")
Back to Journals » OncoTargets and Therapy » Volume 12
Postchemotherapy sarcoma as a somatic-type malignancy derived from the gonadal yolk sac tumor in a patient with 46, XY pure gonadal dysgenesis
Authors Zong X, Yang JX, Zhang Y, Cao DY, Shen K, You Y, Guo LN
Received 24 October 2018
Accepted for publication 20 February 2019
Published 28 March 2019 Volume 2019:12 Pages 2365—2372
DOI https://doi.org/10.2147/OTT.S192111
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Sanjay Singh
Xuan Zong,1 Jia-Xin Yang,1 Ying Zhang,1 Dong-Yan Cao,1 Keng Shen,1 Yan You,2 Li-Na Guo2
1Department of Obstetrics and Gynecology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China; 2Department of Pathology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China
Abstract: 46, XY pure gonadal dysgenesis (PGD) is characterized as a female phenotype with strip-like gonads, which has a high tendency to develop into gonadal tumors. Somatic-type malignancies of germ cell tumors (SMs of GCTs) refer to the presence of malignant non-germ cell histologies admixed with GCTs, which are usually chemoresistant and indicate poor prognosis. This case report aimed to analyze the special histological type of GCTs and the importance of salvage surgery in the treatment of refractory GCTs. We report a unique case of gonadal yolk sac tumor (YST) transformed into SMs in a patient with 46, XY PGD. This 18-year-old woman underwent laparoscopic pelvic tumor resection, considered her first surgery, 2 years ago, and pathology revealed YST with initial alpha-fetoprotein (AFP) level measuring >3,000 ng/mL. She underwent seven cycles of chemotherapy, and the AFP level decreased to within a normal range after the second cycle. However, a computed tomography scan after the seventh cycle revealed abdominal and pelvic metastases, and vaginal bleeding was continuously observed. Laparoscopic exploration and laparotomy with tumor subtotal resection were performed. A pathology report showed SMs (sarcoma) derived from YST. Whole exome sequencing demonstrated that the main somatic mutation was a non-synonymous mutation of KRAS (c.182A>G), and this result did not show any indications for targeted drugs. She received three cycles of PEI (cisplatin, etoposide, and ifosfamide) chemotherapy but showed no response. She refused to undergo further treatment and has been alive with the disease for 7 months. This suggests that SMs may be one of the reasons for chemoresistance of refractory GCTs, and salvage surgery may be one of the most effective treatments for this patient. Targeted therapy may be a new choice for chemoresistant GCTs, but drug selection must be based on gene sequencing, and its efficacy still needs to be verified by further study.
Keywords: disorder of sex development, germ cell tumor, somatic-type malignancy, salvage surgery
Introduction
46, XY pure gonadal dysgenesis (PGD), also known as Swyer syndrome, is estimated to have an incidence of 1/80,0001 and is characterized as a female phenotype with strip-like gonads, which have a 15%–35% risk of developing into tumors.2,3 Gonadoblastoma is the most common one, which is associated with the presence of Y-chromosome material.4 Although gonadoblastoma is a benign tumor, it has a high tendency to progress into germ cell malignancies, the majority of which are dysgerminomas.5 Others include immature teratoma, embryonal carcinoma, and yolk sac tumor (YST).6 Previous studies have demonstrated that testis-specific protein Y-linked (TSPY) gene expression and sex-determining region Y (SRY), Wilms tumor 1 (WT1), and SRY box 9 (SOX9) gene mutations are associated with tumor development.4,7–9 Hence, it is recommended that patients with 46, XY PGD undergo preventive bilateral gonadectomy at the time of diagnosis.2
YST is one of the most common malignant ovarian germ cell tumors (OGCTs) and has a relatively good prognosis owing to its sensitiveness to chemotherapy such as PEB (cisplatin, etoposide, and bleomycin) or PVB (cisplatin, vincristine, and bleomycin).10 Alpha-fetoprotein (AFP) is its typical tumor marker, which can be used not only in establishing diagnosis but also in the surveillance of treatment effect and tumor recurrence.11 In our previous study, the cure rates following the initial treatment and 5-year disease-free survival for all malignant OGCTs were 85.02% and 86%, respectively, and the independent prognostic factors for relapse included the level of the medical institution and the extent of surgery and disease.12 There is no standard treatment for recurrent OGCTs, and the prognosis is much worse even after secondary cytoreductive surgery with salvage chemotherapy.12,13 According to National Comprehensive Cancer Network (NCCN) Guidelines, the tumor molecular genetic testing performed before the beginning of therapy for recurrent epithelial ovarian cancer has not been regularly recommended for recurrent OGCTs. In addition, genomic features contributing to chemoresistance of OGCTs remain to be further investigated.
