")
Back to Journals » International Medical Case Reports Journal » Volume 11
Postsplenectomy Kawasaki disease in a 17-year‑old male
Authors Gharebaghi N, Aghdashi M , Seyed Mokhtari SA
Received 9 May 2018
Accepted for publication 2 August 2018
Published 30 October 2018 Volume 2018:11 Pages 287—292
DOI https://doi.org/10.2147/IMCRJ.S173605
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Naser Gharebaghi,1 Miramir Aghdashi,2 Seyed Arman Seyed Mokhtari3
1Department of Infectious Disease, Faculty of Medicine, Urmia University of Medical Science, Urmia, Iran; 2Department of Rheumatology and Internal Medicine, Faculty of Medicine, Urmia University of Medical Science, Urmia, Iran; 3Student Research Committee, Urmia University of Medical Sciences, Urmia, Iran
Abstract: Kawasaki disease (KD) is characterized with an acute systemic vasculitis of the medium- and small-sized vessels. This disease mainly involves children within the age of 6 months to 5 years and it is often self-limited and patients seem to recover well; however, it may lead to devastating and fatal cardiovascular complications such as coronary artery aneurysm. Thus, early diagnosis and appropriate management of this disease have a significant effect on improving the prognosis and preventing its serious complications. Adult-onset KD (AKD) is rare and often misdiagnosed. Here we report a rare case of KD that occurred in a 17-year-old young adult who presented 4 weeks post splenectomy, with clinical signs and symptoms consistent with AKD. This may potentially highlight the association of AKD with infectious etiologies.
Keywords: Kawasaki disease, adult, splenectomy
Introduction
Kawasaki disease (KD) also known as mucocutaneous lymph node syndrome is one of the most common forms of vasculitis in children.1–3 The characteristic signs are skin rash, skin desquamation of the peripheral limbs, conjunctival congestion, erythema of the oral cavity, lips, and palms, and cervical lymphadenopathy.3 The disease was first reported in 1967 in Japan by Tomisaku Kawasaki.4 In vast majority of the cases, this syndrome occurs in children younger than 5 years and it rarely occurs in adults.1,5,6 The etiology of this disease remains unknown, but some of the available evidence suggests a potential role for infectious agents and associated immunological processes.7,8 Herein, we describe a rare case of KD that occurred in a 17-year-old young adult who presented 4 weeks post splenectomy, with clinical signs and symptoms consistent with adult-onset KD (AKD).
Case presentation
A 17-year-old young adult originally presented to a community hospital following a car accident. At the time of presentation, he was diagnosed with a number of trauma-related etiologies including hemothorax, potential splenic rupture, head trauma, and traumatic seventh nerve palsy. Following that he underwent splenectomy and was referred to a community hospital for further neurological observation and monitoring as a cautionary measure in regard to his potential head trauma. His hospital stay, which lasted for a week, was unremarkable and uneventful. During his hospital stay, seizure prophylactic treatment with phenytoin was started and continued.
Four weeks post splenectomy (and 3 weeks following his discharge), he presented with high fever, general body pain, and severe sore throat. Following his presentation, he was diagnosed with potential postsplenectomy sepsis and was hospitalized for a total of 4 days. At that time, he was treated with intravenous (IV) antibiotics, namely vancomycin and ceftriaxone. With partial improvement in his clinical symptoms, he was discharged on oral antibiotics (clindamycin and ciprofloxacin), with appropriate outpatient follow-ups. The decrease in body temperature was only relative and short as a result of antibiotic treatments.
Within 36 hours of his discharge from the hospital, he once again presented with fever, generalized maculopapular erythrodermic rash, oral lesions, sore throat, swollen neck, malaise, and myalgia.
At the time of presentation, patient’s vital signs were as follows: heart rate, 128 bpm; blood pressure, 110/90 mmHg; respiratory rate, 26 breaths per minute; and body temperature, 39.5°C.
Patient’s physical examination showed the following pertinent findings: diffused maculopapular erythrodermic rash and scaling in the distal extremities and around the nails (Figure 1A). Furthermore, patient’s oral and pharyngeal mucosa and lips were severely inflamed but showed no exudates. He had severe conjunctivitis and edema of the periorbital areas and the eyelids. Cracking and edema of the lips was also evident (Figure 1B). There was multiple, painful lymphadenopathy in the anterior and posterior cervical and axillary regions and maximum diameter of 17 mm was reported to be present among the cervical lymph nodes. With the exception of tachycardia, the physical examination of the heart, lungs, and abdomen were all unremarkable. No clinical signs of infection in the surgical site were present. There were no clinical signs and symptoms of meningeal irritation. Neurological examination only revealed the peripheral paralysis of the seventh right cranial nerve.
