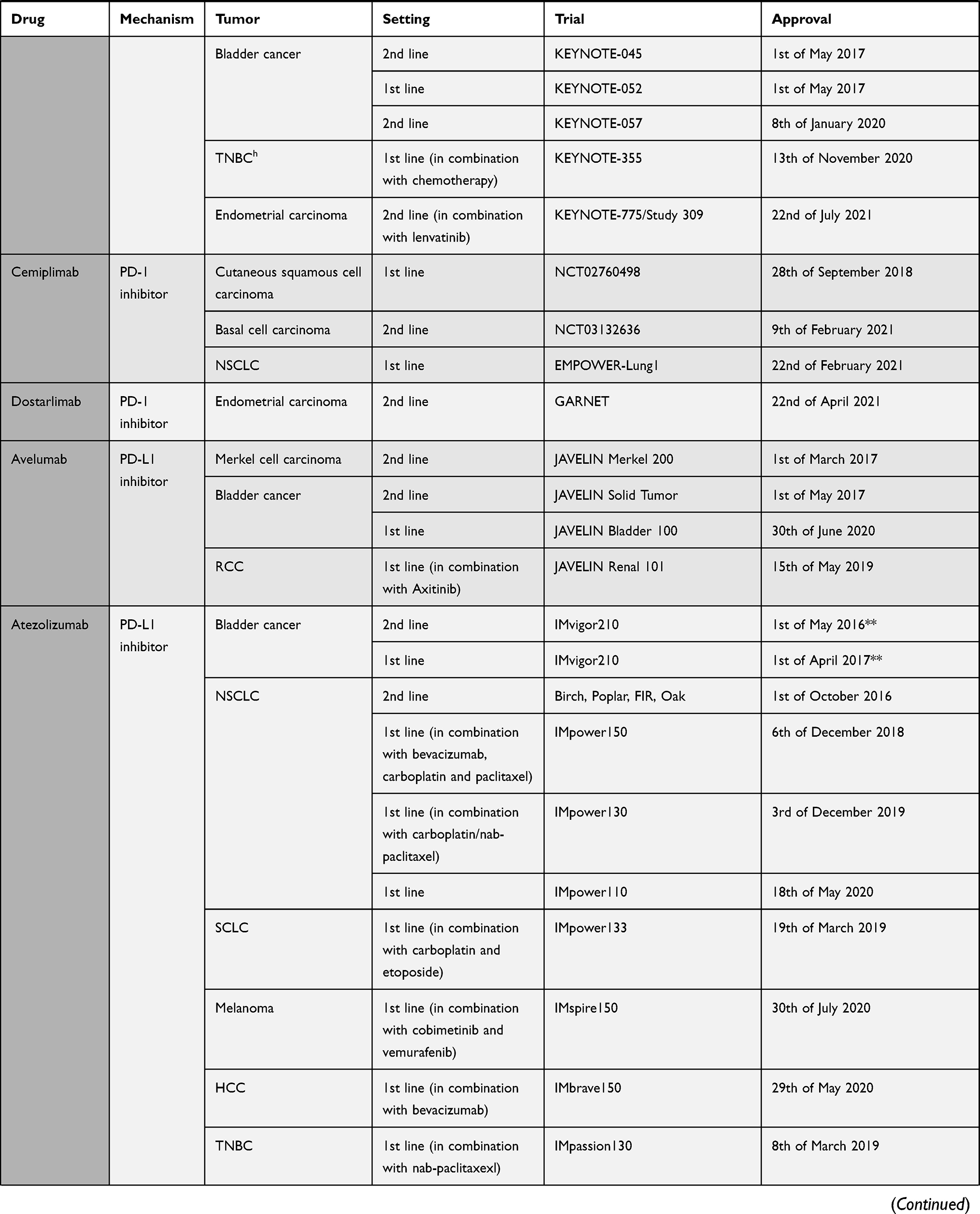 | 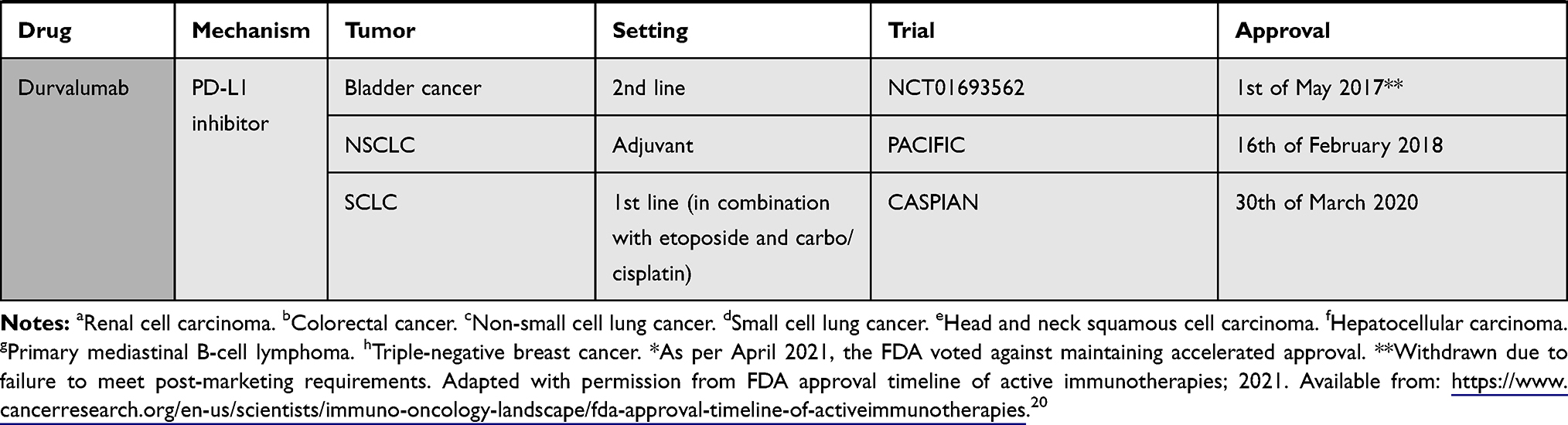 |
Table 1 Immune Checkpoint Inhibitors Approved by FDA by 22nd of July 202120 |
The PD-1-PD-L1/2 Axis
PD-1 (CD279) is a surface protein, a member of the CD28/CTLA4 family, found on activated T-cells, B cells and myeloid cells.15,17,22 PD-1 is a receptor for its two ligands, PD-L1 (CD274) and PD-L2 (CD273), both B7 protein family members which share a sequence homology.23,24 When triggered, this signaling axis behaves as an inhibitory by down-regulating T-cell signaling, effector function and killing ability.12 Tumor cells can exploit these inhibitory signals by expressing PD-1 ligands on their surface. There are two mechanisms known to be involved in PD-1 ligand expression by tumor cells: one is constitutive, secondary to genomic alterations, whilst the other is inducible, regarded as an adaptive immune resistance:12,25 tumor cells escape cytotoxic T-cell destruction through a natural regulatory pathway, up-regulating PD-L1 expression after the release of interferon (IFN)-γ from T-cells upon tumor recognition.26 Consequently, a PD-L1/PD-1 mediated tumor-T-cell signaling takes place, leading to T-cell exhaustion and apoptosis, favoring tumor growth and progression.27 Blocking this interaction has emerged as an efficient immune response-boosting action.