Somatic-type malignancies of germ cell tumors (SMs of GCTs) refer to the presence of malignant non-germ cell histologies admixed with GCTs.14–16 Traditionally, SMs are attributed to the “malignant transformation” of prior or concomitant teratoma.17 However, in recent studies, some patients with SMs did not have teratoma, and YST has been suggested as another origin.18–20 Normally, SMs are categorized into the following two broad categories: sarcomatoid neoplasms and epithelioid neoplasms. With the application of more sensitive and specific immunohistochemistry (IHC) markers, sarcomatoid neoplasms can be classified into sarcomatoid yolk sac tumor (SYST), sarcomatoid carcinoma, and sarcoma. SYST is positive for both pancytokeratin (AE1/AE3) and glypican-3 (GPC3), whereas sarcoma is negative for AE1/AE3 and GPC3.21 SMs have been thought to be chemoresistant and predicted to have less favorable prognosis in cases of GCTs.21–23 However, there have been no studies on the association between SMs of GCTs and prognosis in OGCTs. Herein, we report a case of an 18-year-old patient with 46, XY PGD who had gonadal YST, which transformed into SMs (sarcoma) after undergoing PEB and cisplatin and etoposide (PE) chemotherapy. To our knowledge, this is the first report of SMs derived from YST in 46, XY PGD.
Case report
The patient is an 18-year-old woman. When she was 13 years old, she visited a doctor because of primary amenorrhea. Chromosome analysis revealed 46, XY karyotype (Figure 1), and she had an artificial menstrual cycle for 2 years. On June 1, 2016, the then 16-year-old woman visited the emergency room following a complaint of acute abdominal pain. A computed tomography (CT) scan revealed a pelvic mass, and the initial AFP level was >3,000 ng/mL. Laparoscopic pelvic tumor resection was performed at a local hospital. Intraoperative observation showed that the patient had typical female external genitalia with sparse pubic hair and a normal clitoris. The mass was approximately 10 cm in diameter and was not connected to the vagina, and the fallopian tubes were discovered on the side of the mass. No other tumors were observed after a comprehensive intraoperative exploration was performed. The mass was completely resected, and the pathology report confirmed that the histological type of the gonadal tumor was YST without other GCT or SM components after extensive sampling. Smooth muscle tissue lined with the endometrium was also observed from certain sections, which indicated that both the uterus and pelvic tumor had been resected. After the surgery, seven cycles of chemotherapy were performed: four cycles with PEB and three cycles with PE. The AFP level was within the normal limits after the second cycle. However, a CT scan after the seventh cycle revealed abdominal and pelvic metastases including liver metastases. Furthermore, continuous vaginal bleeding occurred after the surgery. Hysteroscopic exploration was performed to search for the cause of vaginal bleeding. According to the local operation recording, a 3 cm neoplasm in the vagina, which was not connected to the pelvic cavity, was noted. The symptom of vaginal bleeding was still observed after the surgery, and she received hemostatic drugs as the symptomatic treatment.
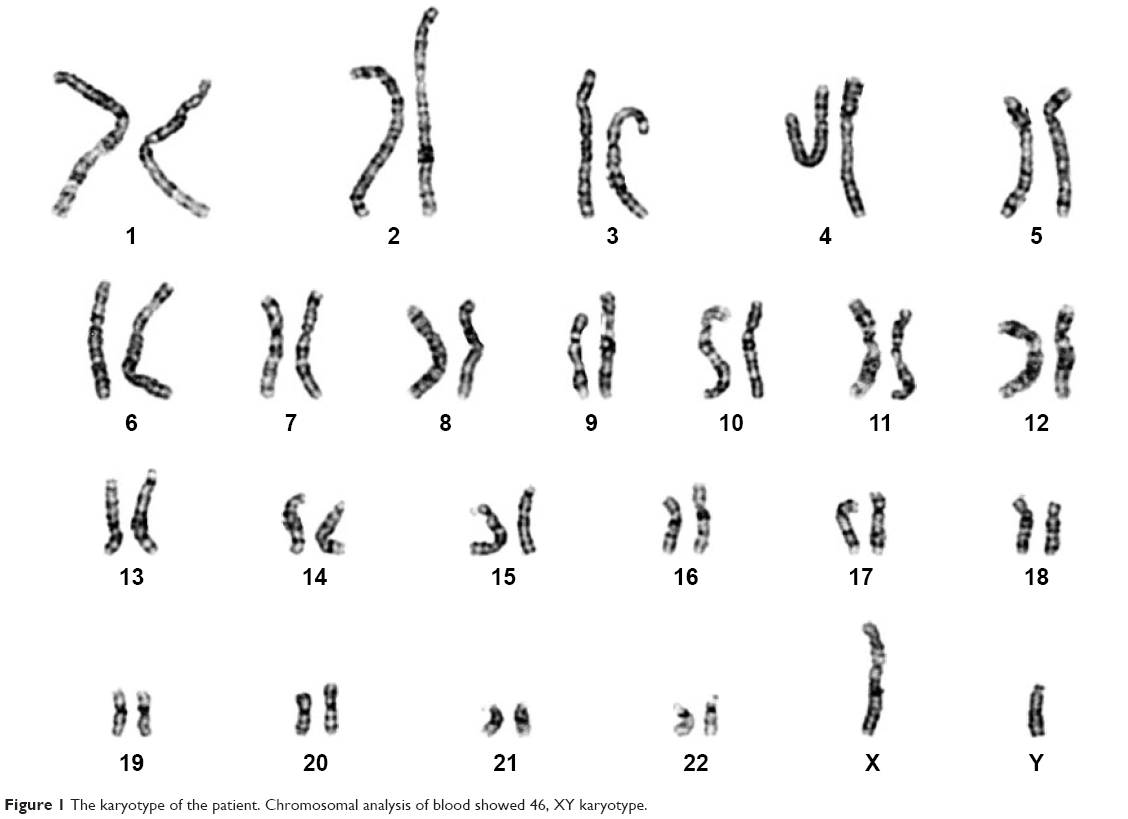 | Figure 1 The karyotype of the patient. Chromosomal analysis of blood showed 46, XY karyotype. |
After almost 1 year since the last chemotherapy was performed, the patient was referred to our hospital because of vaginal bleeding and abdominal distension. A whole-body tomography-computed tomography (PET-CT) scan revealed multiple metastases in the abdominal wall and pelvic cavity, and on the surface of the liver and intestine. All tumor markers including AFP were within normal ranges. Consultant pathology reports revealed YST in the first surgery, but SMs derived from YST for the second surgery.