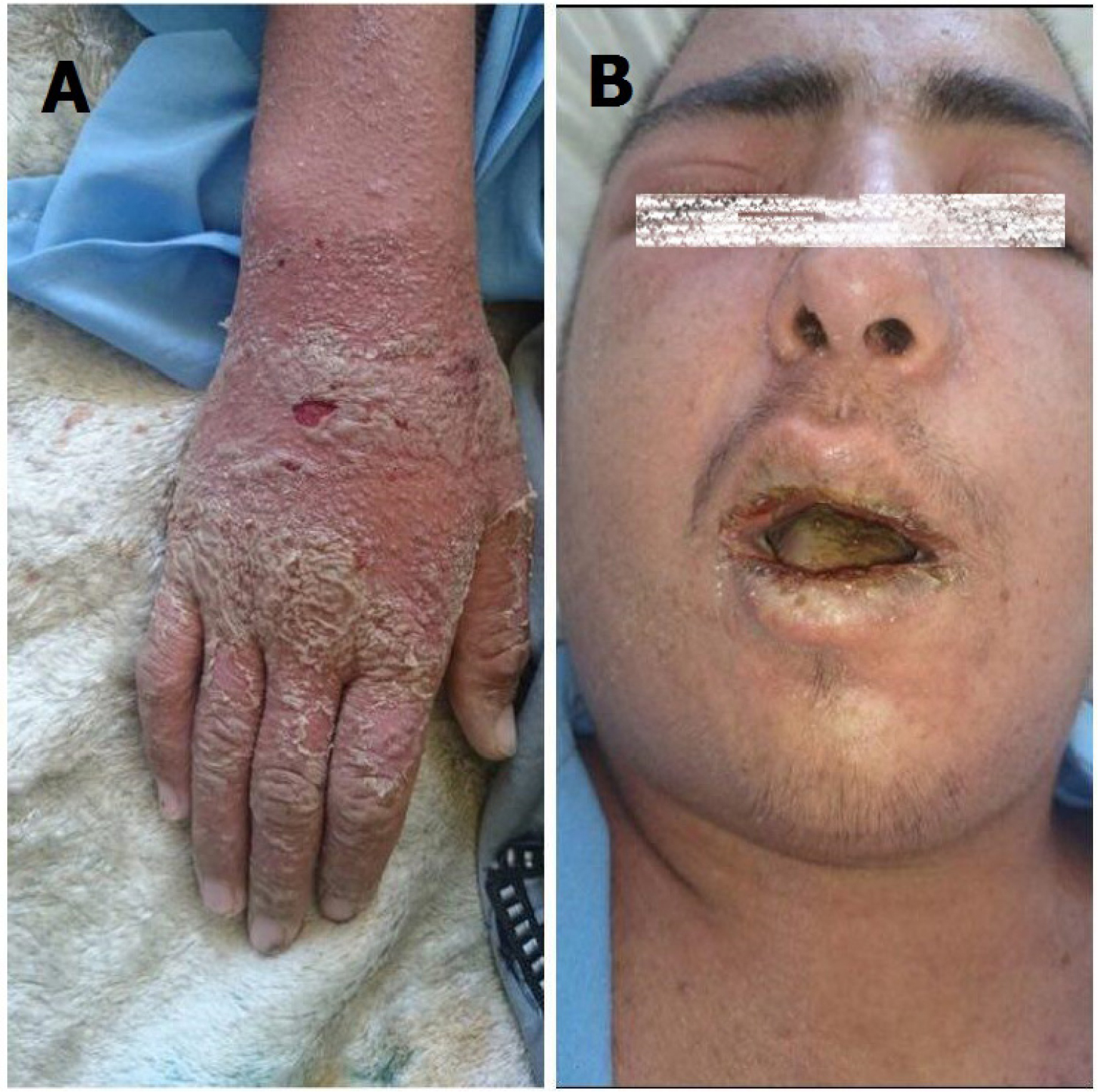 | Figure 1 The physical examination of patient showed diffused maculopapular erythrodermic rash, and scaling in the distal extremities and around the nails (A) and severe conjunctivitis of the periorbital and the eyelids and also cracking and edema of the lips (B). |
Laboratory and imaging findings
The patient’s laboratory findings were recorded as shown in Table 1.