Biomarkers of Response to ICI – Paving the Way to Personalized Immunotherapy
One way of systematizing potential biomarkers is to follow their connection with components of the cancer immunity cycle. Meyers and Banerji analyzed biomarkers of response to ICI according to 3 fundamental elements of the cancer immunity cycle: immune stimulus, immune response and immune modulators.28 Some biomarkers could be considered host-related (Figure 1), whilst others as tumor-associated. Taking the aforementioned elements into account, PD-L1 expression in tumors is one of the most widely investigated predictive biomarkers for the efficacy of immunotherapy. Microsatellite instability (MSI) status and deficient mismatch repair (dMMR) are two further biomarkers currently used in ICI therapeutic approaches. Tumor mutational burden (TMB) status, tumor infiltrating lymphocytes (TILs) and, more recently, tertiary lymphoid structures (TLS), POLE and POLD1 as DNA repair genes, cancer neoantigens or even RNA-signatures and the gut microbiome are emerging, potentially crucial biomarkers to be validated (Table 2). Although extensively investigated, the majority of these biomarkers are not in routine use in the clinical setting, where there is a substantial unmet need for predictive markers.
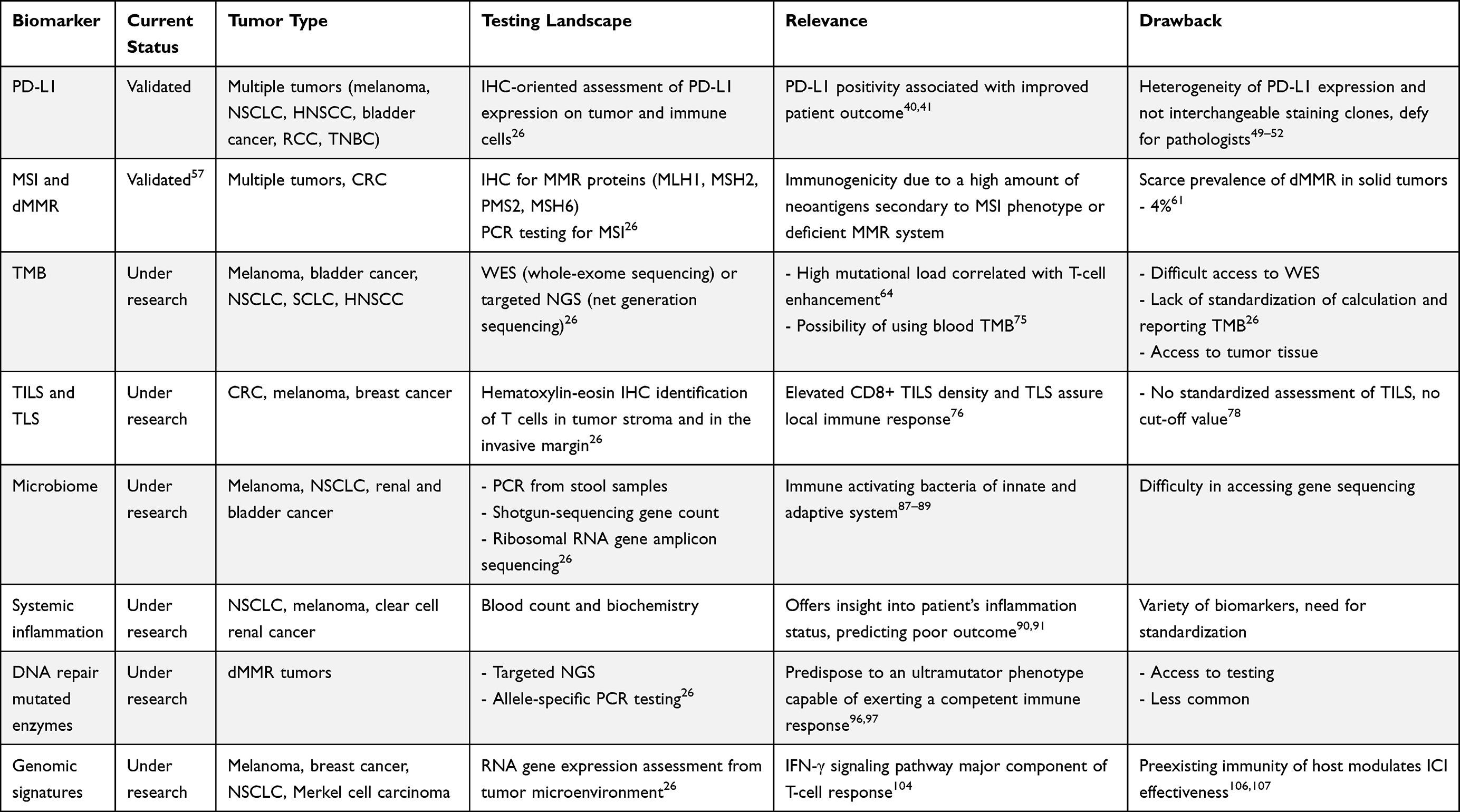 |
Table 2 Status of Current and Emerging Biomarkers |
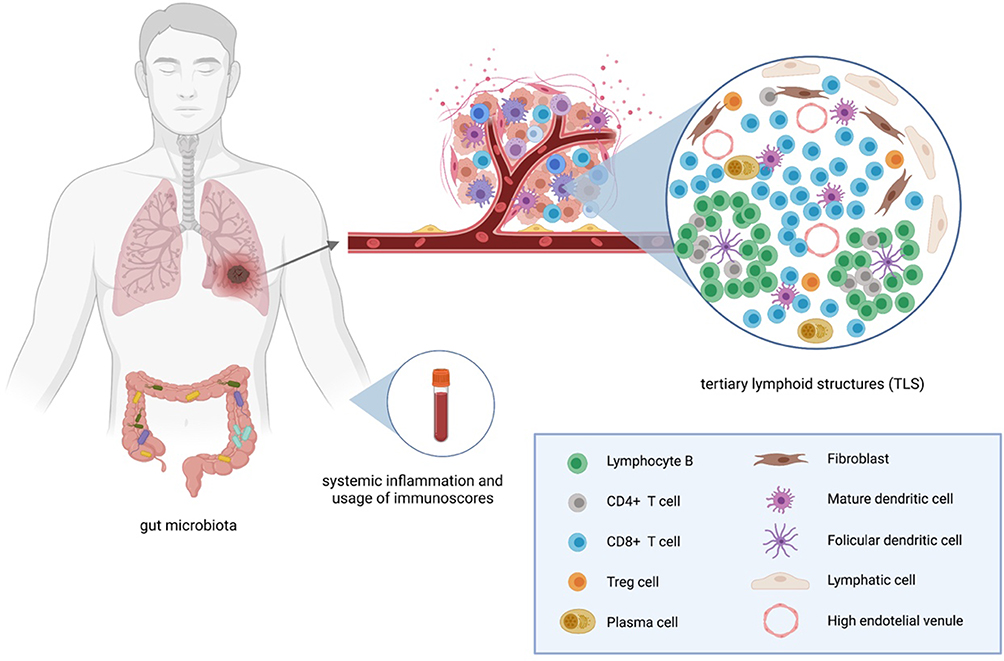 |
Figure 1 Host-related biomarkers. Created by https://www.Biorender.com. |
Biomarkers in Use – Status Approved
PD-L1 Expression
Being expressed on a wide range of cells from B and T lymphocytes to dendritic cells, macrophages and, not lastly, cancer cells, PD-L1 normally plays a role in preventing autoimmunity and redundant inflammation. Censoring immunogenicity, PD-L1 signaling is a target of anti-PD-1/PD-L1 mAbs, being the most investigated predictive biomarker for this therapy to date.