Owing to the different pathology reports and continuous vaginal bleeding, we performed laparoscopic exploration and examination under anesthesia. Intraoperative findings included the following: 1,500 mL bloody ascitic fluid, multiple gray solid tumor nodules on the omentum majus and liver surface, and an approximately 3 cm bleeding nodule on the anterior abdominal wall (Figure 2A and B). There was a bleeding neoplasm in the vagina, and after its resection, ulcer tissues could be seen at the deep end of the vagina. After exploratory surgery, the platelet level gradually declined for an unknown reason and decreased to 30×109/L on the third day after the surgery. The patient presented with symptoms of hemorrhagic shock, and we had to perform emergency exploratory laparotomy on that day owing to acute intraperitoneal hemorrhage. The operation (right streak gonad resection, omentectomy, and tumor resection) was performed after obtaining informed consent from the patient. The primary purpose of the second surgery in our hospital was hemostasis, and it was impossible to perform satisfactory tumor reduction at that time (remaining tumor >1 cm).
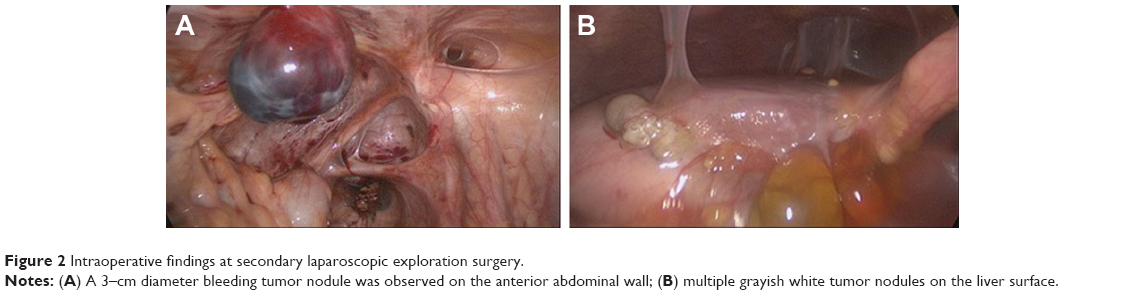 | Figure 2 Intraoperative findings at secondary laparoscopic exploration surgery. |
The pathology report confirmed SMs derived from YST, and the specific histological type of SM was sarcoma. Histopathological analysis of liver metastatic tumors revealed a lacunar, microcystic, or reticular pattern. Irregular or fusiform tumor cells were variably sized with severe nuclear atypia. Mitotic figures were observed. Pleomorphic tumor giant cells could be seen and had a scattered distribution. The stroma was edematous with multifocal hemorrhage (Figure 3A and B). IHC staining showed that tumor cells were focally positive for AE1/AE3 (Figure 3C), but negative for GPC3 and Sal-like protein 4 (SALL4) (Figure 3D and E), and the Ki-67 labeling index was approximately 10% (Figure 3F). Histopathological analysis of the vaginal tumor revealed the same pattern (Figure 4A). IHC staining showed that tumor cells were negative for AE1/AE3, GPC3, and SALL4 (Figure 4B–D). Histopathological analysis from the right gonad sections revealed fibrous tissue with no germ cells, which indicated PGD (Figure 5A), and IHC staining was negative for octamer-binding transcription factor 3/4 (OCT3/4) (Figure 5B). All micrographs were taken using an Olympus Camera U-TV0.5×C.