 | Table 1 Obtained tests during the first 48 hours of hospitalization Abbreviations: INR, international normalized ratio; EBV, Epstein-Barr virus; PMN, polymorphonuclear leukocytes. |
Diagnostic imaging
The patient’s Chest X-Ray revealed no specific pathology at the time of admission. Ultrasonography of the abdomen and neck 2 days after hospitalization showed bilateral neck and axillary lymphadenopathy in addition to mild ascites. The size of the liver and pancreas were normal. No pathology in the para-aortic lymph nodes was detected. Echocardiography that was carried out during his second day of hospital stay demonstrated only traces of pericardial fluid, and did not exhibit any signs of cardiac contusion or valvular abnormalities. The left ventricular ejection fraction at the time was estimated to be 70%.
Management and course in the hospital
The patient received supportive care, which included IV normal saline, IV antibiotics (vancomycin and imipenem), antihistamine (loratadine), and acetaminophen. Two days into the treatment, the above-mentioned treatments had no measurable effect on the patient’s clinical status, and it did not alter either his body temperature or skin rash. In addition, his erythroderma and scaling were further intensified (Figure 1A).
Although we continued the patient’s supportive care and antibiotics, given the lack of any significant improvement in the patient’s clinical status, other potential diagnoses including AKD were entertained. The AKD diagnosis in our patient was confirmed based on the American College of Cardiologist’s Criteria.9
Consequently, this patient was treated with 6 vials of 30 g/d Intravenous immunoglobulin for 4 days, 80 mg/kg aspirin for 6 days, and then 80 mg/d after this period. Prior to IVIG administration and despite the treatment with acetaminophen and antibiotics, the patient remained pyretic. After 2 days of IVIG treatment, the fever as well as the pain in the lymph nodes