Several studies have concluded that patients with PD-L1 positive tumors benefit from a better response to anti-PD-1/PD-L1 antibodies as compared to those with PD-L1 negative tumors.29 In the KEYNOTE-001 trial where pembrolizumab, an anti-PD-1 mAb was investigated as monotherapy in melanoma patients, the objective response rate (ORR) was 57% in PD-L1 positive tumors versus 8% ORR in PD-L1 negative patients.30 However, a meta-analysis of eight prospective randomized clinical trials which included 4174 patients with advanced or metastatic tumors showed, compared to conventional therapeutic strategies, a prolonged overall survival in both patients that were PD-L1 positive (n=2254, hazard ratio 0.66, 95% confidence interval 0.59 to 0.74) and PD-L1 negative (1920, 0.80, 0.71 to 0.90).31 Nevertheless, in this meta-analysis, the effectiveness of PD-(L)1 blockade was significantly better in PD-L1 positive patients as compared to PD-L1 negative ones, using a cut-off of 1% for defining PD-L1 positivity. In the CheckMate 067 trial where nivolumab plus ipilimumab were compared to nivolumab alone in advanced melanoma, outcomes were independent of PD-L1 status and best responses were seen in the combination arm.32 Consequently, in melanoma, none of the presently approved ICIs requires PD-L1 status assessment.
In the non-small cell lung cancer (NSCLC) phase III trials CheckMate 017 and CheckMate 057 there was an improved overall survival (OS) for nivolumab compared to docetaxel in the second line setting, after platinum doublet chemotherapy.33–35 A 20% ORR in the nivolumab arm was noted in the CheckMate 017 trial, seemingly independent of the PD-L1 status of the tumor, while in the CheckMate 057 trial the reported ORR was 19%, with improved OS in the nivolumab arm of 12.2 months versus 9.4 months; p=0.002, HR: 0.73. Although on subgroup analysis there is a good correlation of response to PD-L1 status as defined by ≥1% tumor membrane expression, with a better rate of response in the nivolumab arm, in less than 5% of PD-L1 positive subjects the OS did not vary between treatment arms. As a result, in the second line setting for all NSCLC subtypes, a PD-L1 status assessment is not mandatory before nivolumab administration. These results were supported by another phase III study, the OAK trial, where both PD-L1 positive and negative subgroups of patients in the atezolizumab arm had a better OS of 20.5 months (HR: 0.41) for the PD-L1 positive group and 12.6 months for the PD-L1 negative group, versus docetaxel.36 The difference in OS according to PD-L1 expression supports its relevance as a predictive biomarker.
In first line setting, in the KEYNOTE-024 phase III trial, patients with ≥50% PD-L1 positivity treated with pembrolizumab had a longer median PFS (10.3 months) and 6 months OS (80.2%; HR: 0.60, 95% CI, 0.41 to 0.89) than those on platinum doublet chemotherapy (median PFS 6 months; OS at 6 months: 72.4%).37 Later, the KEYNOTE-042 trial found an extended OS in the pembrolizumab arm versus platinum-based chemotherapy treated patients in locally advanced or metastatic NSCLC patients having ≥1% PD-L1 positive cell staining.38 However, in untreated locally advanced or metastatic NSCLC patients, in the CheckMate 026 study, which used a threshold for PD-L1 expression of ≥5%, there was no significant difference in PFS or OS with nivolumab as monotherapy.39 As a consequence, the FDA approved the anti-PD-L1 atezolizumab (based on the Impower110 trial)40 and, more recently, in February 2021, the antiPD-1 in cemiplimab (based on the EMPOWER-Lung 1 trial)41 in the first line setting as monotherapy, conditional on ≥50% PD-L1 expression. In the PACIFIC trial, stage III NSCLC patients treated with durvalumab as consolidation therapy after chemoradiation experienced a significant improvement of the OS versus placebo independent of the tumor PD-L1 positivity status.42
In head and neck squamous cell and urothelial cancer patients, high levels of PD-L1 expression in tumor cells were associated with durable responses and improved OS after pembrolizumab, respectively avelumab.43,44 A correspondence between PD-L1 expression in infiltrating immune cells and response to atezolizumab in urothelial cancer and atezolizumab plus chemotherapy in triple-negative breast cancer was also reported.45,46
All aforementioned data considered, PD-L1 is a biomarker of variable relevance, neither excluding nor warranting response to PD-1/PD-L1 blockade across different tumor types including melanoma,30,47 squamous cell NSCLC33–35 and renal cell cancer.48 There are, however, situations where PD-L1 positivity can predict a better response to PD-1/PD-L1-blocking mAbs. The issue to be addressed is PD-L1 testing scores and defining the cut-off value for positivity. The vast diversity of expression of PD-L1, both intratumoral and intertumoral, is a challenge for pathologists since there are 4 FDA-approved staining assays (22C3, 28-8, SP142, SP263), one more to be validated (73-10 PD-L1 clone) and each with different staining affinity for PD-L1 on tumor and immune cells. There are several studies, like the Blueprint Project,49,50 that showed that these clones are not interchangeable in terms of staining, consequently being discordant and with statistically significant differences at 1% and 50% cutoff values.51,52 The most important concern is comparing the concordance of these assays in terms of primary and secondary endpoints. By doing so, several PD-L1 assays with different cut-offs for a vast majority of PD-L1 inhibitors could be validated, assuring a precise selection of patients.
MSI and dMMR
Microsatellites are repetitive short sequences of DNA about 1 to 6 nucleotides long, appearing throughout the genome. These tandem sequences are located in both genes and intergenic non-coding regions, usually occurring in promoter and terminal regions, introns and in coding exons.53,54 When the genome gains or loses ≥1 repeat during DNA replication, MSI (microsatellite instability) occurs. These faults are normally repaired by the mismatch repair (MMR) system that comprises 4 key proteins: MLH1, MSH2, PMS2 and MSH6.53,54 A defective or deficient MMR system (dMMR) appears if there is a mutation in any of these genes or hypermethylation in the promoter of the MLH1 gene. Consequently, errors during DNA replications cannot be corrected, resulting in a MSI phenotype. According to the number of distressed microsatellites, tumors can be classified as: MSI high (MSI-H), MSI low and microsatellite stable (MSS).55 Therefore, MMR deficiency is a mechanism that predisposes to accumulation of mutations, increasing the probability of neoantigen expression as compared to MMR-proficient tumors and consequently generating immunogenicity.