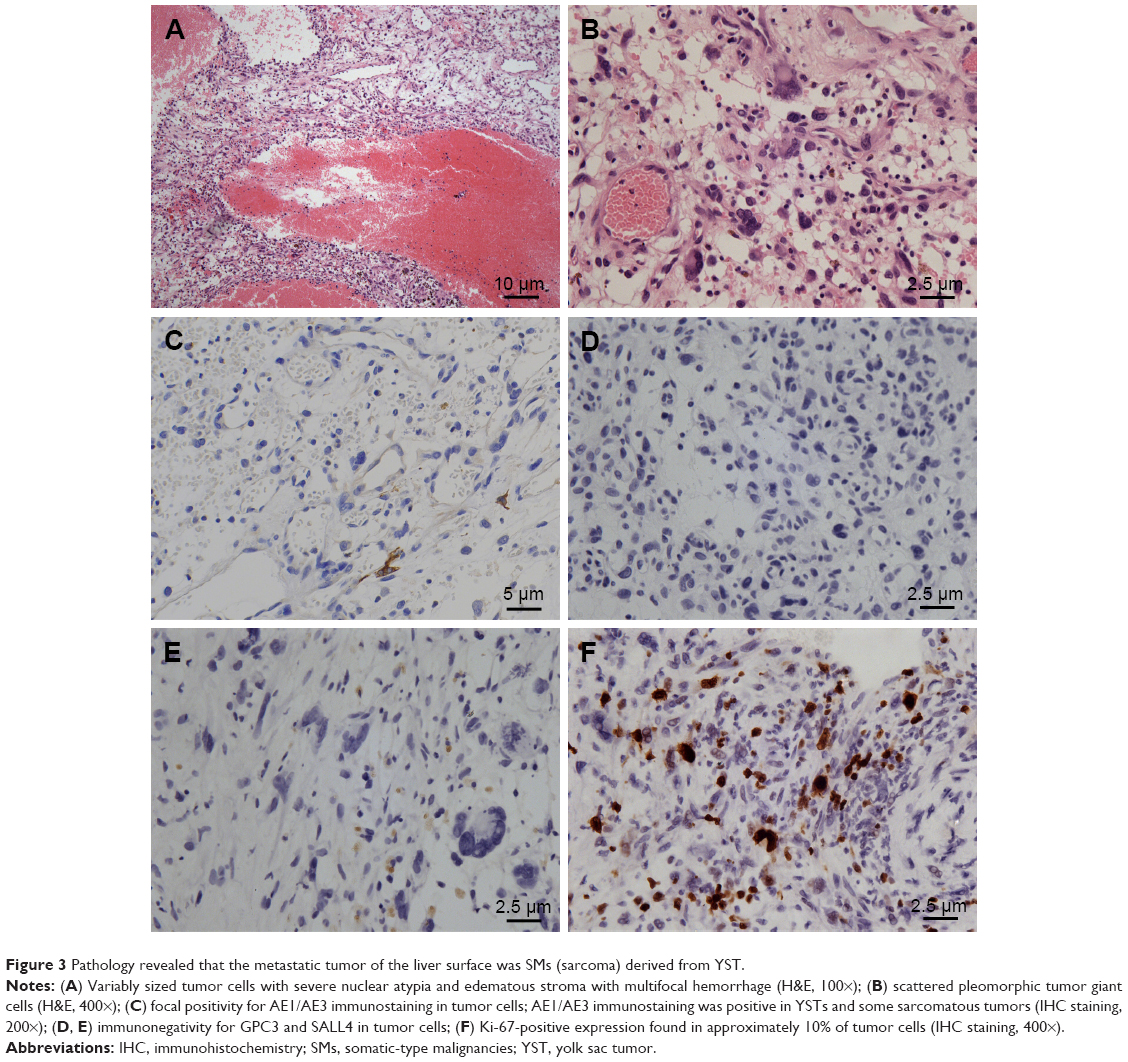 | Figure 3 Pathology revealed that the metastatic tumor of the liver surface was SMs (sarcoma) derived from YST. |
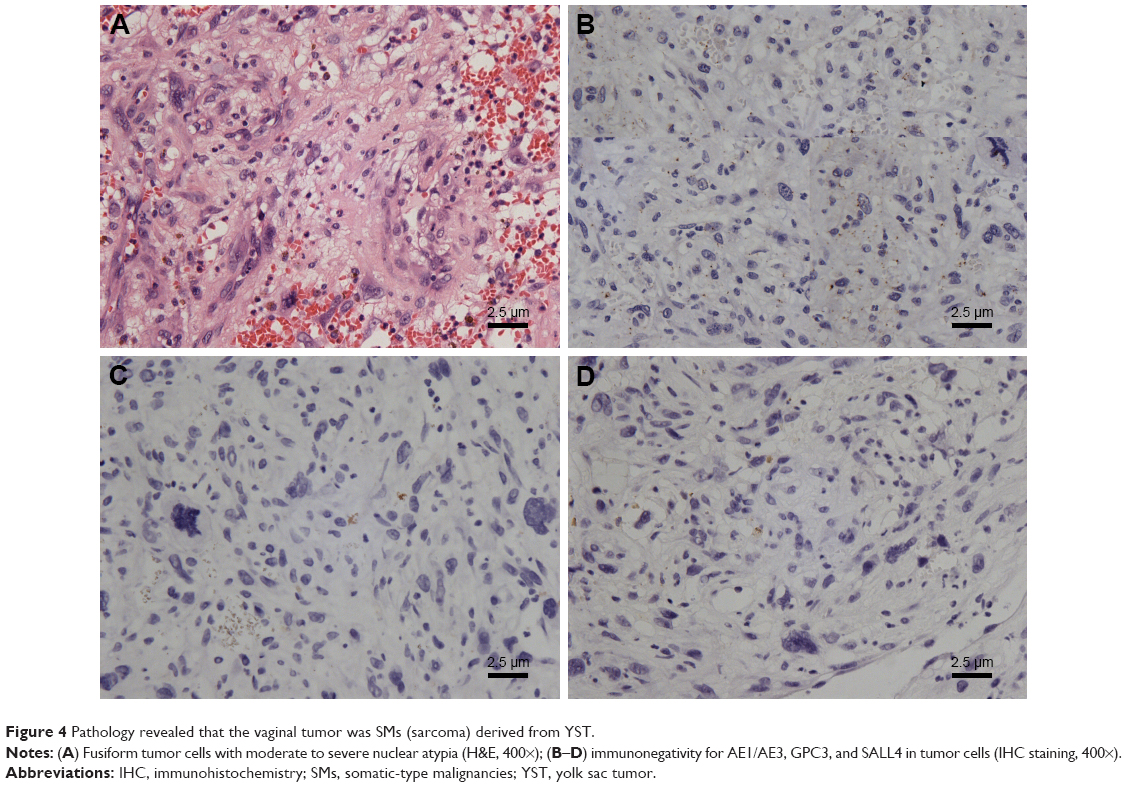 | Figure 4 Pathology revealed that the vaginal tumor was SMs (sarcoma) derived from YST. |
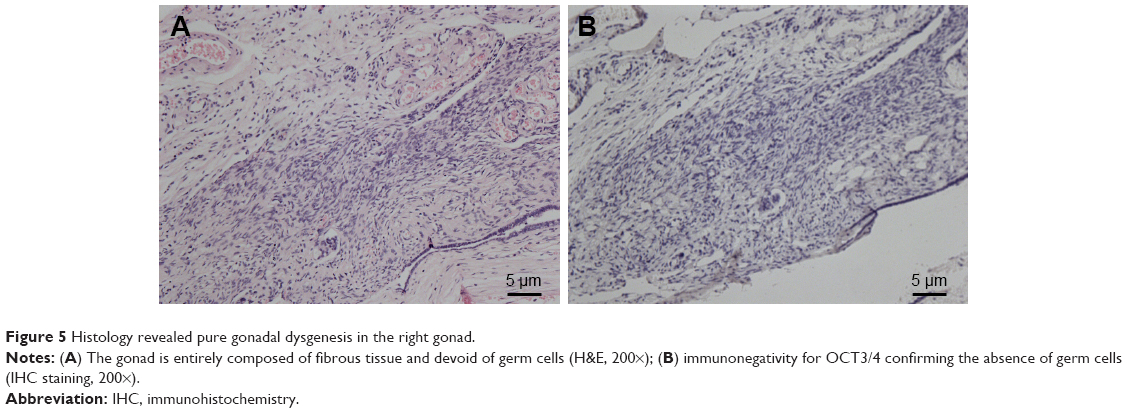 | Figure 5 Histology revealed pure gonadal dysgenesis in the right gonad. |
Considering the sarcomatous components of the tumor, we performed three cycles of PEI (cisplatin, etoposide and ifosfamide) chemotherapy. However, pelvic ultrasound revealed that the pelvic residual lesions did not reduce and vaginal bleeding was still observed, while all the tumor markers were within normal ranges. We also performed whole exome sequencing, and the germline mutation results demonstrated copy-number variations of FGF9 gene (chr13:21948488-25914325)×1 and frameshift mutation of MAP3K1 gene c.2822_2827delCAACAA (p.Thr941_943del); somatic gene mutation of samples from the primary gonadal tumor and metastatic tumors revealed a non-synonymous mutation of KRAS (c.182A>G). There was no indication for currently targeted drugs.
After communicating with the patient, she refused to undergo further treatment. At the time of this writing this report, the patient has been alive with the disease 7 months after the final PEI chemotherapy was administered.
Discussion