subsided. After 4 days and a total received dosage of 120 g, IVIG administration was discontinued. These treatments led to significant improvements in the patient’s clinical status, his fever subsided, erythroderma was reduced, and the pain and swelling in his lymph nodes were significantly decreased. Given his remarkable clinical improvement, after 4 days, his IVIG treatment was discontinued. At the sixth day of hospitalization, the patient complained of bilateral mild hearing loss which was improved after reducing the dose of aspirin to 80 mg/d.
Ten days after admission, the patient’s temperature normalized and his lymphadenopathies resolved (Figure 2). His echocardiogram and coronary angiography that were carried out on the 14th and 20th day of his admission, respectively, reported normal and showed no signs of coronary artery aneurysm (Figure 3).
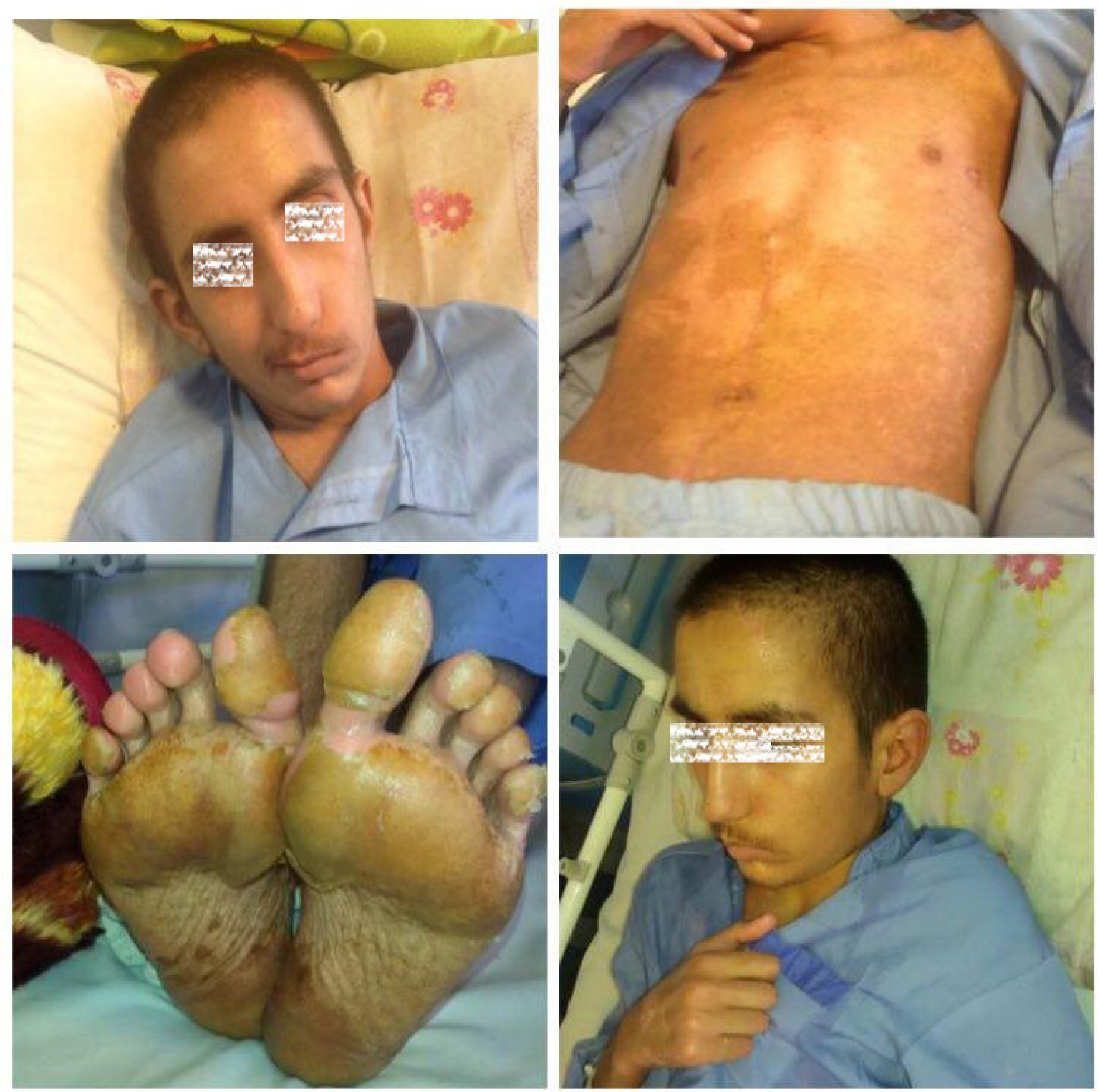 | Figure 2 The physical examination of patient was normal 10 days after admission and his lymphadenopathies resolved. |
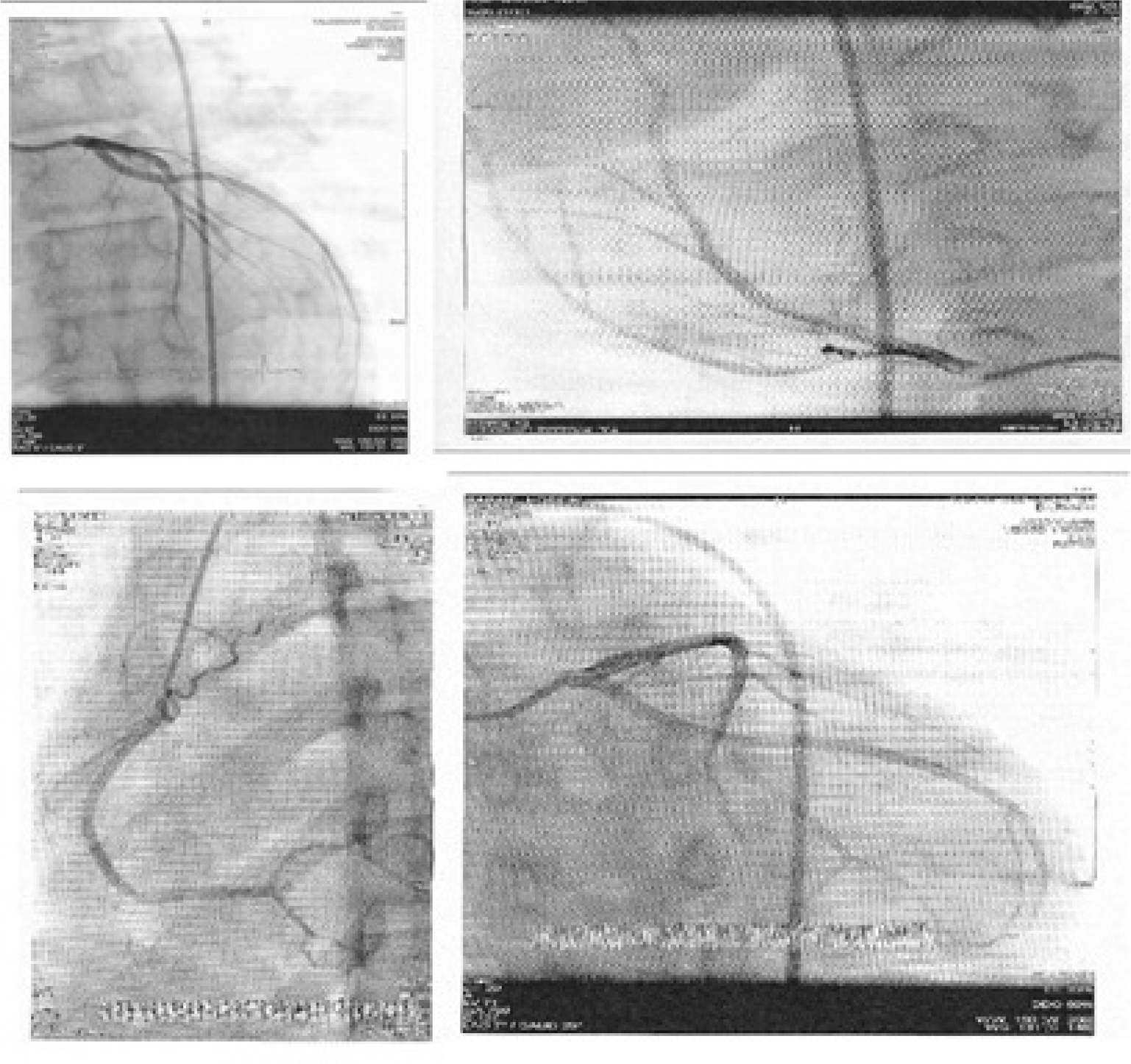 | Figure 3 Echocardiogram and coronary angiography of patient reported normal and showed no signs of coronary artery aneurysm. |
Written informed consent of the patient was obtained to publish this case report. In addition, this case report was approved by the Institutional Review Board of the Urmia University of Medical Sciences.
Discussion
Simultaneous occurrence of fever and skin rash is a common and challenging clinical presentation in both pediatric and adult patients. Although the differential diagnosis is very broad, adequate history and physical examination may help the clinician to narrow down a potential list of probable etiologies. Since the spectrum of diagnosis can include mild self-limiting condition to life-threatening disease, clinical discretion remains imperative. Given that both infectious and noninfectious conditions can underline fever and rash, in an optimal clinical