To investigate the MSI issue, pembrolizumab was administered to a heterogeneous cohort of 41 metastatic, previously treated, patients, irrespective of tumor localization, with or without dMMR.56 There was a substantial difference in the ORR in dMMR versus proficient MMR (pMMR) patients: 40% ORR in dMMR colorectal (CRC) patients, 78% ORR in dMMR non-CRC and 0% ORR in pMMR patients, respectively. In first line setting, in the KEYNOTE-177 phase III trial, pembrolizumab was superior to chemotherapy in metastatic MSI-H-dMMR CRC patients, with a median PFS of 16.5 months vs 8.2 months (HR: 0.60, 95% CI, 0.45 to 0.80; p=0.0002), becoming a new standard of care.57
As a premiere, the FDA granted approval of pembrolizumab in MSI-H/dMMR previously treated metastatic or unresectable solid tumors, regardless of tumor type, making it the first ICI to be validated based on a biomarker, based on ORR of pembrolizumab of 39.6% among 149 patients with 15 different MSI-H/dMMR tumor types and a 7% complete response rate.58 Other ICIs to be approved in this setting were nivolumab (anti-PD-L1) and the combination of nivolumab plus ipilimumab, for MSI-H/dMMR metastatic CRC patients.59,60
Only a small percentage of tumors are MSI-H or harbor a dMMR system. The prevalence of dMMR is only 4% in all adult solid tumors,61 a deficient MMR mechanism having been reported only in endometrial, gastric, small bowel, colorectal, cervical, prostate, bile duct, liver and thyroid carcinomas, neuroendocrine tumors and uterine sarcomas. A form of inherited dMMR exists, in the form of the genetic condition called Lynch syndrome. Knowing the predilection of Lynch syndrome to CRC and endometrial cancer (52–82%, respectively; 25–60% lifetime risk), it is mandatory to identify all cases of Lynch syndrome, not only for the patient himself, but also for his family members. Consequently, it is highly advised that all CRC and endometrial tumors undergo screening for defective MMR in order to benefit from ICI.
In Need of Validation – Emerging Biomarkers
TMB
TMB relates to the prevalence of somatic mutations in the genome, leading to nonsynonymous single-nucleotide variants, consequently increasing the capacity of a tumor to create neoantigens. A high TMB enables tumor cells to create unique peptides, expressing them on their surface as a major histocompatibility complex-associated neoantigen. Being interpreted as “non-self”, these neoantigens are capable of generating T-cell responses. Consequently, the higher the TMB, the more immunogenic the tumor is and, as a result, the more susceptible to responding to immunotherapy.
It is well known that TMB differs across tumor types, with melanoma, lung and bladder cancer holding the highest mutation prevalence62 with a consistent response to ICI.63,64 In melanoma, in a CTLA-4 blockade setting by ipilimumab and tremelimumab, high mutational load was associated with a persistent clinical benefit of more than 6 months and a significant improvement of OS (p=0.04).65 In this study, TMB cut-off was defined as more than 100 mutations per sample as identified throughout whole exome sequencing (WES).
In an NSCLC study, Rizvi et al used a cut-off of >200 mutations per sample as determined by WES, showing that high TMB is correlated with an elevated tumor objective response (63% vs 0%, p=0.03) and PFS (14.5 months vs 3.7 months, p=0.01, HR 0.19) after PD-1 blockade with pembrolizumab.66 A distinct perspective revealed by this study is that effectiveness of antiPD-1 was associated with a molecular smoking signature, independent of smoking history, higher neoantigen burden and DNA repair pathway, raising awareness concerning other predictive biomarkers.
Besides melanoma and NSCLC, an elevated mutational load was a predictor of response to checkpoint blockade in many other tumor types such as urothelial cancer (atezolizumab),45 small-cell lung cancer (nivolumab alone or in combination with ipilimumab)67 and head and neck squamous cell cancer negative for human papilloma virus (antiPD-1/PD-L1).68
Frequently, however, rather modest responses to immunotherapy are noted in cases with a high mutational load and there are limited responses to immunotherapy, especially in proficient mismatch repair colorectal cancer.69 Conversely, patients with a modest TMB sometimes benefit from ICI.70,71 Many studies identified TMB and PD-L1 expression as independent predictive biomarkers, both of them associated with a better response. In this regard, in the CheckMate 026 study, patients with advanced NSCLC with nivolumab as first line treatment and having both high TMB and PD-L1 expression had a substantial response rate of 75% versus 34% for patients with only high PD-L1, respectively 32% for high TMB alone, and 16% for those with none of the two markers.72 Also, patients with a significant mutational load and positive PD-L1 expression (>1%) treated with nivolumab and ipilimumab had an improved response and PFS in the CheckMate 227 and 012 trials.73,74
Limitations in validating TMB as a biomarker are difficult access to WES, lack of standardization of the assessment and reporting of TMB and, last but not least, access to tumor tissue. As an alternative to tumor tissue assessment, Gandara et al75 used peripheral blood TMB, with a cut-off of 16 or more mutations, which was relevant for PFS, irrespective of PD-L1 status, in patients treated with atezolizumab versus docetaxel chemotherapy, making blood TMB assessment an innovative approach to be considered when tumor tissue is not available.
TILS and TLS
Tumor-associated lymphocytes are circulating lymphocytes that migrate from blood to tumor across the tumor endothelial barrier.76 TILs depict an active, inflamed tumor microenvironment with the capacity of modulating the T-cell response in accordance to the tumor neoantigens, consequently being a good candidate for a predictive biomarker.
There are 3 distinct types of CD-8+ T cells infiltrating tumors, as described by Naito et al77 after examining 131 patients with resected colorectal cancer: T-cells infiltrated within cancer nests, in the cancer stroma and at the junction between tumor and host (invasive margin). In this study, only the TILS within the cancer nest had a significant independent correlation with patient survival (p=0.016; HR: 0.52) due to the fact that earlier stages were associated with a greater infiltration.
TILs’ density, both at the invasive margin and within the cancer nest, was considered a predictive biomarker in the baseline biopsies of melanoma patients responding to pembrolizumab versus the progressing group.25 Also, an elevated density of CD8+, CD3+ and CD45RO+ T-cells in pretreatment melanoma patients was associated with a better response to CTLA-4 blockade.78 However, in both of these studies, there was no clear cut-off value for TILs to distinguish between responders and non-responders to ICI therapy. Moreover, Chen et al78 demonstrated that there is also an enrichment of T-cells in the tumor center versus the margin in responders compared to non-responders, indicating an ICI-induced tumor infiltration.
Also, in breast cancer trials the presence of TILs was associated with an immune checkpoint inhibitor response. In the phase IB/II PANACEA trial, the use of pembrolizumab-trastuzumab in HER2 positive metastatic patients was associated with a better ORR and longer period of disease control in the presence of TILs at baseline.79
Furthermore, there are studies that evaluated residual disease (RD) TILs after neoadjuvant chemotherapy in triple-negative breast cancer, revealing there is a positive correlation between RD TILs and CD8+ T-cell density, with an enhanced OS of these patients.80,81 Consequently, RD TILs could be a independent biomarker, serving as a predictive one in the adjuvant setting.