This rare case is worth reporting for the following reasons. First, the key lesson we learned from it is the importance of early diagnosis of 46, XY PGD and the necessity of undergoing prophylactic gonadectomy as soon as possible. However, this can be difficult because of the presence of normal female external genitalia. Most patients are diagnosed only when they present with primary amenorrhea.
SRY, MAP3K1, and FGF9 genes have been identified as the causative genes in 46, XY PGD.24 Although the mechanism of tumorigenesis in 46, XY PGD is still unclear, studies have found that tumor development is associated with TSPY gene expression and certain gene mutations, such as SRY, SOX9, and WT1.2,4,7,8 In this report, germline gene mutation demonstrated copy-number variations of the FGF9 gene and frameshift mutation of the MAP3K1 gene, which may explain why this patient had the diagnosis of 46, XY PGD. Previous studies have reported that the risk of GCTs in patients with 46, XY PGD was estimated to be 15%–35%,1 and a recent study in our hospital showed the incidence to be 23.33% (21/90).25 According to this patient’s medical history and the intraoperative observation, her left gonad had developed into a malignant tumor, and the primary tumor was confined without gross metastasis. However, with regard to genes associated with gonadal tumorigenesis, the molecular genetic testing revealed that the only pathogenic driver gene mutation was in KRAS.