approach, a patient’s history of the presenting illness and the physical examination findings remain crucial. These important aspects of history and physical examination may include distribution, morphology as well as progression of rash, manner of resolution, timing of fever, patient’s age, travel history, geographic location, exposure history, medication, and immunization.
In our patient, the presentation of fever and mucocutaneous rash, postsplenectomy, led to clinical suspicion for the diagnosis of sepsis and toxic shock syndrome. Yet, the negative result of blood cultures and lack of response to antibiotic treatments as well as clinical features not consistent with infectious etiologies led to the consideration of other alternative diagnoses.
Moreover, the use of phenytoin in the patient, led to the consideration of drug reaction as an alternative diagnosis, which could have presented in a clinical spectrum from mild-to-severe reaction, such as that seen in Steven–Johnson syndrome (SJS). However, the presence of thrombocytosis and diffuse desquamation was against this diagnosis. The diagnosis of SJS or toxic epidermal necrolysis is not plausible. Fever is not a primary feature of these diseases and systemic reactions, such as increased white blood cell count, elevated ESR, positive CRP levels, and thrombocytosis are not expected to occur with this degree and severity; edema of the extremities, scaling, and the absence of target lesions are supportive of the diagnoses other than SJS.10
When this diagnostic challenge was met in a renewed assessment of patient, it was noticed that patient’s overall constellation of signs and symptoms were fulfilling the diagnostic criteria of AKD.
Thus, by considering all of the signs and symptoms in the patient and fulfillment of the diagnostic criteria for the Kawasaki syndrome, clinical diagnosis was made, and patient was started on appropriate therapy, which led to an uneventful course of recovery.