All these studies demonstrate the existence of TILs as a noteworthy cancer immunotherapy biomarker candidate; however, like with other predictive aforementioned biomarkers, there is an unmet need for a standardized assessment, with a quantification of TILs using a homogenous scoring, not a manual IHC interpretation. Nowadays, in the artificial intelligence era, machine learning-based algorithms could be the key in identifying the best subset of immunoresponsive patients.
Lately, besides TILs, tertiary lymphoid structures (TLS) are of extreme interest as potential biomarkers. TLS are defined as de novo ectopic lymphoid structures that develop in non-lymphoid tissues because of chronic exposure to inflammatory signals mediated by chemokines and cytokines82 such as IL-7 and CXCL13.83 TLS are usually found in the peritumoral stroma, invasive margin, and/or core of different tumor types, forming a unique lymphoid structure similar to secondary lymphoid organoids, resembling a B-cell follicle, with a germinal center and an enriched T-cell area with mature dendritic cells.84 TLS assure a local immune response by the induction of effector functions, antibody generation, and clonal expansion.76 Therefore, TLS are associated with favorable prognosis and sustained response to ICI, especially in melanoma patients where they were correlated with increased survival after CTLA-4 blockade.85 Seeing the strong link between TLS and the antitumor immune response, there is ongoing research on the therapeutic induction of TLS, combined with immune checkpoint blockade.82
Microbiome
Microorganisms found in the gastrointestinal tract, on the skin and on mucosal surfaces represent the human microbiome. Their immunomodulatory role has been recognized since the 2013 description by Viaud et al of gut microbiota modulating the antitumor immune response and influencing the efficacy of cyclophosphamide.86 In the PD-1/PD-L1 setting, Routy et al87 described an interaction between gut microbiota and ICIs, showing that patients with NSCLC, renal and urothelial cancer treated with broad spectrum antibiotics prior to antiPD-1/PD-L1 therapy had a significantly shorter OS and PFS. Furthermore, shotgun sequencing showed that fecal samples from PD-1 blockade responders were more abundant in a particular commensal bacteria, Akkermansia muciniphila. They concluded that bacterial diversity and specific bacteria such as A. muciniphila restore barrier integrity, reduce systemic inflammation and enhance immunosurveillance.87
In the same regard, Matson et al88 found 8 bacterial species to be more abundant in metastatic melanoma patients responding to PD-L1 blockade, and 2 species to be more abundant in non-responders. The authors even proposed a ratio between beneficial immune-activating bacteria and deleterious bacteria that negatively regulate innate and adaptive immunity as a predictive biomarker for immunotherapy. Another study reported an association between CD8+ tumor T-cell infiltrates and the richness of Fecalibacterium genus, Ruminococcaceae family and Clostridiales order in the gastrointestinal tract in responder metastatic melanoma patients, proposing the hypothesis of an improved antitumor systemic response due to increased antigen presentation and refined T-cell function.89
All of the above considered, the microbiome, through its diversity and composition, is a strong candidate for an immune predictive biomarker, though it requires clinical validation.
Systemic Inflammation and Immunoscores
Blood biomarkers are preferred over tumor tissue biomarkers because of their better availability and their objective reflection of the patient’s systemic inflammation status. High levels of serum lactate dehydrogenase (LDH), associated with an elevated tumor load and cellular turnover, were predictive of poor outcome in NSCLC and melanoma patients.90,91 In this setting, one experimental study investigated in vitro the antitumor effects of an LDH A inhibitor that suppresses tumor growth through apoptotic cell death,92 proposing combination approaches including PD-1/PD-L1 ICIs. Another study investigated soluble forms of immune checkpoint molecules in metastatic clear cell renal carcinoma patients treated with nivolumab, showing that high plasma levels of sPD-1, sPD-L1 and BTN3A1 were correlated with a longer PFS.93
Additionally, there are some immunoscores that could help clinicians in identifying subsets of patients with a worse outcome. For example, lung immune prognostic index (LIPI), based on a derived neutrophils/ leukocytes minus neutrophils ratio (dNLR) greater than 3 and LDH greater than the upper limit of normal permitted to distinguish 3 prognostic groups of patients: good (0 factors), intermediate (1 factor) and poor (2 factors).91 To date, PILE is another immunoscore based on pan-immune inflammation value (PIV), a recently developed peripheral blood count biomarker, lactate dehydrogenase level, and Eastern European Oncology Group performance status.94 The higher the immunoscore, the poorer OS noted in ICI-treated patients.
Arbour et al95 showed that immunosuppressive agents such as steroids, used at the beginning of PD-1/PD-L1 therapy, were significantly associated with a decreased PFS (p=0.03; HR: 1.3) and OS (p=0.001; HR: 1.7). Even though this study was conducted on a small subset of patients, systemic corticoid use is generally discouraged in the antiPD-1/PD-L1 setting, especially in the absence of irAE.
POLE and POLD1 Along with Other DNA Repair Mutated Enzymes
Polymerase ε (POLE gene) and δ (POLD1 gene) are DNA polymerase enzymes participating in DNA replication in the S phase of the cell cycle. Through their exonuclease domain, the enzymes allow excision and replacement of incorrect bases, ensuring a correct DNA replication. Mutations of the exonuclease domain lead to an accumulation of mutations in the genome, promoting an ultramutator phenotype.96
POLD1 mutations are less common than POLE mutations and occur mainly in dMMR tumors, but nevertheless can lead to an ultramutator phenotype.97 POLE mutations are mainly found in MSS/pMMR tumors but some cases were described in MSI patients with unexplained Lynch syndrome.98 POLE mutations associated with an ultramutator phenotype have a plethora of mutations across the genome, an inflamed tumor microenvironment and up-regulated PD-L1; consequently, they were linked to a better prognosis, similar to dMMR tumors.26 In this regard, there is a strong scientific rationale to assess these mutations with targeted NGS or allele-specific PCR testing not only for POLE/POLD1, but also for other DNA repair enzymes and mechanisms such as MGMT, homologous recombination, base excision repair and nucleotide excision repair71 since they can significantly influence the response to immunotherapy. Further clinical studies are awaited for validation.
Genomic Signatures Responsible for Immune Feedback
Several studies used RNA-based gene expression profiling to identify underlying mechanisms of tumor response to immunotherapy. Taube et al, by a whole genome sequencing approach, showed that factors like IL-10 and IL-32 gamma induce PD-L1 expression on monocytes but not tumor cells, in melanoma patients, as an adaptive immune regulatory mechanism.99 Adaptive PD-L1 expression was also observed in other tumors such as NSCLC,100 breast cancer,101 and Merkel cell carcinoma.102 The IFN-γ signaling pathway is considered a sine qua non component of a pertinent T-cell response. This is in accordance with the fact that Janus kinase (JAK) 1 and JAK 2 immunotherapy-resistant mutations prevent up-regulation of IFN-γ target genes.103
Gene signatures such as an IFN-γ 10-gene and expanded-immune 28-gene signatures described by Ribas et al104 were correlated with an improved ORR and PFS in melanoma patients treated with pembrolizumab. In the POPLAR study, NSCLC patients with IFN-γ gene signature and 8 gene T-effector had a better OS when treated with atezolizumab versus docetaxel.105
As stated, these specific mutational signatures shape the response to PD-1/PD-L1 blockade and point out interactions between the host and the tumor. For example, NSCLC patients with tumors having molecular smoking signatures had higher response rates compared to those without such mutational patterns.66,106 Another gene signature associated with durable clinical benefit in NSCLC patients treated with anti-PD-1 therapy, was the apolipoprotein B mRNA editing enzyme catalytic polypeptide-like (APOBEC) mutation.107 Patients with head and neck squamous cell carcinoma and bladder cancer with APOBEC-related mutational signature responded to immune checkpoint blockade.106 In melanoma patients, those with mutational signatures related to ultraviolet exposure or prior treatment with alkylating agents had a consistent clinical benefit versus other dominant mutational signatures.106
These studies suggest that preexisting host-related immune status plays a role in predicting the effectiveness and benefit of immune checkpoint blockade.