The gonadoblastoma is the most common tumor in 46, XY PGD, and YST is rare and has usually been reported as a coexistent tumor with other GCTs in such patients.25–29 In this case, the gonadal tumor in the first surgery was YST without other GCT or SM elements, and more specifically, it transformed into SMs during the disease process. To our knowledge, this is the first report of SMs derived from YST in 46, XY PGD. SMs of GCTs are mainly reported in testicular GCTs. In 1984, Ulbright et al first summarized 11 cases of non-germ cell malignancies within GCTs, and these somatic malignancies were considered to arise from teratomatous elements.14 However, more recent studies have found that not all patients with SMs have teratoma; YST and malignant progenitor cells have also been proposed as possible origins of SMs.16,20,21,23 Howitt et al summarized 22 cases of SYSTs from 14 patients, of which at least four patients lacked teratoma in primary testicular masses.23 Kum et al performed interphase fluorescence in situ hybridization (FISH) to analyze chromosome 12p overexpression and isochromosome 12p (i12p) in 27 pairs of teratoma and SMs in metastatic lesions, and the results demonstrated that 78% (21/27) of pairs showed the same pattern, which indicated that GCTs and SMs may develop from the same progenitor cell.30 In this case, we performed the molecular genetic testing for samples of the primary tumor and the metastatic tumors, and the results revealed the same non-synonymous KRAS gene mutation. Based on this result, we propose that, in this patient, the YST and the SMs are clonally related and may have the same origin. During the clinical course of tumor progression, the YST gradually developed into the SMs, and the specific histological type of the SMs for this patient was sarcoma. Its morphology observed under a microscope showed irregular and fusiform tumor cells with severe nuclear atypia, and both AE1/AE3 and GPC3 staining results were negative. Above all, according to the genetic testing results, morphological and IHC features, and the patient’s medical history, once again, this case indicated that SMs such as sarcoma could indeed be derived from YST.
Sarcomatoid transformation has a low incidence and mostly appears in metastases after chemotherapy, especially in late recurrences.31,32 Magers et al summarized 124 cases of SMs of GCTs, and they believed that the final pathological diagnosis of SMs was based on morphological and IHC features, instead of serum tumor markers.21 In fact, many patients with SMs derived from YSTs have normal AFP levels.21,23,33 One assumption is that the parietal cells of the yolk sac are chemosensitive and can produce AFP, and these cells are killed during the chemotherapy, while the remaining mesenchymal cells acquire genetic changes and gradually develop into tumors.18,20 However, we also need to mention that some SMs were found to be coexistent with primary GCTs before any treatment. For example, Rice et al summarized that 26.45% (32/121) of patients were found to have SMs at the first occurrence of GCTs.22 That is, chemotherapy is definitely not an essential condition for sarcomatous transformation. The pathogenesis of SMs needs to be further investigated. In this case, sarcoma metastasis, which is characterized as AFP negative, was diagnosed after chemotherapy. It developed immediately after or perhaps during the chemotherapy, not in late recurrence, which is contrary to the previous literature reports.
SMs of GCTs have the following characteristics. They are mainly chemoresistant and have worse prognosis than GCTs.21,23 Tumor grade is an important prognostic factor in sarcomatoid transformation.22 There have been no reports about SMs in recurrent OGCTs. Here, we propose that some refractory or recurrent OGCTs may transform into SMs, which are resistant to former chemotherapy such as PEB or PVB therapy. Regarding chemorefractory disease, surgery with complete resection seems to be the only way to achieve complete remission.21,22 In the present case, it was impossible to perform radical surgery, especially for the liver metastases. However, this salvage surgery prolonged the patient’s survival. Three cycles of PEI chemotherapy after the surgery showed little effect, as the remaining tumors did not reduce, but all the serum tumor markers were within normal ranges.
Ras genes are the most common target of somatic mutations in human cancers, of which KRAS is the most frequently mutated isoform.34 The signaling pathway upstream of KRAS comprises a series of receptor tyrosine kinases including the epidermal growth factor receptor (EGFR), and the downstream signal pathways include the Ras-Raf-MEK-EFK (MAPK) pathway and PI3K-AKT-mTOR pathway.35,36 With regard to therapeutic strategies, first, Ras activation could reduce the clinical efficacy of EGFR inhibitors;37 that is, mutant KRAS is not suitable for EGFR inhibitors. Secondly, there are no currently available drugs that directly target KRAS. Thirdly, regarding the inhibitors targeting the downstream signaling pathway players, such as MEK inhibitors or ERK inhibitors, on the one hand, there are no available clinical trials about these drugs being administered for GCTs and, on the other hand, the results of most current clinical trials on these inhibitors have not strongly revealed improved outcomes for patients with colorectal, pancreatic, or other tumors.37–39 Considering this information, we did not recommend any targeted drugs for this patient.
In the case of this patient with a rare disease, several discussions took place in a multidisciplinary team (MDT). The MDT involves a multifaceted team of health care professionals such as gynecological pathologists and gynecological oncologists. Each case of rare disease can receive an individualized and optimized diagnosis and treatment strategy through the MDT model.
In summary, we report a unique case of gonadal YST transformed into SMs in a patient with 46, XY PGD. Prophylactic gonadectomy is highly recommended for this patient. Apart from teratoma, YST can be one of the origins of SMs of GCTs. Regarding treatment, owing to the chemoresistant characteristics of the tumor, radical surgery is the recommended treatment. Targeted therapy may be considered, but drug selection must be based on gene sequencing outcomes, and its efficacy needs to be verified by further study.