Given that prevalence of Kawasaki syndrome remains low, high index of clinical suspicion and application of the diagnostic criteria for this potentially treatable disease remain invaluable. Early diagnosis and effective therapy with IVIG have been shown to significantly reduce the morbidity and mortality from this disease.11–13
In our patient, given that the diagnosis of Kawasaki syndrome was made postsplenectomy, it once again raises the question in regard to the potential association of this condition with infectious etiologies. While a body of evidence supports an association between Kawasaki syndrome and infectious agents, the exact mechanisms of this association remain unclear.14
A number of clinical features of KD support the notion that infectious etiologies are capable of triggering the onset of the disease.7,8,14 These features include the following aspects of KD: there is an overlap of the clinical picture of KD and other infectious diseases,14 such as adenovirus and scarlet fever. It has been known that KD has a seasonal clustering in the winter and spring, which is similar to the other infectious diseases. There are also temporal clusters of epidemics which have been so far reported in Japan, the United States, Canada, and Finland.8 Indeed, an outbreak in Japan began in an area and spread throughout the country in a period of 3 months. Finally, the low incidence in the first 3 months of life may suggest a partial protection from transplacental antibodies.7,8 Our case report which highlights the unique occurrence of KD in a young adult postsplenectomy patient may further support a potential role for infectious etiology in the onset of KD.
The spleen plays a central role in host immunity against infectious agents, by combining the innate and adaptive immune system in a uniquely organized way. This function, in combination with a highly organized lymphoid compartment, makes the spleen the most important organ for antibacterial and antifungal immune reactivity. Indeed, splenectomy is known to cause greatly diminished immune function against infectious agents.15
Conclusion
While one is tempted to speculate an association between our patient’s splenectomy and his presentation with Kawasaki syndrome, currently this potential link remains unknown. Indeed our manuscript is a rare, and to the best of our knowledge, the first case report about the presentation of Kawasaki syndrome in a postsplenectomy young adult. Our case report further strengthens the already established association of Kawasaki syndrome with infectious etiologies and it may also heighten clinical discretion in approach to “fever and rash” in postsplenectomy patients. It may also guide the future research on the complicated and currently unknown association between human immune response and Kawasaki syndrome.
Author contributions
All the authors contributed toward data analysis, drafting, and revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Maddox RA, Person MK, Joseph LI, et al. Monitoring the occurrence of Kawasaki syndrome in the United States. Proceedings of the Eleventh International Kawasaki Disease Symposium; 3–6 February 2015; Honolulu, Hawaii. Abstract O.03, p. 28. | ||
Gomard-Mennesson E, Landron C, Dauphin C, et al. Kawasaki disease in adults: report of 10 cases. Medicine. 2010;89(3):149–158. | ||
Mccrindle BW, Rowley AH, Newburger JW, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. 2017;135(17):e927–999. | ||
Kawasaki T. Acute febrile muco-cutaneous lymph node syndrome in young children with unique digital desquamation. Jpn J Allergol. 1967;16:178–222. | ||
Muta H, Ishii M, Yashiro M, Uehara R, Nakamura Y. Late intravenous immunoglobulin treatment in patients with Kawasaki disease. Pediatrics. 2012;129(2):e291–297. | ||
Sève P, Stankovic K, Smail A, et al. Adult Kawasaki disease: report of two cases and literature review. Semin Arthritis Rheum. 34(6):2005:785–792. | ||
Burns JC, Cayan DR, Tong G, et al. Seasonality and temporal clustering of Kawasaki syndrome. Epidemiology. 2005;16(2):220–225. | ||
Burgner D, Harnden A. Kawasaki disease: what is the epidemiology telling us about the etiology? Int J Infect Dis. 2005;9(4):185–194. | ||
Newburger JW, Takahashi M, Burns JC. Kawasaki disease. J Am Coll Cardiol. 2016;67(14):1738–1749. | ||
Rowley AH, Shulman ST. Kawasaki syndrome. Pediatr Clin North Am. 1999;46(2):313–329. | ||
Furusho K, Kamiya T, Nakano H, et al. High-dose intravenous gammaglobulin for Kawasaki disease. Lancet. 1984;2(8411):1055–1058. | ||
Tse SM, Silverman ED, Mccrindle BW, Yeung RS. Early treatment with intravenous immunoglobulin in patients with Kawasaki disease. J Pediatr. 2002;140(4):450–455. | ||
Kuo HC, Yang KD, Chang WC, Ger LP, Hsieh KS. Kawasaki disease: an update on diagnosis and treatment. Pediatr Neonatol. 2012;53(1):4–11. | ||
Greco A, de Virgilio A, Rizzo MI, et al. Kawasaki disease: an evolving paradigm. Autoimmun Rev. 2015;14(8):703–709. | ||
Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. 2005;5(8):606–616. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.