Lately, there has been a focus of attention on the role of long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) in regulating PD-L1/PD-1 axis signaling.108 Both lncRNAs and oncogene miRNAs are upstream modulators of the axis with the ability of hampering anti-tumor immunity, including molecular pathways such as STAT3,109 PI3K/Akt110 and MAPK.111 Consequently, these molecules became potential targets for a genetic therapeutic approach.
Conclusion
As with other anticancer treatments, the availability of reliable biomarkers predictive for the efficacy of ICI remains a cobsiderable challenge. One crucial concern regards the significant proportion of patients with concomitant driver alterations and PD-L1 expression, raising questions regarding patients’ optimally tailored treatment.112 To date, only two biomarkers have been validated for clinical use: PD-L1 expression in selected tumor types and MSI-H/dMMR for all types of solid tumor.
There are many other promising biomarkers such as TMB, TILS and TLS, and the microbiome, which have to be validated. In this regard, the development of robust, appropriate assays for their investigation is mandatory. We believe that the impressive efficacy seen with ICIs in selected patients warrants the effort to identify biomarkers for predicting treatment efficacy. Besides this, predictors of non-response may also prove of huge clinical relevance, allowing for the avoidance of submitting patients to an ineffective, but not innocuous, treatment course. Finally, predictors for the risks of side effects are also of potentially great clinical benefit. Therefore, a systematic recording of patient and tumor-related characteristics in both clinical trials and real life is crucial, allowing for the retrospective identification of correlations with treatment outcome.
In the years to come, this setting will be a thriving territory of research and validation.
Disclosure
Prof. Dr. Mihnea Zdrenghea received clinical study funding from GlaxoSmithKline, personal fees from Amgen, non-financial support from Astra Zeneca, personal fees from Bristol Myers Squibb, personal fees from Novartis, personal fees from Janssen, personal fees from Pfizer, grants and personal fees from Roche, personal fees from Sanofi, personal fees from Servier and personal fees from Takeda during the conduct of the study.
The authors report no conflicts of interest in this work.
References
1. Bretscher PA. The history of the two-signal model of lymphocyte activation: a personal perspective. Scand J Immunol. 2019;89(6):0–2. doi:10.1111/sji.12762
2. Huang PW, Chang JWC. Immune checkpoint inhibitors win the 2018 Nobel Prize. Biomed J. 2019;42(5):299–306. doi:10.1016/j.bj.2019.09.002
3. Borcoman E, Kanjanapan Y, Champiat S, et al. Novel patterns of response under immunotherapy. Ann Oncol. 2019;30(3):385–396. doi:10.1093/annonc/mdz003
4. Frelaut M, du Rusquec P, de Moura A, Le Tourneau C, Borcoman E. Pseudoprogression and hyperprogression as new forms of response to immunotherapy. BioDrugs. 2020;34(4):463–476. doi:10.1007/s40259-020-00425-y
5. Stephen Hodi F, Ballinger M, Lyons B, et al. Immune-modified response evaluation criteria in solid tumors (imrecist): refining guidelines to assess the clinical benefit of cancer immunotherapy. J Clin Oncol. 2018;36(9):850–858. doi:10.1200/JCO.2017.75.1644
6. Carretero-González A, Lora D, Ghanem I, et al. Analysis of response rate with anti-PD1/PDL1 antibodies in advanced solid tumors: a meta-analysis of randomized clinical trials (RCT). J Clin Oncol. 2017;35(15_suppl):e14576. doi:10.1200/jco.2017.35.15_suppl.e14576
7. Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139–148. doi:10.1016/j.ejca.2015.11.016
8. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi:10.1016/j.immuni.2013.07.012
9. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi:10.1038/nature21349
10. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. doi:10.1016/j.ccell.2015.03.001
11. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–214. doi:10.1016/j.cell.2015.03.030
12. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi:10.1038/nrc3239
13. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. doi:10.1126/science.271.5256.1734
14. Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med. 2012;366(26):2517–2519. doi:10.1056/NEJMe1205943
15. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi:10.1146/annurev.immunol.23.021704.115611
16. Ville S, Poirier N, Blancho G, Vanhove B. Costimulatory blockade of the CD28/CD80-86/CTLA-4 balance in transplantation: impact on memory T cells? Front Immunol. 2015;6:1–11. doi:10.3389/fimmu.2015.00411
17. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. doi:10.1084/jem.192.7.1027
18. Teng F, Meng X, Kong L, Yu J. Progress and challenges of predictive biomarkers of anti PD-1/PD-L1 immunotherapy: a systematic review. Cancer Lett. 2018;414:166–173. doi:10.1016/j.canlet.2017.11.014
19. Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers. 2020;12(3):738. doi:10.3390/cancers12030738
20. FDA approval timeline of active immunotherapies; 2021. Available from: https://www.cancerresearch.org/en-us/scientists/immuno-oncology-landscape/fda-approval-timeline-of-active-immunotherapies.