Consent
Written informed consent was obtained from the patient for publishing this case report and any accompanying images. The consent was given in the standardized informed consent form of the participating institution. The project and consent process were approved by the Ethics Committee of Peking Union Medical College Hospital, Beijing.
Acknowledgments
This work was supported by the Chinese Academy of Medical Sciences Initiative for Innovative Medicine (CAMS-2017-12M-1-002). We are grateful for the help provided by the Scientific Precision Company in the interpretation of genetic testing.
Disclosure
The authors report no conflicts of interest in this work.
References
Michala L, Goswami D, Creighton SM, Conway GS. Swyer syndrome: presentation and outcomes. BJOG. 2008;115(6):737–741. doi:10.1111/j.1471-0528.2008.01703.x | ||
King TF, Conway GS. Swyer syndrome. Curr Opin Endocrinol Diabetes Obes. 2014;21(6):504–510. doi:10.1097/MED.0000000000000113 | ||
Liu AX, Shi HY, Cai ZJ, et al. Increased risk of gonadal malignancy and prophylactic gonadectomy: a study of 102 phenotypic female patients with Y chromosome or Y-derived sequences. Hum Reprod. 2014;29(7):1413–1419. doi:10.1093/humrep/deu109 | ||
Cools M, Wolffenbuttel KP, Drop SL, Oosterhuis JW, Looijenga LH. Gonadal development and tumor formation at the crossroads of male and female sex determination. Sex Dev. 2011;5(4):167–180. doi:10.1159/000329477 | ||
McCann-Crosby B, Mansouri R, Dietrich JE, et al. State of the art review in gonadal dysgenesis: challenges in diagnosis and management. Int J Pediatr Endocrinol. 2014;2014(1):4. doi:10.1186/1687-9856-2014-4 | ||
Pauls K, Franke FE, Buttner R, Zhou H. Gonadoblastoma: evidence for a stepwise progression to dysgerminoma in a dysgenetic ovary. Virchows Arch. 2005;447(3):603–609. doi:10.1007/s00428-005-1272-9 | ||
Hersmus R, Stoop H, van de Geijn GJ, et al. Prevalence of c-KIT mutations in gonadoblastoma and dysgerminomas of patients with disorders of sex development (DSD) and ovarian dysgerminomas. PLoS One. 2012;7(8):e43952. doi:10.1371/journal.pone.0043952 | ||
Lau YF, Li Y, Kido T. Gonadoblastoma locus and the TSPY gene on the human Y chromosome. Birth Defects Res C Embryo Today. 2009;87(1):114–122. doi:10.1002/bdrc.20144 | ||
Tsuchiya K, Reijo R, Page DC, Disteche CM. Gonadoblastoma: molecular definition of the susceptibility region on the Y chromosome. Am J Hum Genet. 1995;57(6):1400–1407. | ||
Brown J, Friedlander M, Backes FJ, et al. Gynecologic Cancer Intergroup (GCIG) consensus review for ovarian germ cell tumors. Int J Gynecol Cancer. 2014;24(9 Suppl 3):S48–S54. doi:10.1097/IGC.0000000000000223 | ||
de la Motte Rouge T, Pautier P, Genestie C, et al. Prognostic significance of an early decline in serum alpha-fetoprotein during chemotherapy for ovarian yolk sac tumors. Gynecol Oncol. 2016;142(3):452–457. doi:10.1016/j.ygyno.2016.07.005 | ||
Zhao Q, Yang J, Cao D, et al. Tailored therapy and long-term surveillance of malignant germ cell tumors in the female genital system: 10-year experience. J Gynecol Oncol. 2016;27(3):e26. doi:10.3802/jgo.2016.27.e26 | ||
Motzer RJ, Mazumdar M, Sheinfeld J, et al. Sequential dose-intensive paclitaxel, ifosfamide, carboplatin, and etoposide salvage therapy for germ cell tumor patients. J Clin Oncol. 2000;18(6):1173–1180. doi:10.1200/JCO.2000.18.6.1173 | ||
Ulbright TM, Loehrer PJ, Roth LM, Einhorn LH, Williams SD, Clark SA. The development of non-germ cell malignancies within germ cell tumors. A clinicopathologic study of 11 cases. Cancer. 1984;54(9):1824–1833. | ||
Lutke Holzik MF, Hoekstra HJ, Mulder NH, Suurmeijer AJ, Sleijfer DT, Gietema JA. Non-germ cell malignancy in residual or recurrent mass after chemotherapy for nonseminomatous testicular germ cell tumor. Ann Surg Oncol. 2003;10(2):131–135. | ||
Malagon HD, Valdez AM, Moran CA, Suster S. Germ cell tumors with sarcomatous components: a clinicopathologic and immunohistochemical study of 46 cases. Am J Surg Pathol. 2007;31(9):1356–1362. doi:10.1097/PAS.0b013e318033c7c4 | ||
Mikuz G, Colecchia M. Teratoma with somatic-type malignant components of the testis. A review and an update. Virchows Arch. 2012;461(1):27–32. doi:10.1007/s00428-012-1251-x | ||