21. Tang J, Shalabi A, Hubbard-Lucey VM. Comprehensive analysis of the clinical immuno-oncology landscape. Ann Oncol. 2018;29(1):84–91. doi:10.1093/annonc/mdx755
22. Liang SC, Latchman YE, Buhlmann JE, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33(10):2706–2716. doi:10.1002/eji.200324228
23. Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2(3):261–268. doi:10.1038/85330
24. Lin DY-W, Tanaka Y, Iwasaki M, et al. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc Natl Acad Sci U S A. 2008;105(8):3011–3016. doi:10.1073/pnas.0712278105
25. Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi:10.1038/nature13954
26. Walk EE, Yohe SL, Beckman A, et al. The cancer immunotherapy biomarker testing landscape. Arch Pathol Lab Med. 2020;144(6):706–724. doi:10.5858/arpa.2018-0584-CP
27. Mandai M. PD-1/PD-L1 blockage in cancer treatment-from basic research to clinical application. Int J Clin Oncol. 2016;21(3):447. doi:10.1007/s10147-016-0969-x
28. Meyers DE, Banerji S. Biomarkers of immune checkpoint inhibitor efficacy in cancer. Curr Oncol. 2020;27(S2):S106–S114. doi:10.3747/co.27.5549
29. Chen Q, Li T, Yue W. Drug response to PD-1/PD-l1 blockade: based on biomarkers. Onco Targets Ther. 2018;11:4673–4683. doi:10.2147/OTT.S168313
30. Daud AI, Wolchok JD, Robert C, et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J Clin Oncol. 2016;34(34):4102–4109. doi:10.1200/JCO.2016.67.2477
31. Shen X, Zhao B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: meta-analysis. BMJ. 2018;362:k3529. doi:10.1136/bmj.k3529
32. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345–1356. doi:10.1056/NEJMoa1709684
33. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi:10.1056/NEJMoa1507643
34. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi:10.1056/NEJMoa1504627
35. Horn L, Spigel DR, Vokes EE, et al. Nivolumab versus docetaxel in previously treated patients with advanced non-small-cell lung cancer: two-year outcomes from two randomized, open-label, Phase III trials (CheckMate 017 and CheckMate 057). J Clin Oncol. 2017;35(35):3924–3933. doi:10.1200/JCO.2017.74.3062
36. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389(10066):255–265. doi:10.1016/S0140-6736(16)32517-X
37. Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi:10.1056/NEJMoa1606774
38. Mok TSK, Wu Y-L, Kudaba I, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. 2019;393(10183):1819–1830. doi:10.1016/S0140-6736(18)32409-7
39. Gettinger S, Rizvi NA, Chow LQ, et al. Nivolumab monotherapy for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol. 2016;34(25):2980–2987. doi:10.1200/JCO.2016.66.9929
40. Herbst RS, Giaccone G, de Marinis F, et al. Atezolizumab for first-line treatment of PD-L1–selected patients with NSCLC. N Engl J Med. 2020;383(14):1328–1339. doi:10.1056/nejmoa1917346
41. Sezer A, Kilickap S, Gümüş M, et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet. 2021;397(10274):592–604. doi:10.1016/S0140-6736(21)00228-2
42. Antonia SJ, Villegas A, Daniel D, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. 2018;379(24):2342–2350. doi:10.1056/NEJMoa1809697
43. Chow LQM, Haddad R, Gupta S, et al. Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: results from the Phase Ib KEYNOTE-012 expansion cohort. J Clin Oncol. 2016;34(32):3838–3845. doi:10.1200/JCO.2016.68.1478
44. Apolo AB, Infante JR, Balmanoukian A, et al. Avelumab, an anti-programmed death-ligand 1 antibody, in patients with refractory metastatic urothelial carcinoma: results from a multicenter, Phase Ib Study. J Clin Oncol. 2017;35(19):2117–2124. doi:10.1200/JCO.2016.71.6795
45. Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, Phase 2 trial. Lancet. 2016;387(10031):1909–1920. doi:10.1016/S0140-6736(16)00561-4
46. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–2121. doi:10.1056/NEJMoa1809615
47. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. doi:10.1056/NEJMoa1412082
48. Motzer RJ, Rini BI, McDermott DF, et al. Nivolumab for metastatic renal cell carcinoma: results of a Randomized Phase II Trial. J Clin Oncol. 2015;33(13):1430–1437. doi:10.1200/JCO.2014.59.0703
49. Hirsch FR, McElhinny A, Stanforth D, et al. PD-L1 Immunohistochemistry assays for lung cancer: results from Phase 1 of the blueprint PD-L1 IHC assay comparison project. J Thorac Oncol. 2017;12(2):208–222. doi:10.1016/j.jtho.2016.11.2228
50. Tsao MS, Kerr KM, Kockx M, et al. PD-L1 immunohistochemistry comparability study in real-life clinical samples: results of Blueprint Phase 2 Project. J Thorac Oncol. 2018;13(9):1302–1311. doi:10.1016/j.jtho.2018.05.013
51. Munari E, Rossi G, Zamboni G, et al. PD-L1 Assays 22C3 and SP263 are not interchangeable in non-small cell lung cancer when considering clinically relevant cutoffs: an interclone evaluation by differently trained pathologists. Am J Surg Pathol. 2018;42(10):1384–1389. doi:10.1097/PAS.0000000000001105
52. Rimm DL, Han G, Taube JM, et al. A prospective, multi-institutional, pathologist-based assessment of 4 immunohistochemistry assays for PD-L1 expression in non-small cell lung cancer. JAMA Oncol. 2017;3(8):1051–1058. doi:10.1001/jamaoncol.2017.0013
53. Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5(6):435–445. doi:10.1038/nrg1348
54. Ryan E, Sheahan K, Creavin B, Mohan HM, Winter DC. The current value of determining the mismatch repair status of colorectal cancer: a rationale for routine testing. Crit Rev Oncol Hematol. 2017;116:38–57. doi:10.1016/j.critrevonc.2017.05.006
55. Imai K, Yamamoto H. Carcinogenesis and microsatellite instability: the interrelationship between genetics and epigenetics. Carcinogenesis. 2008;29(4):673–680. doi:10.1093/carcin/bgm228
56. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi:10.1056/NEJMoa1500596
57. André T, Shiu -K-K, Kim TW, et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med. 2020;383(23):2207–2218. doi:10.1056/NEJMoa2017699
58. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res. 2019;25(13):3753–3758. doi:10.1158/1078-0432.CCR-18-4070
59. Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36(8):773–779. doi:10.1200/JCO.2017.76.9901
60. Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–1191. doi:10.1016/S1470-2045(17)30422-9
61. Bonneville R, Krook MA, Kautto EA, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol. 2017;2017. doi:10.1200/PO.17.00073
62. Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi:10.1038/nature12477
63. Büttner R, Longshore JW, López-Ríos F, et al. Implementing TMB measurement in clinical practice: considerations on assay requirements. ESMO Open. 2019;4(1):e000442. doi:10.1136/esmoopen-2018-000442
64. Samstein RM, Lee C-H, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202–206. doi:10.1038/s41588-018-0312-8
65. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. doi:10.1056/NEJMoa1406498
66. Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. 2015;348(6230):124–129. doi:10.1126/science.aaa1348
67. Hellmann MD, Callahan MK, Awad MM, et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell. 2019;35(2):329. doi:10.1016/j.ccell.2019.01.011
68. Hanna GJ, Lizotte P, Cavanaugh M, et al. Frameshift events predict anti-PD-1/L1 response in head and neck cancer. JCI Insight. 2018;3(4). doi:10.1172/jci.insight.98811
69. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377(25):2500–2501. doi:10.1056/NEJMc1713444
70. McGrail DJ, Federico L, Li Y, et al. Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers. Nat Commun. 2018;9(1):1317. doi:10.1038/s41467-018-03730-x
71. Mouw KW, Goldberg MS, Konstantinopoulos PA, D’Andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017;7(7):675–693. doi:10.1158/2159-8290.CD-17-0226
72. Carbone DP, Reck M, Paz-Ares L, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. 2017;376(25):2415–2426. doi:10.1056/NEJMoa1613493
73. Hellmann MD, Ciuleanu T-E, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378(22):2093–2104. doi:10.1056/NEJMoa1801946
74. Hellmann MD, Nathanson T, Rizvi H, et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell. 2018;33(5):843–852.e4. doi:10.1016/j.ccell.2018.03.018
75. Gandara DR, Paul SM, Kowanetz M, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24(9):1441–1448. doi:10.1038/s41591-018-0134-3
76. Paijens ST, Vledder A, de Bruyn M, Nijman HW. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol Immunol. 2021;18(4):842–859. doi:10.1038/s41423-020-00565-9
77. Naito Y, Saito K, Shiiba K, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58(16):3491–3494.
78. Chen P-L, Roh W, Reuben A, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6(8):827–837. doi:10.1158/2159-8290.CD-15-1545
79. Loi S, Giobbie-Hurder A, Gombos A, et al. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): a single-arm, multicentre, phase 1b-2 trial. Lancet Oncol. 2019;20(3):371–382. doi:10.1016/S1470-2045(18)30812-X
80. Luen SJ, Salgado R, Dieci MV, et al. Prognostic implications of residual disease tumor-infiltrating lymphocytes and residual cancer burden in triple-negative breast cancer patients after neoadjuvant chemotherapy. Ann Oncol. 2019;30(2):236–242. doi:10.1093/annonc/mdy547
81. Park YH, Lal S, Lee JE, et al. Chemotherapy induces dynamic immune responses in breast cancers that impact treatment outcome. Nat Commun. 2020;11(1):6175. doi:10.1038/s41467-020-19933-0
82. Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19(6):307–325. doi:10.1038/s41568-019-0144-6
83. Lee Y, Chin RK, Christiansen P, et al. Recruitment and activation of naive T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity. 2006;25(3):499–509. doi:10.1016/j.immuni.2006.06.016
84. Sautès-Fridman C, Lawand M, Giraldo NA, et al. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol. 2016;7:407. doi:10.3389/fimmu.2016.00407
85. Cabrita R, Lauss M, Sanna A, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–565. doi:10.1038/s41586-019-1914-8
86. Viaud S, Saccheri F, Mignot G, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342(6161):971–976. doi:10.1126/science.1240537
87. Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–97. doi:10.1126/science.aan3706
88. Matson V, Fessler J, Bao R, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104–108. doi:10.1126/science.aao3290
89. Gopalakrishnan V, Spencer CN, Nezi L, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103. doi:10.1126/science.aan4236
90. Buder-Bakhaya K, Hassel JC. Biomarkers for clinical benefit of immune checkpoint inhibitor treatment-a review from the melanoma perspective and beyond. Front Immunol. 2018;9:1474. doi:10.3389/fimmu.2018.01474
91. Mezquita L, Auclin E, Ferrara R, et al. Association of the lung immune prognostic index with immune checkpoint inhibitor outcomes in patients with advanced non-small cell lung cancer. JAMA Oncol. 2018;4(3):351–357. doi:10.1001/jamaoncol.2017.4771
92. Kim E-Y, Chung T-W, Han CW, et al. A novel lactate dehydrogenase inhibitor, 1-(phenylseleno)-4-(trifluoromethyl) benzene, suppresses tumor growth through apoptotic cell death. Sci Rep. 2019;9(1):3969. doi:10.1038/s41598-019-40617-3
93. Incorvaia L, Fanale D, Badalamenti G, et al. Baseline plasma levels of soluble PD-1, PD-L1, and BTN3A1 predict response to nivolumab treatment in patients with metastatic renal cell carcinoma: a step toward a biomarker for therapeutic decisions. Oncoimmunology. 2020;9(1):1832348. doi:10.1080/2162402X.2020.1832348
94. Guven DC, Yildirim HC, Bilgin E, et al. PILE: a candidate prognostic score in cancer patients treated with immunotherapy. Clin Transl Oncol. 2021;23(8):1630–1636. doi:10.1007/s12094-021-02560-6
95. Arbour KC, Mezquita L, Long N, et al. Impact of baseline steroids on efficacy of programmed cell death-1 and programmed death-ligand 1 blockade in patients with non-small-cell lung cancer. J Clin Oncol. 2018;36(28):2872–2878. doi:10.1200/JCO.2018.79.0006
96. Heitzer E, Tomlinson I. Replicative DNA polymerase mutations in cancer. Curr Opin Genet Dev. 2014;24(100):107–113. doi:10.1016/j.gde.2013.12.005
97. Mertz TM, Baranovskiy AG, Wang J, Tahirov TH, Shcherbakova PV. Nucleotide selectivity defect and mutator phenotype conferred by a colon cancer-associated DNA polymerase δ mutation in human cells. Oncogene. 2017;36(31):4427–4433. doi:10.1038/onc.2017.22
98. Jansen AM, van Wezel T, van den Akker BE, et al. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur J Hum Genet. 2016;24(7):1089–1092. doi:10.1038/ejhg.2015.252
99. Taube JM, Young GD, McMiller TL, et al. Differential expression of immune-regulatory genes associated with PD-L1 display in melanoma: implications for PD-1 pathway blockade. Clin Cancer Res. 2015;21(17):3969–3976. doi:10.1158/1078-0432.CCR-15-0244
100. Velcheti V, Schalper KA, Carvajal DE, et al. Programmed death ligand-1 expression in non-small cell lung cancer. Lab Invest. 2014;94(1):107–116. doi:10.1038/labinvest.2013.130
101. Cimino-Mathews A, Thompson E, Taube JM, et al. PD-L1 (B7-H1) expression and the immune tumor microenvironment in primary and metastatic breast carcinomas. Hum Pathol. 2016;47(1):52–63. doi:10.1016/j.humpath.2015.09.003
102. Lipson EJ, Vincent JG, Loyo M, et al. PD-L1 expression in the Merkel cell carcinoma microenvironment: association with inflammation, Merkel cell polyomavirus and overall survival. Cancer Immunol Res. 2013;1(1):54–63. doi:10.1158/2326-6066.CIR-13-0034
103. Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–829. doi:10.1056/NEJMoa1604958
104. Ribas A, Robert C, Hodi FS, et al. Association of response to programmed death receptor 1 (PD-1) blockade with pembrolizumab (MK-3475) with an interferon-inflammatory immune gene signature. J Clin Oncol. 2015;33(15_suppl):3001. doi:10.1200/jco.2015.33.15_suppl.3001
105. Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837–1846. doi:10.1016/S0140-6736(16)00587-0
106. Miao D, Margolis CA, Vokes NI, et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat Genet. 2018;50(9):1271–1281. doi:10.1038/s41588-018-0200-2
107. Wang S, Jia M, He Z, Liu X-S. APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene. 2018;37(29):3924–3936. doi:10.1038/s41388-018-0245-9
108. Ashrafizadeh M, Zarrabi A, Hushmandi K, et al. PD-1/PD-L1 axis regulation in cancer therapy: the role of long non-coding RNAs and microRNAs. Life Sci. 2020;256:117899. doi:10.1016/j.lfs.2020.117899
109. Song TL, Nairismägi M-L, Laurensia Y, et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood. 2018;132(11):1146–1158. doi:10.1182/blood-2018-01-829424
110. Li M, Sun X-H, Zhu X-J, et al. HBcAg induces PD-1 upregulation on CD4+T cells through activation of JNK, ERK and PI3K/AKT pathways in chronic hepatitis-B-infected patients. Lab Investig. 2012;92(2):295–304. doi:10.1038/labinvest.2011.157
111. Stutvoet TS, Kol A, de Vries EGE, et al. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J Pathol. 2019;249(1):52–64. doi:10.1002/path.5280
112. Mazieres J, Drilon A, Lusque A, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. 2019;30(8):1321–1328. doi:10.1093/annonc/mdz167
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.