Michael H, Ulbright TM, Brodhecker CA. The pluripotential nature of the mesenchyme-like component of yolk sac tumor. Arch Pathol Lab Med. 1989;113(10):1115–1119. | ||
Ulbright TM, Michael H, Loehrer PJ, Donohue JP. Spindle cell tumors resected from male patients with germ cell tumors. A clinicopathologic study of 14 cases. Cancer. 1990;65(1):148–156. | ||
Nogales FF, Preda O, Nicolae A. Yolk sac tumours revisited. A review of their many faces and names. Histopathology. 2012;60(7):1023–1033. doi:10.1111/j.1365-2559.2011.03889.x | ||
Magers MJ, Kao CS, Cole CD, et al. “Somatic-type” malignancies arising from testicular germ cell tumors: a clinicopathologic study of 124 cases with emphasis on glandular tumors supporting frequent yolk sac tumor origin. Am J Surg Pathol. 2014;38(10):1396–1409. doi:10.1097/PAS.0000000000000262 | ||
Rice KR, Magers MJ, Beck SD, et al. Management of germ cell tumors with somatic type malignancy: pathological features, prognostic factors and survival outcomes. J Urol. 2014;192(5):1403–1409. doi:10.1016/j.juro.2014.05.118 | ||
Howitt BE, Magers MJ, Rice KR, Cole CD, Ulbright TM. Many postchemotherapy sarcomatous tumors in patients with testicular germ cell tumors are sarcomatoid yolk sac tumors: a study of 33 cases. Am J Surg Pathol. 2015;39(2):251–259. doi:10.1097/PAS.0000000000000322 | ||
Ono M, Harley VR. Disorders of sex development: new genes, new concepts. Nat Rev Endocrinol. 2013;9(2):79–91. doi:10.1038/nrendo.2012.235 | ||
Huang H, Wang C, Tian Q. Gonadal tumour risk in 292 phenotypic female patients with disorders of sex development containing Y chromosome or Y-derived sequence. Clin Endocrinol (Oxf). 2017;86(4):621–627. doi:10.1111/cen.13255 | ||
Morsy AH, al-Fadly A, Mokhtar S, el-Aasar EM, Farag TI. Swyer syndrome: an unusual presentation. Int J Gynaecol Obstet. 1995;49(2):185–186. | ||
Madiwale CV, Fernandes GC, Pandit AA, Kane SV. Gonadoblastoma with distinctly unusual pattern of yolk sac tumour overgrowth. J Postgrad Med. 2003;49(2):175–176. | ||
Mathur RV, Kumar S, Aparicio S, Johnson PW, Newstead CG. Haemolytic uraemic syndrome following chemotherapy for an unusual germ-cell tumour. Nephrol Dial Transplant. 1999;14(7):1786–1788. | ||
Zhu HL, Bao DM, Wang Y, Shen DH, Li Y, Cui H. Swyer’s syndrome with mixed ovarian malignant germ cell tumor and ovarian gonadoblastoma. Chin Med J (Engl). 2016;129(14):1752–1754. doi:10.4103/0366-6999.185864 | ||
Kum JB, Ulbright TM, Williamson SR, et al. Molecular genetic evidence supporting the origin of somatic-type malignancy and teratoma from the same progenitor cell. Am J Surg Pathol. 2012;36(12):1849–1856. doi:10.1097/PAS.0b013e31826df1ab | ||
Little JS Jr, Foster RS, Ulbright TM, Donohue JP. Unusual neoplasms detected in testis cancer patients undergoing post-chemotherapy retroperitoneal lymphadenectomy. J Urol. 1994;152(4):1144–1149. | ||
Michael H, Lucia J, Foster RS, Ulbright TM. The pathology of late recurrence of testicular germ cell tumors. Am J Surg Pathol. 2000;24(2):257–273. | ||
Guo CC, Punar M, Contreras AL, et al. Testicular germ cell tumors with sarcomatous components: an analysis of 33 cases. Am J Surg Pathol. 2009;33(8):1173–1178. doi:10.1097/PAS.0b013e3181adb9d7 | ||
Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72(10):2457–2467. doi:10.1158/0008-5472.CAN-11-2612 | ||
McCormick F. K-Ras protein as a drug target. J Mol Med (Berl). 2016;94(3):253–258. doi:10.1007/s00109-016-1382-7 | ||
Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25(3):272–281. doi:10.1016/j.ccr.2014.02.017 | ||
Knickelbein K, Zhang L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. 2015;2(1):4–12. doi:10.1016/j.gendis.2014.10.002 | ||
Van Cutsem E, Hidalgo M, Canon JL, et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int J Cancer. 2018;143(8):2053–2064. doi:10.1002/ijc.31603 | ||
Zimmer L, Barlesi F, Martinez-Garcia M, et al. Phase I expansion and pharmacodynamic study of the oral MEK inhibitor RO4987655 (CH4987655) in selected patients with advanced cancer with RAS-RAF mutations. Clin Cancer Res. 2014;20(16):4251–4261. doi:10.1158/1078-0432.CCR-14-0341 |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.